

Abstract
Long-chain fatty acids amplify insulin secretion from the pancreatic beta cell. The G protein-coupled receptor GPR40 is specifically expressed in beta cells and is activated by fatty acids. Loss of function of GPR40 was shown to markedly inhibit fatty-acid stimulation of insulin secretion in vitro. However, the role of GPR40 in acute regulation of insulin secretion in vivo remains unclear. To this aim, we generated GPR40 knock-out (KO) mice and examined glucose homeostasis, insulin secretion in response to glucose and Intralipid in vivo, and insulin secretion in vitro after short- and long-term exposure to fatty acids. Our results show that GPR40 KO mice have essentially normal glucose tolerance and insulin secretion in response to glucose. Insulin secretion in response to Intralipid was reduced by approximately 50%. In isolated islets, insulin secretion in response to glucose and other secretagogues was unaltered, but fatty-acid potentiation of insulin release was markedly reduced. Islets from GPR40 KO mice were as sensitive to fatty-acid inhibition of insulin secretion upon prolonged exposure as islets from wild-type animals. We conclude that GPR40 contributes approximately half of the full insulin secretory response to fatty acids in mice, but does not play a role in the mechanisms of lipotoxicity.
Long-chain fatty acids are essential regulators of normal pancreatic beta-cell function, and are likely to play a role in the pathogenesis of beta-cell dysfunction in type 2 diabetes (reviewed in (1)). Under normal circumstances, fatty acids do not initiate insulin release, but amplify glucose-stimulated insulin secretion (GSIS) (2–5). Fatty-acid potentiation of insulin secretion has physiological implications, particularly after a period of fasting (6). Until recently, the prevailing model postulated that the effects of fatty acids on the beta cell were mediated by their intracellular metabolism and the generation of lipid derived signals which, in turn, potentiate GSIS (2; 7). According to this hypothesis, fatty acids are transported across the plasma membrane and activated into their long-chain coenzyme A esters, which in turn modulate a number of intracellular targets that influence insulin secretion. Moreover, evidence suggests that intracellular fatty-acid metabolism is a key component of both nutrient- and non-nutrient-induced insulin secretion (7). In contrast to their acute, stimulatory effect on GSIS, prolonged exposure to elevated levels of fatty acids impairs beta-cell function, a phenomenon referred to as lipotoxicity (reviewed in (1)). The mechanisms of lipotoxicity remain poorly understood but have been proposed to also involve intracellular metabolism of fatty acids and the generation of lipid-derived metabolites (8).
The models described above have been challenged by the observation that fatty acids activate the G-protein coupled receptor (GPCR) GPR40 (9–11), also referred to as the free fatty-acid 1 receptor (FFA1R) (12; 13). GPR40 belongs to a class of GPCR with high structural conservation, of which GPR41 and GPR43 (FFA2R) are activated by short-chain fatty acids (14; 15). GPR40 is highly expressed in pancreatic beta cells and insulin-secreting cell lines (9–11), but not in any other tissues tested except the ileum (9) and some areas of the brain (10). Using a ligand-fishing strategy, it was shown that fatty acids increase intracellular calcium concentrations ([Ca2+]i) in GPR40-expressing cells. A wide range of saturated fatty acids from C6 to C20, unsaturated fatty acids from C18 to C22, and some ecosanoids, elicited a calcium response, with an EC50 in the range of 1–100 μM. In general, fatty acids that activated GPR40 in transfected cells also induced an increase in [Ca2+]i and amplified GSIS in the MIN6 pancreatic beta-cell line. A physiological role for GPR40 is suggested by the observations that loss-of-function of GPR40 via small interfering RNA (9; 16), antisense oligonucleotides (13), pharmacological inhibition (17), or gene deletion in the mouse (18) suppresses fatty-acid potentiation of GSIS in vitro. However, whether GPR40 signaling is important for stimulation of insulin secretion by fatty acids in vivo remains unknown. Using mice with targeted deletion of GPR40, the present study was aimed to examine the role of GPR40 in insulin secretion in vivo and in vitro as well as its implication in the mechanisms of lipotoxicity.
Research Design and Methods
Generation and genotyping of GPR40−/− animals
GPR40−/− (KO) mice on a mixed C57Bl/6 /129 background were generated by homologous recombination in embryonic stem cells at Lexicon Genetics Incorporated (The Woodlands, TX). Exon 2 of the GPR40 gene was replaced with a LacZ gene (Figure 1A). Pups were screened by PCR of genomic DNA as shown in Figure 1B. Wild-type (WT) littermates were used as controls. For experiments shown in Figure 4C, 8–10-week old male C57Bl/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed on a 12-h light/dark cycle with free access to water and standard laboratory chow. All procedures using animals were approved by the Institutional Animal Care and Use Committee.
Figure 1. Generation of GPR40 KO mice.
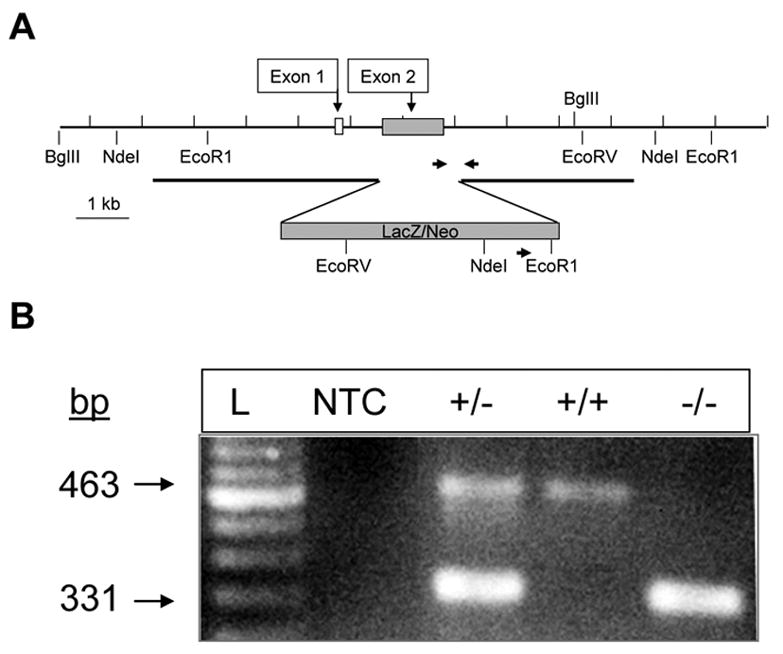
Structure of the targeting vector (A) and representative PCR analysis of tail DNA (B). Bp: base pairs; L: molecular weight marker; NTC: no-template control.
