

Abstract
Pathological agents such as ionizing radiation and oxidative free radicals can cause breaks in both strands of the DNA at a given site (double-strand break). This is the most serious type of DNA damage because neither strand is able to provide physical integrity or information content, as would be the case for single-strand DNA damage where one strand of the duplex remains intact. The repair of such breaks usually results in an irreversible alteration of the DNA. Two physiological forms of intentional double-strand (ds) DNA breakage and rejoining occur during lymphoid differentiation. One is V(D)J recombination occurring during early B and T cell development, and the other is class switch recombination, occurring exclusively in mature B cells. The manner in which physiological and most pathological double-strand DNA breaks are rejoined to restore chromosomal integrity are the same. Defects during the phases in which pathological or physiological breaks are generated or in which they are joined can result in chromosomal translocations or loss of genetic information at the site of breakage. Such events are the first step in some cancers and may be a key contributor to changes in DNA with age. Inherited defects in this process can result in severe combined immune deficiency. Hence, pathological and physiological DNA double-strand breaks are related to immune defects and cancer and may be one of the key ways in which DNA is damaged during aging.
Among the macromolecules that compose our cells, irreversible damage to DNA has the greatest probability of having pathological consequences. Damage to the DNA can come from radiation, endogenous DNA enzymes, mistakes during replication, and chemicals (both exogenous and endogenous) (Table 1) ▶ . One can broadly classify the types of DNA repair as those dedicated to repair of damage to a single DNA strand (single-strand break repair) or those dedicated to repair of damage where both DNA strands are broken (double-strand break repair) (Figure 1 ▶ , Table 2 ▶ ).
Table 1.
Major Sources of DNA Damage
| Sources of DNA damage | Nature of the damage |
|---|---|
| Radiation | |
| UV | ssb |
| Ionizing radiation (x-ray, γ-rays) | dsb and ssb |
| Chemicals | |
| Alkylating agents | ss damage |
| Bleomycin and oxidative free radicals | dsb and ssb |
| Replication errors | ss mismatches, deletions, and insertions |
| Endogenous DNA enzyme errors | |
| V(D)J recombination | ssb and dsb |
| Class switch recombination | dsb |
| Other enzymes | ssb and dsb |
ssb, single-strand break; dsb, double-strand break.
Figure 1.

Diagrams of the major categories of DNA damage. DNA can be damaged on one strand (single-strand damage) or both strands (double-strand damage). Damage due to single-strand breaks or alkylation events are handled by excision repair (nucleotide and base excision pathways). Mismatches from DNA replication are handled by the mismatch repair system. Double-strand breaks are repaired by either homologous recombination (HR) or nonhomologous DNA end joining (NHEJ). In HR, the information at b is copied from the intact chromosome such that the sequence information a-b-c is restored; this process requires at least a 2N (diploid) DNA content. In NHEJ, even though the cell is diploid, a is joined directly to c, leaving the information at b deleted from the chromosome.
Table 2.
Major Types of DNA Repair
| Type of DNA Repair |
|---|
| Excision repair |
| Base excision repair (BER) |
| Nucleotide excision repair (NER) |
| Mismatch repair (MMR) |
| Double-strand break repair |
| Homologous recombination (HR) |
| Nonhomologous DNA end joining (NHEJ) |
Single-strand DNA damage has the other anti-parallel strand to provide physical integrity and information content to direct the accurate repair of the defective strand. However, double-strand breaks are sites where the DNA has lost both physical integrity and information content on both strands. Such a cell will have permanently lost information on that particular chromosome, even if it manages to put the two DNA ends back together.
Single-celled eukaryotes, such as yeast, can use the other chromosome when they are in their diploid phase to copy the information by a process called homologous recombination (HR). However, unlike yeast, the genomes of higher eukaryotes have an abundance of repetitive DNA, with similar or identical repeats scattered over many different chromosomes. HR under such circumstances may be difficult because of the abundance of similar sequences throughout the genome.
Instead, cells of multicellular eukaryotes rely on a process where the two broken DNA ends are joined back together, even if information between the two broken ends is lost. This process is called nonhomologous DNA end joining (NHEJ) to distinguish it from HR.