Figure 4. Insulin response to intravenous glucose and Intralipid.
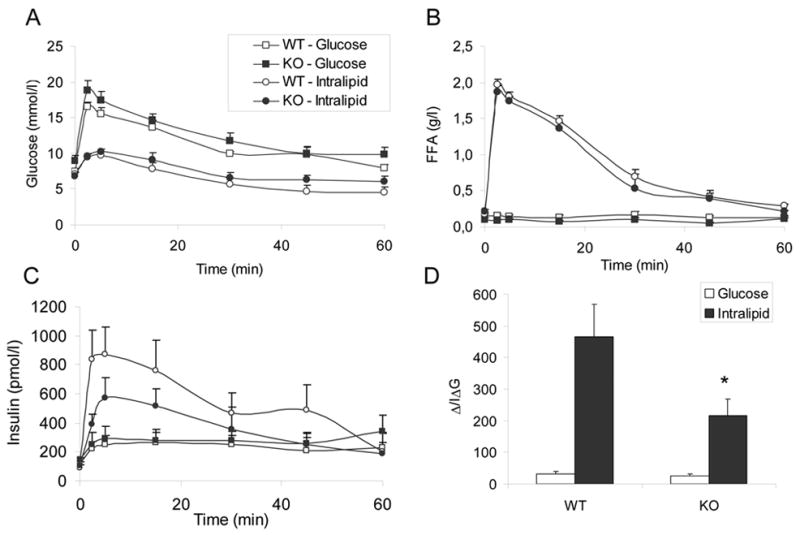
Glucose (0.5g/kg) or Intralipid (100 μL of a 20% solution preceded by 20U of heparin) was administered to overnight-fasted, 13-week old male mice by intravenous injection at time 0. Plasma glucose (A), FFA (B), and insulin (C) levels were measured at times 0, 2.5, 5, 15, 30, 45, and 60 min. The insulinogenic indexes of glucose and Intralipid (D) were calculated as the ratio between the increment in secreted insulin from 0 to 30 min and the increment in plasma glucose during the same period. N=5–7 animals per group. *: P<0.05.
Assessment of glucose homeostasis
A cohort of male and female KO and WT mice were studied longitudinally from 6 to 24 weeks of age. Weight and fasting blood glucose were measured weekly. Oral glucose tolerance was assessed at 6 and 13 weeks of age in overnight-fasted animals by measuring tail blood glucose 0, 15, 30, 60, 90, and 120 minutes after oral administration of 1g/kg glucose by gavage. Insulin tolerance was measured at 8 and 14 weeks of age in overnight-fasted animals by measuring tail blood glucose 0, 30, 60, and 120 minutes after intraperitoneal injection of 0.75 U/kg insulin. Fasting blood samples were taken at 8, 14, and 24 weeks of age for plasma insulin and free fatty acid (FFA) determinations. At 24 weeks of age the animals were sacrificed and their pancreas removed for histological examination. Insulin secretion in response to intravenous glucose or Intralipid administration was examined on a second group of 13-week old animals. Mice were anesthetized by intraperitoneal injection of pentobarbital (75 mg/kg), and a catheter was inserted into the right jugular vein. Thirty minutes after the catheterization, a bolus of glucose (0.5 g/kg) or Intralipid (Fresenius Kabi AB, Uppsula, Sweden; 100 μL of a 20% solution preceded by 20 U of heparin) was injected into the jugular catheter. Blood samples were drawn 0, 2.5, 5, 15, 30, 45, 60 min after the injection. Red blood cells were re-suspended with sterile saline containing 5U/ml heparin and re-injected. Plasma glucose was measured using a portable meter (Accucheck Advantage, Roche, Manheim, Germany). Plasma FFA were measured using the Half-Micro test (Roche). Plasma insulin was measured using the Ultra Sensitive Rat Insulin ELISA Kit (Crystal Chem, Downers Grove, IL) with mouse insulin standards.
Measurements of beta-cell surface area
Pancreata were fixed in 4% buffered paraformaldehyde and embedded in paraffin. Sections were rehydrated, blocked, and immunostained with guinea pig anti-insulin IgG (Linco Research, St Chalres, MO) followed by donkey anti–guinea pig IgG-alkaline phosphatase (Jackson Immunoresearch, West Grove, PA). Following development with Vector Red substrate (Vector Laboratories, Burlingame, CA), sections were counterstained with hematoxylin, cleared, and mounted in Permount (Fisher Scientific, Worcester, MA). The proportion of islet beta-cell surface area versus surface area of the whole pancreas was determined as described (19).