Steps and Proteins Involved in the Joining of Broken DNA Ends
When double-strand breaks occur in DNA, the biochemical configuration of the broken ends can be any of a large number of possibilities. Hence, putting the two ends back together cannot usually be achieved by a simple ligation step.
For the two DNA ends to be processed so that they can be joined, they must be maintained in physical proximity (Figure 2) ▶ . The term synapsis can be used to describe this step. Concurrently, proteins may bind that signal that there is a double-strand break, a step that can be referred to as end activation. One of the first proteins to bind is Ku. Ku binds to DNA ends and can diffuse to internal positions from the end. It is not yet clear whether Ku is responsible for synapsis of the DNA ends, as some reports have suggested, 2,3 or whether additional proteins are involved.
Figure 2.

Model for the steps in nonhomologous DNA end joining. When a DNA break occurs, the ends must be held in proximity to permit subsequent repair steps to proceed and to align the two ends. This first step can be referred to as synapsis. Ku and DNA-dependent protein kinase (DNA-PK) bind to DNA ends during this initial phase, although it is not precisely clear how the synapsis occurs or what proteins specifically carry out this function. End activation can be used to refer to the signaling that recruits other repair components, configures the DNA termini for the subsequent steps, and alters the cell cycle until the repair is complete. End alignment can occur if there is terminal microhomology of one to four nucleotides, typically, between the two ends. This is an optional aspect, as NHEJ occurs regardless of microhomology. End processing refers to the removal of DNA by nucleases and the filling in of gaps by polymerases. Ligation is the final step, and it requires a ligatable nick on each strand. Some of the proteins known or suspected at each step are listed to the left of the arrows.
Next, a 470-kd protein called DNA-dependent protein kinase (DNA-PK) binds to the Ku:DNA complex. DNA-PK is the only known protein kinase that requires a double-strand DNA end as an essential cofactor. That is, DNA-PK is inactive except when there is a DNA double-strand break. Hence, DNA-PK is the ideal alarm system for the cell to signal that there is a double-strand break. DNA-PK can phosphorylate itself or Ku at serines or threonines. It is not yet clear what other physiological targets DNA-PK has. Current research is likely to soon define the dynamics of DNA-PK and Ku at broken DNA ends to determine how the steps of synapsis and end activation can progress to subsequent steps.
When the two broken DNA ends are brought into proximity, the exact point of joining can occur at any base pair. There is a propensity for the joining to occur at one to four nucleotides that are complementary between the two ends. Therefore, if the DNA end sequence at one end is —-TTGGT* (where the dashes represent the rest of the chromosome and the asterisk represents the break site) and the other end begins as *aggcc—, then the sequence at the point of joining has a higher probability of being —TTGGcc—-. This means that the T at one end and the a at the other end were removed, and the joining occurred at the two base pairs GG or gg, where the two ends shared these two nucleotides of microhomology. The reason for this preference at the junction is because this is where the two DNA ends can base pair. This tendency is called microhomology usage. It is not an essential feature, because two ends that do not share microhomology can still be joined at normal efficiencies. Rather, the microhomology usage is something that occurs at a much higher frequency than random positioning of the join site. This tendency for microhomology usage is seen in general DNA end joining and in specialized physiological forms of double-strand break repair, such as V(D)J recombination. 4
When the two DNA ends do undergo alignment at points of microhomology, then there is often either excess DNA beyond the point of alignment that must be removed by nucleases or there are gaps that must be filled by polymerases. We call this the DNA end processing step. The intriguing feature about this step is that the two DNA ends must be held in proximity to prevent them from diffusing away from one another, and yet the means by which those ends are held cannot prevent access by the nucleases and polymerases that must process the DNA termini. The nucleases and polymerases involved at this step are not yet defined. However, we have identified a structure-specific nuclease called FEN-1 (flap endoNuclease-1) that cleaves flap structures at DNA branch points. Such a nuclease may account for removal of the type of overhang shown in Figure 2 ▶ (X. Wu, T. Wilson, and M. Lieber, submitted). Regarding polymerases, we have some data suggesting that polymerase β accounts for some of the filling of gaps, but polymerases β, δ, and ɛ may all contribute (T. Wilson and M. Lieber, submitted).
The last step in restoring chromosomal integrity at a DNA break site is ligation of each of the two DNA strands. Recently, we determined that the DNA ligase responsible for repairing double-strand breaks is DNA ligase IV. In mammalian cells, DNA ligase IV exists in a complex with XRCC4. Without XRCC4, mammalian DNA ligase IV is unstable and has much lower activity.
In eukaryotic cells, absence of any of the components along this pathway results in an inability to repair some or most double-strand breaks caused by ionizing radiation and oxidative agents (such as bleomycin) and breaks caused by physiological processes such as V(D)J recombination and class switch recombination.
DNA Breakage and Rejoining Phases of V(D)J Recombination
All vertebrate animals have a specific immune system in addition to the nonspecific one that all animals possess. The central feature of the specific immune system is its ability to generate an enormous repertoire of antigen receptors by a process called V(D)J recombination. During this process, any one from an array of V (variable) gene segments can be joined to one of many D (diversity) or J (joining) gene segments to generate a new exon encoding the antigen-binding domains of immunoglobulins or T-cell receptors (Figure 3) ▶ . The V(D)J recombination process happens millions of times each day in the proliferating pool of lymphoid precursors of vertebrates. The cis elements mediating the site specificity of the recombination reaction are recombination signal sequences (RSSs) flanking the V, D, and J gene segments. They consist of a palindromic heptamer separated from an A/T-rich nonamer by a spacer of either a 12- or 23-bp length. 5 A single recombination event is directed by a pair of these joining signals with different spacer lengths, a restriction referred to as the 12/23-bp rule. The recombination process is initiated by two lymphoid-specific proteins that are encoded by the RAG1 and -2 genes. 6,7 Remarkable progress has been made recently in understanding the mechanism of V(D)J recombination.
Figure 3.

Summary of components and steps of V(D)J recombination. A single V and J are depicted, although combinatorial diversity is achieved using numerous V and J segments. A signal sequence is adjacent to each V or J coding segment. Within each signal, the nonamer of each signal is located furthest from its coding segment, whereas the heptamer is directly adjacent to the coding segment. Subsequent figures (4 to 6) ▶ ▶ ▶ provide more detail for all of the correspondingly numbered steps. Ku is the Ku70/86 heterodimer. TdT is not an essential component for V(D)J recombination but is a lymphoid-specific enzyme that is present in pre-B and pre-T cells during most of the period in which they carry out this process. The nucleases that remove excess DNA at the junction are still being defined, but FEN-1 is one candidate. The polymerases that fill in gaps at the junction are also still being defined, but polymerase β is one candidate. In the bottom line of the diagram, the coding joint has dashed lines on each side of the junction to depict nucleotide loss and addition, resulting in junctional diversity.
Cleavage Phase of V(D)J Recombination
The first cell-free V(D)J cutting activity has been described by van Gent and co-workers in the Gellert laboratory using nuclear extracts from lymphoid cells supplemented with a purified, recombinant core RAG1 protein. 8 They later showed that the RAG proteins alone were sufficient to catalyze DNA cleavage at RSSs (Figure 4) ▶ . 9 The reaction occurs by a two-step mechanism, during which one DNA strand is initially nicked at the heptamer of the RSS. The 3′OH group thus generated attacks the phosphodiester bond of the anti-parallel strand in a direct transesterification reaction 10 leaving hairpinned coding ends and blunt signal ends (with 5′P and 3′OH). This is reminiscent of the chemistry involved in nicking and strand transfer seen in reactions of transposable elements in prokaryotes and in retrotransposons (including retroviruses) in eukaryotes. 11 Nicking and hairpin formation at isolated signals occurs in a stable RAG-1:RAG-2 DNA complex that disassembles after completion of the reaction. 12,13
Figure 4.