Islet isolation and culture
Islets were isolated from 8–10-week old animals by collagenase digestion and handpicking under a stereomicroscope as described (20). For experiments shown in Figure 5, islets were cultured for 1 h in RPMI 1640 containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 11.1 mmol/l glucose prior to assessment of insulin secretion. For experiments shown in Figure 6, islets were cultured overnight in the medium described above, then suspended in fresh media and incubated in various experimental conditions as described in Results. Preparation of culture media containing fatty acids was as described (21). The final molar ratio of fatty acid:bovine serum albumin (BSA) was 5:1. All control conditions contained the same amount of BSA and vehicle (EtOH:H2O; 1:1) as those with fatty acids.
Figure 5. Insulin secretion in freshly isolated islets.
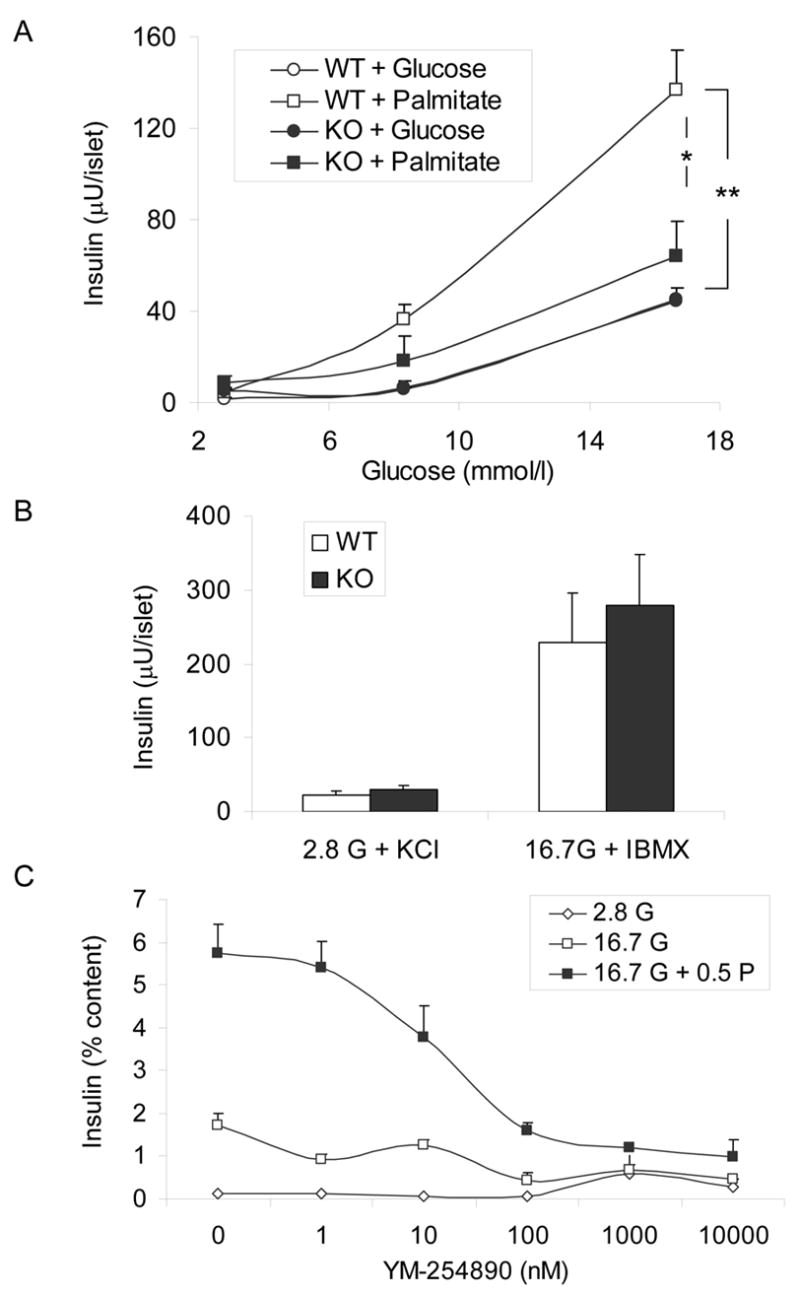
A: Insulin secretion was assessed in 1-h static incubations in the presence of 2.8, 8.3, or 16.7 mmol/l glucose with or without 0.5 mmol/l palmitate. N=2–10 replicate experiments per condition. *: P<0.05; **: P<0.001. B: Insulin secretion was assessed in 1-h static incubations in the presence of 2.8 mmol/l glucose + 40 mmol/l KCl or 16.7 mmol/l glucose + 0.1 mmol/l IBMX. N=4 replicate experiments. C: Insulin secretion was assessed in 1-h static incubations in the presence of 2.8 mmol/l glucose, 16.7 mmol/l glucose, or 16.7 mmol/l glucose + 0.5 mmol/l palmitate, with increasing concentrations of YM-254890. Data are expressed as the percentage of secreted insulin / insulin content. N=2–9 replicate experiments per condition.
Figure 6. Effects of prolonged exposure to fatty acids on insulin secretion.
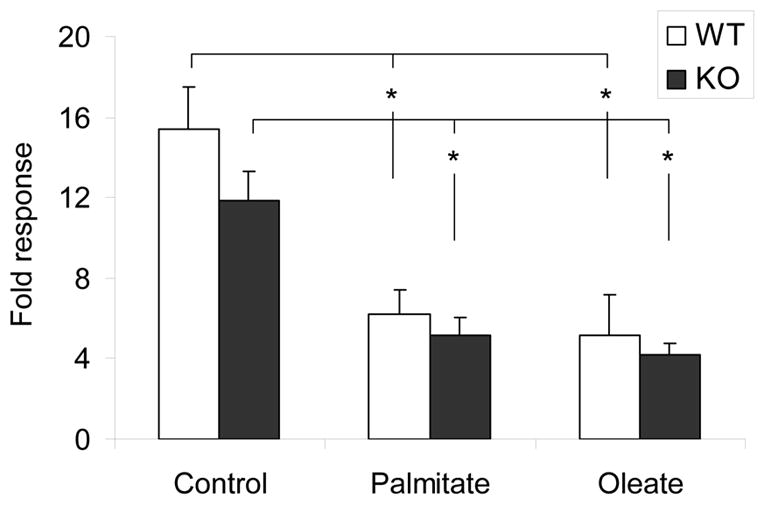
Isolated islets were cultured for 72 h in the presence of 16.7 mmol/l glucose with or without 0.5 mmol/l palmitate or oleate. Insulin secretion was assessed in 1-h static incubations. Data are expressed as the ratio of insulin secreted at 16.7 mmol/l glucose / insulin secreted at 2.8 mmol/l glucose (fold response). N = 7 (WT) and 8 (KO) replicate experiments. *: P<0.01.