Model for the nicking and hairpin formation phases of V(D)J recombination. Symbols are the same as Figure 3 ▶ except that the both strands of the DNA are depicted and the 12-signal is depicted with horizontal hatches and the 23-signal with vertical hatches. The RAG1,2 complex is depicted as a single gray oval. At the cleavage step, the signal ends are blunt and have a 5′ phosphate and 3′ hydroxyl (see Refs. 8-10 and citations therein). The coding ends after step 4 are hairpinned at their termini. The signals are depicted in synapsis in a parallel configuration. 15,71
Although it has been demonstrated that concerted cleavage at two synapsed RSSs in accordance with the 12/23-bp paradigm can be achieved with RAG-1 and -2 proteins alone, 14 the efficiency of the reaction appears to be greatly enhanced by other nuclear components, 15,16 including the DNA-bending protein HMG1. 16,17
There are still many remaining questions regarding the initiation of V(D)J recombination. First, is the 12/23 rule enforced at the nicking or hairpinning steps? Second, which regions of RAG-1 and RAG-2 carry out which aspects of the reaction, including binding to the signal, nicking, hairpinning, and 12/23 rule enforcement? Two studies employing either an indirect yeast-one hybrid system or surface plasmon resonance to measure DNA protein interaction suggest that a region in RAG-1 with homology to bacterial Hin recombinases is required for binding to the nonamer of the RSS. 18,19 Both studies did not detect DNA-binding activity for RAG-2, and the significance of the highly conserved heptamer sequence of the signals for interaction with RAGs remains unclear. Third, how does the nucleosome affect binding, nicking, and hairpinning? This is critical to understanding how chromatin structure affects V(D)J recombination. A crucial aspect of the regulation of V(D)J recombination is that some antigen receptor loci remain inaccessible whereas others are recombined. Although transcription itself may not control the accessibility for V(D)J recombination, 20-23 enhancers clearly do. 24-26 Enhancers may direct changes in nucleosome binding and positioning. They may also affect CpG methylation 27 and histone acetylation, which have major effects on determining accessibility. 28-30 Initial progress has been made at studying the action of exogenous RAG proteins on genomic DNA sites in isolated nuclei. 31,32 Cell-free V(D)J recombination using recombinant RAG proteins and nuclei prepared from cells representing different developmental stages indeed recapitulates the tight developmental regulation of antigen receptor rearrangements observed in vivo. Fourth, mutations in the RAG-1 and -2 genes have been shown to account for ∼14% of human SCID. 33 It will be interesting to see whether components in the joining phase of V(D)J recombination can account for SCID as well.
Rejoining Phase of V(D)J Recombination
After generation of the DNA ds break at RSS, hairpinned coding ends must be opened to permit their processing and joining (Figure 5) ▶ . Analysis of V(D)J recombination intermediates in scid mice has demonstrated that these mice are unable to resolve hairpinned coding end structures at their antigen receptor loci. 34 Scid cells display no DNA-dependent kinase activity, due to a carboxyl-terminal truncation of the 470-kd DNA-PK protein encoded by the scid gene. 35,36 One possibility is that DNA-PK is needed to phosphorylate the hairpin endonuclease to activate it for hairpin opening. Possible candidates for the hairpin endonuclease are the RAG proteins themselves, because they are the only known components required for V(D)J recombination that display nuclease activity. A requirement for RAGs for later stages of V(D)J recombination has been documented and will be discussed below. 37,38
Figure 5.
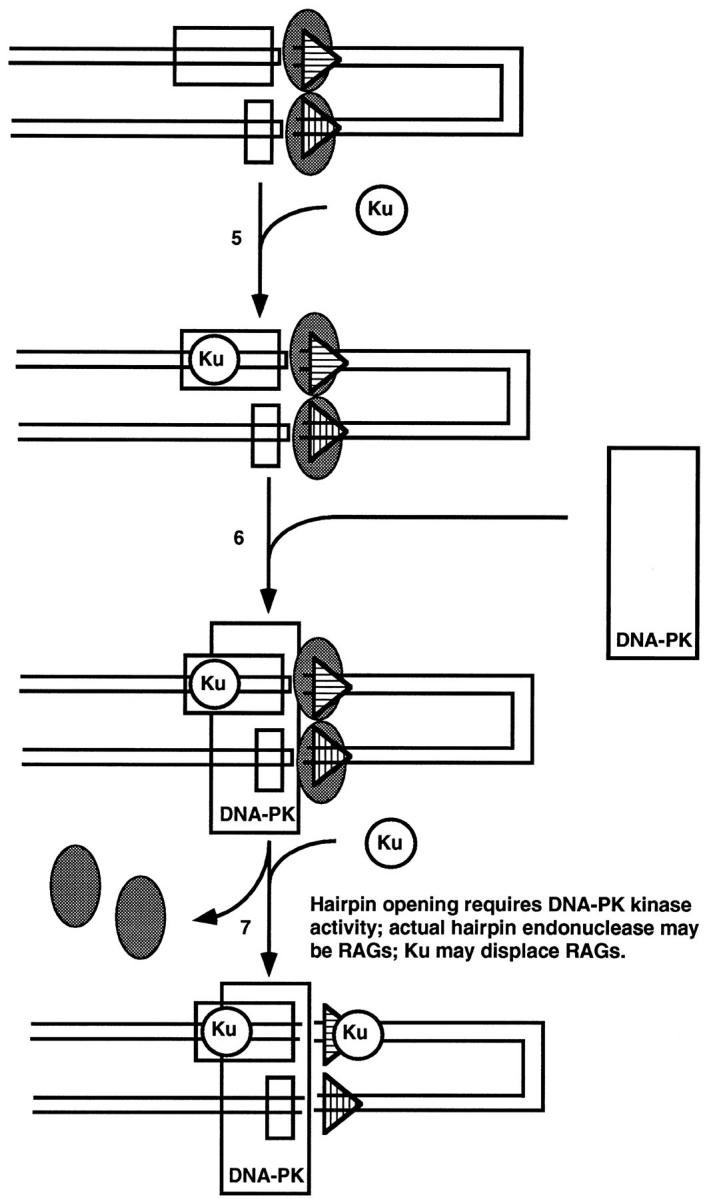
Model for roles of Ku and DNA-PK in the end joining of V(D)J recombination. The order and physical location at which Ku loads on relative to the signal and coding ends and relative to the RAG complex is not clear at the time of this writing. In step 6, DNA-PK is recruited by Ku but must contact the DNA directly. In step 7, Ku may displace the RAG complex at the signals in addition to functioning at the coding ends with DNA-PK.
As discussed above for general NHEJ, DNA-PK kinase activity is stimulated by a heterodimeric complex of Ku70 and -86 proteins. 39 From genetic studies using x-ray-sensitive CHO cell lines and the recent generation of Ku70- and Ku86-deficient mice, 40-43 it is clear that the Ku70/86 heterodimer is important for the formation of coding and signal joints. However, some level of normal coding joint formation does occur in the Ku70 knockout mice beyond that seen in scid mice or in the Ku86 knockout mice. 42,43 Ku can load onto DNA at DNA termini and slide to internal positions. 44 Some authors view Ku as the essential DNA-binding domain for the 470-kd DNA-PK, referring to the kinase as a subunit of a Ku:DNA-PKcs holoenzyme (the subscript cs denotes catalytic subunit). According to this model, DNA-PKcs is inactive without Ku. 39 Other data indicate that the 470-kd kinase has DNA binding and significant kinase activity on its own, even in the absence of detectable Ku, 45-47 perhaps explaining why Ku70 can be absent and still result in more leakiness than when the 470-kd DNA-PK kinase activity itself is absent. 42,43 In either case, Ku clearly stimulates DNA-PK activity, implying some allosteric effect of Ku on the 470-kd kinase once both are in proximity of the DNA. 39,46,48,49
Ku may serve additional functions in V(D)J recombination beyond stimulation of DNA-PK activity. This is suggested by the different phenotypes of mutants in DNA-PK, ie, scid mice, as opposed to Ku-deficient animals. Absence of DNA-PK activity results in failure to form coding joints, but signal joints form at near normal efficiencies. 50 In contrast, mutations in Ku affect both signal and coding joints. 40 One possible explanation of this is that Ku may dislodge the RAG postsynaptic complex from the signal ends to allow them to be ligated. 40 There is precedent for postsynaptic cleavage complexes in prokaryotic recombination systems requiring chaperone proteins to assist in debinding from the signal sequences. 51
Once the hairpins are opened, all four DNA ends are available for modification 52 (Figure 6) ▶ . Terminal deoxynucleotidyl transferase (TdT) is an optional lymphoid-specific component that can add nucleotides to coding ends to generate most of the junctional diversity that is a critical extension to the combinatorial diversity of antigen receptor assembly. 53 A point of mechanistic interest is that TdT can add N-nucleotides at signals ends as well, even though those signal ends are not subject to base loss. 52 This suggests that the RAG postsynaptic complex protects them from nuclease action but not from TdT template-independent nucleotide addition. It has been shown that a post-cleavage complex protects signal ends in vitro from exonuclease digestion. 54 In contrast, the coding ends are available for both TdT addition as well as nuclease action. The nucleases responsible for end nibbling have not yet been identified with certainty but may include FEN-1. The modification of all four ends is likely to occur in a complex where all four ends are in proximity to each other. This is suggested by the occurrence of nonphysiological alternative products called hybrid joints. 55 These involve the mistaken joining of a coding end to the other signal end, instead of to the other coding end. Such an occurrence would require that all four ends remain in proximity until ligation. Using a cell-free system for concerted cleavage at two signals, it has now been demonstrated, in crude extracts, that the signals remain synapsed in a post-cleavage complex. 54