Insulin secretion and insulin content in isolated islets
lnsulin secretion was assessed in 1-h static incubations as described (20). Briefly, batches of 10 islets each were washed twice in Krebs-Ringer Buffer (KRB) containing 0.1% BSA and 2.8 mmol/l glucose for 20 min at 37°C, then incubated for 1 h in the presence of various secretagogues as described in Results. Each condition was run in triplicate. Intracellular insulin content was measured after acid-alcohol extraction. Insulin was measured by radioimmunoassay (Linco Research Inc., St. Charles, MO).
Expression of data and statistics
Data are expressed as mean ± SE. Intergroup comparisons were performed by ANOVA followed by two-by-two comparisons with post-hoc adjustments, or Student’s paired t-test, where appropriate. P<0.05 was considered significant.
Results
Phenotypic characterization of GPR40 KO mice
GPR40−/− animals had no apparent growth defect, clinical signs, or morphological abnormalities. As shown in Figure 2, deletion of GPR40 had no significant effect on weight (Figure 2A&B), fasting blood glucose (Figure 2C&D), plasma FFA levels (Figure 2E&F), or insulinemia (Figure 2G&H) in either males or females up to 24 weeks of age. Glucose clearance curves after oral glucose administration were not different between KO and WT mice at 6 weeks of age (Figure 3A&B). Male KO animals developed a slight but significant glucose intolerance at 13 weeks of age (area under the glucose curve: 25900 ± 1000 vs. 21800 ± 400 arbitrary units in KO vs. WT, respectively; n=6–8; P<0.01; Figure 3C). Glucose tolerance remained identical in 13-week old KO and WT female mice (Figure 3D). Intraperitoneal insulin tolerance tests performed at 14 weeks of age did not reveal any differences between WT and KO in either male (Figure 3E) or female (Figure 3F) mice. Beta-cell surface area estimated at 24 weeks of age was not significantly different between KO and WT animals in either males (0.73 ± 0.14 % vs. 0.80 ± 0.11 %; n=6, NS) or females (0.60 ± 0.04 % vs. 0.88 ± 0.15 %; n=6, NS).
Figure 2. Metabolic characterization of GPR40 KO mice.
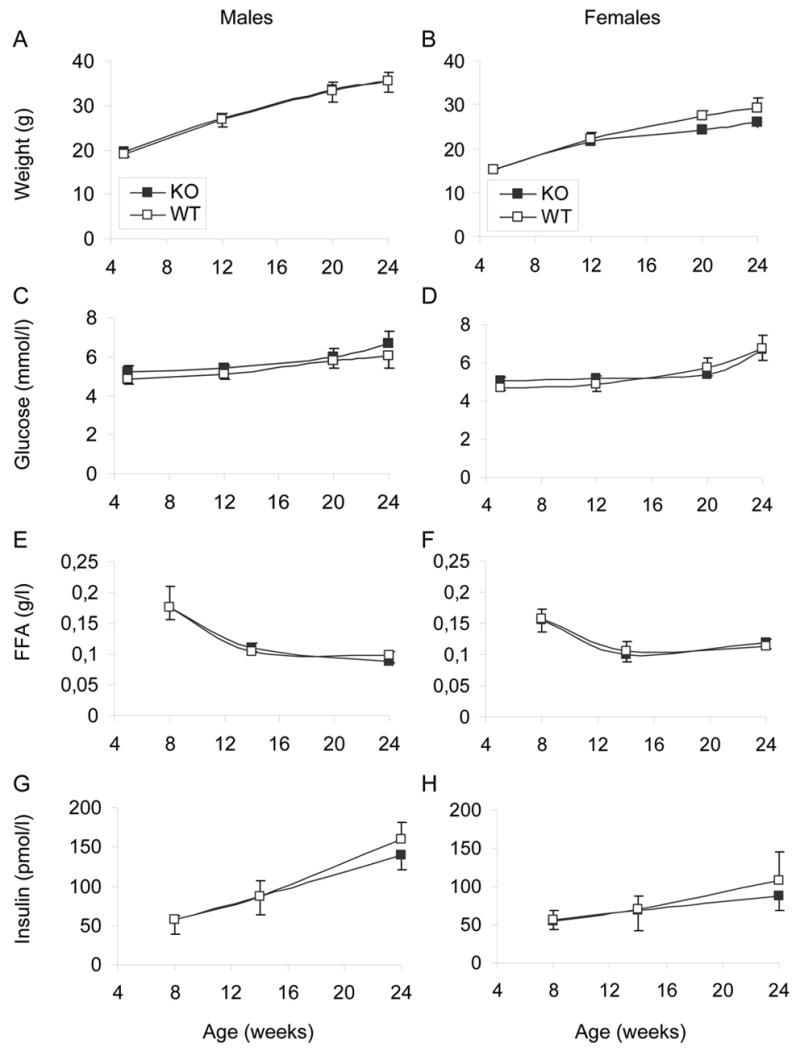
A cohort of male (A, C, E, G) and female (B, D, F, H) GPR40 KO and WT littermates was followed from 5 to 24 weeks of age. Weight (A, B), Fasting blood glucose (C, D), plasma free-fatty acids (FFA) (E, F), and plasma insulin levels (G, H) were measured throughout the study. N= 6–8 animals per group.