Figure 6.

Model for the DNA end joining phase of V(D)J recombination. TdT template-independent nucleotide addition can occur at the 3′OH of any of the four DNA ends but is relevant to the immune system only at the coding ends where it contributes as a major factor to junctional diversification. In addition to RAG1 and -2, TdT is the only other lymphoid-specific component, but unlike RAGs, TdT is not essential for the cutting and joining to occur. The nucleases and polymerases in DNA end joining are not yet known, but polymerase β and FEN-1 are suspected. Alignment of the coding ends (step 9) is known to occur at regions of microhomology, but this, conceivably, could precede the nucleolytic processing or occur after it. The blunt signal ends are ligated together and the aligned coding ends are ligated together. XRCC4 forms a complex with DNA ligase IV, and this is believed to be the major ligase of the reaction.
It is known that the coding ends align at short regions of microhomology. 53 This point in the V(D)J recombination process and all subsequent steps appear to be identical with general NHEJ discussed earlier. As mentioned, it is not clear what the order of events is in this portion of the reaction. For example, do the coding end regions of microhomology align, followed by nuclease trimming of the excess DNA beyond the points of microhomology? Or are the coding ends subjected to random nuclease action, followed by alignment and ligation? The ligase complex of DNA ligase IV and XRCC4 that functions for general NHEJ also appears to function for V(D)J recombination, because cells mutant for XRCC4 fail to carry out V(D)J recombination or repair x-rays.
Class Switch Recombination
Just as V(D)J recombination can be divided into the cutting phase and the joining phases, class switch recombination can also be dissected into at least these two stages (Figure 7) ▶ . Concerning the joining phase, there are indications that this process is like that for general NHEJ. There are indications of the involvement of DNA-PK in class switch recombination because cells mutant for DNA-PK (murine scid cells) fail to carry out class switch recombination in a primary cell culture system. 56 More recent data show that class switch fails to occur in cells missing Ku. 73 Along with previous data demonstrating deleted circular DNA in switching cells, the requirement for DNA-PK is compelling evidence that the joining phase of class switch recombination is that of general NHEJ.
Figure 7.

Class switch recombination at the immunoglobulin heavy chain locus. Unlike V(D)J recombination, which occurs at all three Ig loci (heavy chain, κ chain, and λ chain loci) and all three TCR loci (α/δ, β, and γ loci), class switch recombination occurs only at the IgH locus. The right-angle arrows represent internal promoters called sterile transcript promoters. The transcripts from these do not encode protein because of stop codons. This suggests that transcription may be important for the mechanism of class switch recombination. The ovals represent the G-rich repetitive class switch sequences. Recombination occurs between switch sequences. Not all of the sterile transcript promoters are active at any one time. The cytokine stimulation of B cells in the peripheral lymphoid tissues determine which promoters are active.
The enzymes responsible for the cutting phase of class switch recombination are unknown. It is clear that RAG-1 and -2 are not required, as class switch recombination can occur normally in cells derived from RAG-2-deficient animals. 56 Some data suggest that the highly repetitive, G-rich switch regions acquire a non-B DNA configuration upon transcription. 57-60 Unlike V(D)J recombination, where transcription is not mechanistically required, transcription is important for class switch. 61
Chromosomal Translocations Arising during the Course of V(D)J Recombination and Class Switch Recombination
Translocations Associated with V(D)J Recombination
Many chromosomal translocations in lymphoid cells arise as mistakes during the process of either V(D)J recombination or class switch recombination. 62 One of the most common of these and most well characterized is one that occurs in essentially all follicular lymphomas, the t(14;18) translocation involving bcl-2 and the IgH locus (Figure 8) ▶ . One scenario for this translocation is as follows (Figure 8A) ▶ . The cutting event at the D to J rearrangement step of the IgH locus fails to complete. Cutting at the bcl-2 locus occurs and may be catalyzed by the RAG proteins inadvertently. Finally, the ends of the break at the bcl-2 locus become joined to the D and J segments instead of the D and J being joined.
Figure 8.
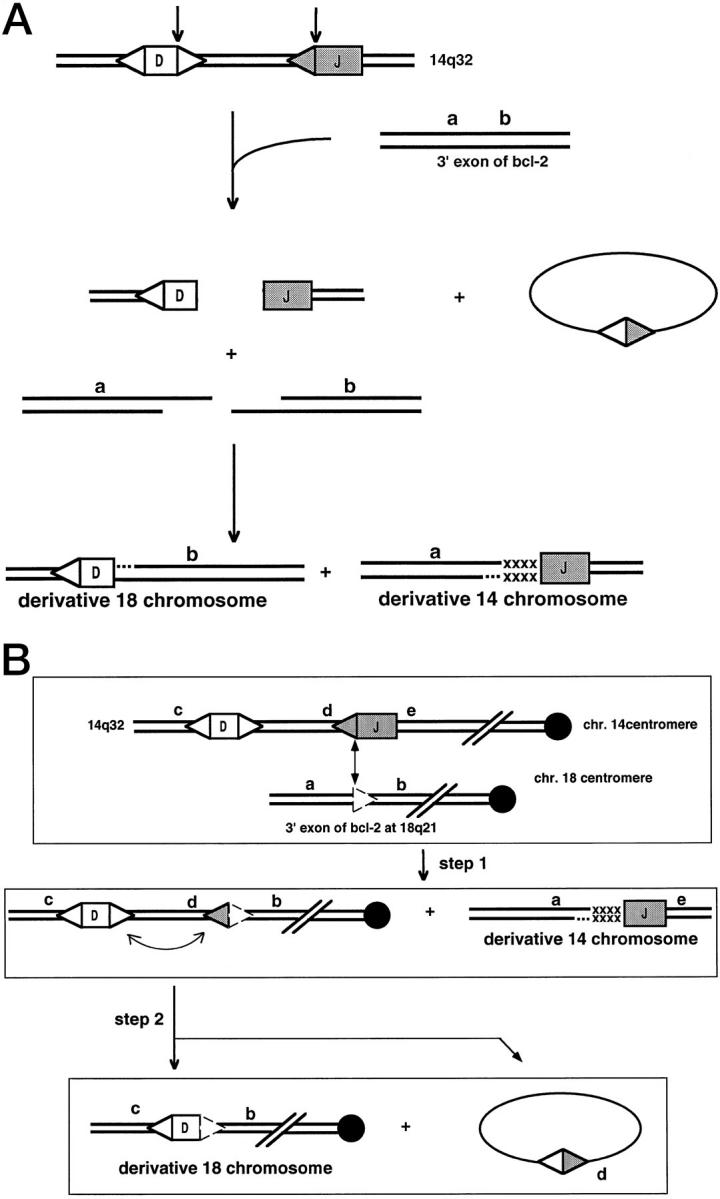
Models for the mechanism of the Bcl-2 translocation. A: One-step model for the t(14;18) translocation. A more detailed view of the likely events in some bcl-2 translocations is shown (based on Ref. 65 ). The letters a and b are simply genetic markers to provide orientation. The x’s represent nucleotide addition of uncertain origin. In this model, an attempt at DH to JH joining fails to be completed. An unrelated break on chromosome 18 at the bcl-2 locus also fails to be rejoined and is, instead, joined to the DH and JH ends to yield the reciprocal translocation. B: Two-step model for the t(14;18) translocation. In this model, the first step involves synapsis of the JH segment to a cryptic site within the bcl-2 gene. Use of cryptic sites having only the CAC match to the optimal signal sequences have been observed (see text). After this V(D)J recombination event between two signal sequences (one of which was cryptic), the second event involves recombination of two standard recombination signals, which modifies the derivative 18 chromosome.