Figure 3. Oral glucose tolerance and intraperitoneal insulin tolerance.
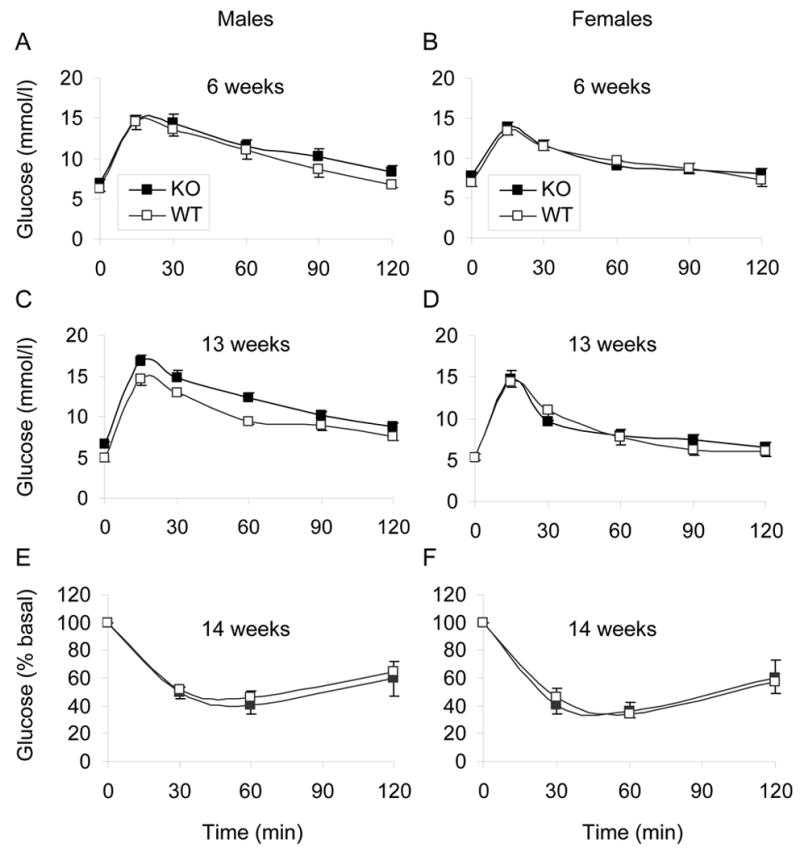
Glucose (1g/kg) was administered orally to overnight-fasted male (A, C) and female (B, D)) mice at 6 (A, B) and 13 (C, D) weeks of age at time 0. Plasma glucose was measured at 0, 15, 30, 60, 90, and 120 min. N= 6–8 animals per group. Insulin (0.75 U/kg) was administered to overnight-fasted, 14-week old male (E) and female (F) mice by intraperitoneal injection at time 0. Plasma glucose was measured at times 0, 30, 60, and 120 min. Data are expressed as percentage of plasma glucose measured at time 0. N=6–8 animals per group.
Insulin secretion in response to glucose and Intralipid in vivo
To examine the effect of GPR40 deletion on insulin secretion in vivo, 13-week old male KO mice and their WT littermates were subjected to an intravenous injection of glucose or Intralipid + heparin (Figure 4). Both glucose (Figure 4A) and FFA levels (Figure 4B) were similar in WT and KO animals during the tests. Glucose clearance curves after an intravenous bolus of glucose was not different in KO vs. WT mice, contrary to the slight glucose intolerance observed after oral glucose administration in mice of the same age (Figure 3C). Insulin secretion in response to glucose was similar in KO and WT animals (Figure 4C). In contrast, insulin secretion in response to Intralipid + heparin was markedly reduced, although not completely abolished, in KO mice (Figure 4C). The insulinogenic index of glucose (ΔI/ΔGglucose), calculated as the ratio between the increment in secreted insulin from 0 to 30 min and the increment in blood glucose during the same period, was not significantly different in KO vs. WT animals (24.4 ± 7.8 vs. 31.6 ± 7.2 respectively; n=7; NS; Figure 4D). In contrast, the insulinogenic index of Intralipid + heparin (ΔI/ΔGIntralipid) was significantly lower in KO than in WT mice (216.6 ± 51.9 vs. 466.1 ± 101.5 respectively; n=6; P<0.05; Figure 4D). Since ΔI/ΔGglucose was similar in both groups, ΔI/ΔGIntralipid represents an estimation of the potentiating effect of Intralipid on insulin secretion, normalized for the confounding effect of variations in blood glucose levels following Intralipid injection. Since ΔI/ΔGIntralipid in KO mice is reduced by approximately 50% as compared to WT mice, we conclude that GPR40 account for approximately half of the potentiating effect of fatty acids on insulin secretion in vivo.
Insulin secretion in response to glucose and fatty acids from isolated islets in vitro
To further explore the effects of GPR40 deletion on insulin secretion in vitro, islets were isolated from 8–10 week-old KO mice and WT littermates and insulin secretion was assessed in 1-h static incubations in response to 2.8, 8.3, or 16.7 mmol/l glucose with or without 0.5 mmol/l palmitate. As shown in Figure 5A, GSIS was similar in KO and WT islets (ANOVA; effect of genotype: NS; n=2–10 per condition). The presence of palmitate in the incubation buffer potentiated GSIS from WT islets at both 8.3 (ANOVA; P<0.01) and 16.7 (ANOVA; P<0.001) mmol/l glucose. In contrast, palmitate did not significantly increase GSIS from KO islets at either 8.3 or 16.7 mmol/l glucose (ANOVA; both NS). In the presence of 16.7 mmol/l glucose and 0.5 mmol/l palmitate, insulin secretion was significantly lower in KO islets than in WT islets (ANOVA; P<0.05). Insulin secretion in response to a depolarizing concentration (40 mmol/l) of potassium chloride was not different between KO and WT islets (n=4; NS; Figure 5B), nor was potentiation of GSIS in the presence of 100 μM isobutyl-methyl xanthine (n=4; NS; Figure 5B). Insulin content was not different between KO and WT islets (2.67 ± 0.21 (n=4) vs. 2.50 ± 0.16 (n=6) mU/islet; NS). These results indicate that deletion of GPR40 specifically impairs the ability of palmitate to potentiate GSIS, whereas insulin secretion in response to glucose, membrane depolarization, or amplification of GSIS by cyclic AMP are not affected.
Based on the lack of sensitivity of GPR40 signaling to pertussis toxin, Itoh et al. (9) and Briscoe et al. (10) have proposed that it might be coupled to the Gαq/11 subunit of heterotrimeric G-proteins. If this is the case, inhibiting Gαq/11 should block fatty-acid potentiation of GSIS. To test this possibility, we used a selective inhibitor of Gαq/11, YM-254890 (Astellas Pharma Inc., Ibaraki, Japan), which blocks the exchange of GDP for GTP at the activation step (22) and was recently shown to inhibit palmitate potentiation of GSIS in INS-1 cells (16). As shown in Figure 5C, YM-254890 dose-dependently inhibited palmitate potentiation of GSIS (ANOVA, P<0.005) in 1-h static incubations of freshly isolated islets from C57Bl/6 male mice without affecting neither GSIS itself (ANOVA; NS) nor basal insulin secretion (ANOVA; NS), at concentrations up to 100 nmol/L. At concentrations >100 nmol/l, the increase in basal insulin release, although not statistically significant by ANOVA, likely reflects non-specific or toxic effects of the inhibitor. The pattern of inhibition of insulin secretion observed with YM-254890 is similar to the effect observed in islets from KO mice (Figure 5A), and suggests that fatty-acid potentiation of GSIS is, at least in part, mediated by GPR40 coupling through Gαq/11.
Islets isolated from GPR40−/− mice are not protected against lipotoxicity
To determine whether deletion of GPR40 protects islets from lipotoxicity, islets were isolated from 8–10 week-old male WT and KO mice and cultured for 72 h in the presence of 16.7 mmol/l glucose with or without 0.5 mmol/l palmitate or oleate, after which insulin secretion in response to 2.8 or 16.7 mmol/l glucose was assessed in 1-h static incubations (Figure 6). As expected, a 72-h exposure to palmitate or oleate inhibited subsequent GSIS in islets from WT animals (ratio of insulin secreted at 16.7 mmol/l glucose /2.8 mmol/l glucose = 15.4 ± 2.1 in control islets vs. 6.2 ± 1.29 in palmitate-cultured islets (n=7; P<0.01) and 5.1 ± 2.1 in oleate-cultured islets (n=7; P<0.01)). Islets from KO animals were similarly affected by both palmitate (5.2 ± 0.9 fold increase vs. 11.9 ± 1.4 fold increase in control islets; n=8; P<0.01) and oleate (4.2 ± 0.6 fold increase vs. 11.9 ± 1.4 fold increase in control islets; n=8; P<0.01). These results indicate that deletion of GPR40 does not protect mouse islets from fatty-acid inhibition of insulin secretion upon prolonged exposure in vitro.
Discussion
The objectives of this study were to investigate the roles of GPR40 in glucose homeostasis and in the regulation of insulin secretion by fatty acids in vivo and in vitro. We observed that deletion of the GPR40 gene in the mouse does not affect normal development and growth (Figure 2), and has minimal effects on glucose homeostasis (Figures 2&3). Although male mice developed oral glucose intolerance at 13 weeks of age (Figure 3C), glucose clearance curves following intravenous glucose administration were not altered in mice of the same age (Figure 4A). Since GPR40 is expressed in the ileum (9), it is conceivable that differences between oral and intravenous glucose tolerance in KO mice may be related to altered intestinal glucose absorption. The essentially normal glucose homeostasis in GPR40 KO mice is consistent with the results of Steneberg et al. (18), who observed normal glucose tolerance after intraperitoneal glucose administration in GPR40 KO mice.
Infusion of Intralipid + heparin in WT mice elicited a marked increase in insulin secretion, as previously reported in dogs (23; 24) and humans (25). Estimation of the potentiating effect of Intralipid by calculating ΔI/ΔGIntralipid indicates that the latter is approximately 15 times greater than ΔI/ΔGGlucose and suggests that Intralipid is a much more potent secretagogue than glucose under our experimental conditions in vivo. In contrast to their normal response to intravenous glucose, GPR40 KO mice had impaired insulin secretion in response to Intralipid (Figure 4C), consistent with a role for GPR40 as a physiologically relevant fatty-acid receptor. However, insulin secretion in response to Intralipid was not completely abolished. In fact, calculation of the insulinogenic indexes of glucose and Intralipid allowed us to estimate the contribution of GPR40 to approximately 50% of the insulin response to Intralipid (Figure 4D). Although this might be artificially due to incomplete deletion of the gene, this possibility is unlikely because GPR40 mRNA was not expressed in islets from GPR40 KO mice (data not shown). Considering the potent stimulation of insulin secretion by Intralipid, it is surprising that a 50% reduction in this response in GPR40 KO mice does not appear to significantly affect glucose homeostasis. This suggests that compensatory mechanisms are sufficient to maintain adequate secretion in response to a lipid load. In fact, the limited response to a lipid load could have beneficial consequences, by dampening the hyperinsulinemia associated with high-fat feeding and therefore the resulting hepatic steatosis and insulin resistance, as shown by Steneberg et al. (18). We hypothesize that the remaining, non GPR40-mediated, insulin secretory response to Intralipid is due to intracellular metabolism of fatty acids, although this remains to be directly examined. Although long-chain fatty-acids can bind to and activate the GPR120 receptor, the residual effect of Intralipid on insulin secretion is unlikely to be mediated via GPR120 since this receptor is not expressed in beta cells (26).
The results of our experiments using islets isolated from GPR40 KO mice are consistent with our in vivo results and further demonstrate that deletion of GPR40 results in a decreased response to fatty acids whereas the responses to glucose and other secretagogues remain normal (Figure 5). Since beta-cell surface area, insulin content, and the response to glucose were unaltered in GPR40 islets, we conclude that the secretory defect is specific for fatty-acid potentiation of insulin secretion. At intermediate glucose levels (8.3 mM), fatty-acid potentiation of insulin secretion was diminished by approximately 50% in islets from GPR40 KO mice (Figure 5A). This degree of inhibition is similar to that observed in vivo (Figure 4C) at a plasma glucose concentration of approximately 10 mM (Figure 4A). Interestingly, insulin secretion in response to palmitate in isolated islets was reduced to a greater extent (approximately 80%) at 16.7 mM glucose than at 8.3 mM glucose (Figure 5A), suggesting that the relative contribution of GPR40- vs. non GPR40-mediated mechanisms to the overall response may vary with the prevailing glucose concentration. Clearly, important differences must be taken into account when comparing in vivo and in vitro situations. First, Intralipid is a soybean oil emulsion which generates a mixture of fatty acids, mostly unsaturated (27), in contrast to the single saturated fatty acid (palmitate) used in in vitro experiments. Second, extrapancreatic (e.g. neuronal (28)) factors contribute to fatty acid stimulation of insulin secretion in vivo but not in isolated islets. These limitations notwhistanding, our results obtained in vivo and in isolated islets in vitro consistently suggest that at intermediate glucose levels, GPR40 contributes to approximately half of the insulin secretory response to fatty acids.
The marked inhibition of palmitate potentiation of GSIS by the inhibitor YM-254890 indicates that it is mediated, at least in part, by signaling through Gαq/11 in mouse islets. If, which has yet to be demonstrated, GPR40 functions as a classical GPCR, one might speculate that binding of fatty acids to GPR40 activates Gαq/11 which in turns stimulates phospholipase C-mediated hydrolysis of phosphatidylinositol 4,5-biphosphate into diacylglycerol and inositol trisphosphate, leading to the respective activation of protein kinase C and calcium mobilization from the endoplasmic reticulum, similar to the mechanisms underlying cholinergic stimulation of insulin release (29). Such hypothesis is supported by the recent observation that fatty-acid induced calcium influx in beta-cells is inhibited by RNA interference against GPR40 (30), and that linoleic acid stimulates phosphatidyl inositol hydrolysis in MIN6 cells (13). In contrast, a previous study failed to detect an increase in inositol triphosphate content in response to palmitate in rat islets (4), and the signaling pathways activated by fatty acids via GPR40 remain to be directly identified.
The results of our experiments examining insulin secretion in vitro and in vivo in response to fatty acids and Intralipid, respectively, in GPR40 KO mice reconcile two apparently conflicting hypothesis regarding the mechanisms of action of fatty acids on insulin release. On the one hand, there is considerable experimental evidence to support the notion that intracellular metabolism of fatty acids is essential for their stimulation of insulin secretion (reviewed in (8)). For instance, reduction of intracellular malonyl-CoA levels by inhibition acyl-CoA synthase with Triacsin C or overexpression of a cytosolic form of malonyl CoA decarboxylase inhibited fatty acid potentiation of insulin secretion in isolated islets (7). On the other hand, near-complete abolition of fatty-acid stimulation of insulin secretion by siRNAs against GPR40 (9) or in isolated islets from GPR40 KO mice (18) suggested a prominent role for a receptor-mediated mode of action of fatty acids on the beta cell. The results of our study demonstrate that, under physiological conditions of stimulation in vivo, the role of GPR40 is important but only partial, indicating that both receptor- and non receptor-mediated effects play a role in the mechanism of action of fatty acids on the beta cell. Under our experimental conditions, each of these mechanism appear to contribute for approximately half of the full response, although their relative contribution may depend on the prevailing glucose concentrations.
Contrary to the defect observed in insulin secretion in response to acute stimulation by fatty acids, islets from GPR40−/− animals were found to be as sensitive to the prolonged, deleterious effects of fatty acids as islets from WT mice. This suggests that GPR40 is not involved in the mechanisms of lipotoxicity, consistent with the large amount of experimental evidence supporting a role for intracellular fatty-acid metabolism in this phenomenon (reviewed in (8)). These findings are in contrast with those of Steneberg et al. (18), who concluded that deletion of GPR40 protects mouse islets from lipotoxicity. The reasons for this discrepancy are unknown, but might be related to differences in fatty-acid concentrations and time of exposure between the two studies.
In conclusion, our results uniquely demonstrate that targeted deletion of GPR40 reduces insulin secretion in response to fatty acids in vivo and in vitro without affecting the response to glucose. Under our experimental conditions, we estimate that GPR40 contributes about half of the full insulin response to fatty acids in the mouse. Our observations indicate that GPR40 plays a role in the regulation of insulin secretion by fatty acids but does not mediate lipotoxicity, thus reinforcing the concept that it might represent a valuable therapeutic target to enhance insulin secretion in type 2 diabetes.
Acknowledgments
This work was supported by the National Institutes of Health (R21-DK70598 to V.P.). V.P. was the recipient of the 2003 Thomas R. Lee Career Development Award from the American Diabetes Association and holds the Canada Research Chair in Diabetes and Pancreatic Beta-cell Function. We thank Drs. M. Taniguchi and J. Takasaki from Astellas Pharma Inc. for generously providing YM-254890, and James Lausier and Violet Roskens for their invaluable technical expertise.
References
- 1.McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42:128–138. doi: 10.1007/s001250051130. [DOI] [PubMed] [Google Scholar]
- 2.Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267:5802–5810. [PubMed] [Google Scholar]
- 3.Warnotte C, Gilon P, Nenquin M, Henquin JC. Mechanisms of the stimulation of insulin release by saturated fatty acids. A study of palmitate effects in mouse beta-cells. Diabetes. 1994;43:703–711. doi: 10.2337/diab.43.5.703. [DOI] [PubMed] [Google Scholar]
- 4.Alcazar O, Qiu-yue Z, Giné E, Tamarit-Rodriguez J. Stimulation of islet protein kinase C translocation by palmitate requires metabolism of the fatty acid. Diabetes. 1997;46:1153–1158. doi: 10.2337/diab.46.7.1153. [DOI] [PubMed] [Google Scholar]
- 5.Parker SM, Moore PC, Johnson LM, Poitout V. Palmitate potentiation of glucose-induced insulin release: a study using 2-bromopalmitate. Metabolism. 2003;52:1367–1371. doi: 10.1016/s0026-0495(03)00279-8. [DOI] [PubMed] [Google Scholar]
- 6.Stein DT, Esser V, Stevenson BE, Lane KE, Whiteside JH, Daniels MB, Chen S, McGarry JD. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest. 1996;97:2728–2735. doi: 10.1172/JCI118727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roduit R, Nolan C, Alarcon C, Moore P, Barbeau A, Delghingaro-Augusto V, Przybykowski E, Morin J, Masse F, Massie B, Ruderman N, Rhodes C, Poitout V, Prentki M. A Role for the Malonyl-CoA/Long-Chain Acyl-CoA Pathway of Lipid Signaling in the Regulation of Insulin Secretion in Response to Both Fuel and Nonfuel Stimuli. Diabetes. 2004;53:1007–1019. doi: 10.2337/diabetes.53.4.1007. [DOI] [PubMed] [Google Scholar]
- 8.Poitout V. Lipid partitioning in the pancreatic beta-cell: physiologic and pathophysiologic implications. Curr Opin Endocrinol Diabetes. 2002;9:152–159. [Google Scholar]
- 9.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 10.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 11.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun. 2003;301:406–410. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 12.Kotarsky K, Nilsson NE, Olde B, Owman C. Progress in methodology. Improved reporter gene assays used to identify ligands acting on orphan seven-transmembrane receptors. Pharmacol Toxicol. 2003;93:249–258. doi: 10.1111/j.1600-0773.2003.pto930601.x. [DOI] [PubMed] [Google Scholar]
- 13.Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C, Olde B. Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res. 2005;322:207–215. doi: 10.1007/s00441-005-0017-z. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun. 2003;303:1047–1052. doi: 10.1016/s0006-291x(03)00488-1. [DOI] [PubMed] [Google Scholar]
- 15.Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun. 2005;335:97–104. doi: 10.1016/j.bbrc.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 17.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, Fornwald JA, Ignar DM, Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metabolism. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Jetton TL, Lausier J, LaRock K, Trotman WE, Larmie B, Habibovic A, Peshavaria M, Leahy JL. Mechanisms of compensatory beta-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes. 2005;54:2294–2304. doi: 10.2337/diabetes.54.8.2294. [DOI] [PubMed] [Google Scholar]
- 20.Poitout V, Rouault C, Guerre-Millo M, Briaud I, Reach G. Inhibition of insulin secretion by leptin in normal rodent islets of Langerhans. Endocrinology. 1998;139:822–826. doi: 10.1210/endo.139.3.5812. [DOI] [PubMed] [Google Scholar]
- 21.Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 23.Crespin SR, Greenough WB, 3rd, Steinberg D. Stimulation of insulin secretion by infusion of free fatty acids. J Clin Invest. 1969;48:1934–1943. doi: 10.1172/JCI106160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madison LL, Seyffert WA, Jr, Unger RH, Barker B. Effect on plasma free fatty acids on plasma glucagon and serum insulin concentrations. Metabolism. 1968;17:301–304. doi: 10.1016/0026-0495(68)90097-8. [DOI] [PubMed] [Google Scholar]
- 25.Boden G, Chen X. Effects of fatty acids and ketone bodies on basal insulin secretion in type 2 diabetes. Diabetes. 1999;48:577–583. doi: 10.2337/diabetes.48.3.577. [DOI] [PubMed] [Google Scholar]
- 26.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 27.Stein DT, Stevenson BE, Chester MW, Basit M, Daniels MB, Turley SD, McGarry JD. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest. 1997;100:398–403. doi: 10.1172/JCI119546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnan C, Collins S, Berthault MF, Kassis N, Vincent M, Gilbert M, Penicaud L, Ktorza A, Assimacopoulos-Jeannet F. Lipid infusion lowers sympathetic nervous activity and leads to increased beta-cell responsiveness to glucose. J Clin Invest. 1999;103:413–419. doi: 10.1172/JCI3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604. doi: 10.1210/edrv.22.5.0440. [DOI] [PubMed] [Google Scholar]
- 30.Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet {beta}-cells: mediation by phospholipase C- and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab. 2005:00035–02005. doi: 10.1152/ajpendo.00035.2005. [DOI] [PubMed] [Google Scholar]