However, an alternative model would be as follows (Figure 8B) ▶ . The RAG proteins may synapse the J segment recombination signal with a cryptic signal at the bcl-2 locus. Recent data indicate that cryptic sites consisting simply of CAC (the essential portion of the heptamer) may be sufficient to permit synapsis with a partner signal at the antigen receptor loci. 63,64 Once the first step is completed, a signal joint has been formed at the derivative 18 chromosome. Signal joints can carry out secondary rounds of rearrangement in which one of the signals (at d in Figure 8B ▶ ) can synapse with the DH segment signal in a second step. The second step would account for the configuration for the derivative chromosome 18 in cases for which this half of the translocation event has been sequenced. 65
The model in Figure 8B ▶ is more consistent with what is currently known about V(D)J recombination and about chromosome breakage. This model would account for the initiation of breakage at the bcl-2 locus. It would also reduce the complexity of the process into two steps rather than trying to explain three concurrent breakages (one at D, one at J, and one at the bcl-2 locus). This model would predict that the derivative chromosome 18 could exist in two states, one before and one after the second step. However, the second step may be quite efficient; hence, it may be difficult to detect the earlier configuration of the derivative 18.
One major uncertainty concerns the predilection for breakage at the bcl-2 locus. The promiscuity of the RAG proteins for cleaving at accessible CAC sites is noteworthy and creates a paradox as to how our lymphoid cells survive such promiscuity. It appears that most of the genome is protected by some degree of chromatin structure. The interplay between the RAG cleavage and chromatin structure is an interesting area for future work. Breaks that do not involve adventitious CAC sites may be caused by other processes. Fragile sites within DNA and chromatin are another area of intense interest. Some of these fragile sites may relate to how the G-rich class switch sequences are cleaved.
Translocations Involving Class Switch Sequences
The best characterized translocation involving class switch sequences is that of c-myc. In sporadic Burkitt’s lymphoma, c-myc typically translocates to the Sμ sequences located upstream of the constant Igμ region. This process appears to arise as a mistake of class switch recombination. Instead of the Sμ and one of the downstream switch regions (such as Sγ, Sα, or Sɛ) recombining, the break site at the Sμ region recombines with a break that arises at the c-myc gene on chromosome 8. A reciprocal translocation results between chromosomes 8 and 14 with most of the c-myc coding region now joined to the IgH locus. A diagram of the translocation process is shown in Figure 9 ▶ (top line). (In endemic Burkitt’s lymphoma, the translocation process typically occurs as a mistake of V(D)J recombination and gives the type of structural outcomes shown in the second through fourth lines of Figure 9 ▶ .) Because the mechanism of switch recombination is not understood, the mechanism of the c-myc translocation process is also not defined.
Figure 9.

The architecture of the common translocations. The chromosomal translocations listed on the far left are diagrammed. The centromere is depicted on the left of the depicted chromosome. The squares with numbers in them are the exons of the designated genes (either the Ig gene, the c-myc gene, or the bcl-2 gene). The rectangle depicts the Ig class switch region. The Ig enhancer is shown as an ellipse. The arrows show the direction of transcription. The Ig chromosome is designated as a solid line; the incoming chromosome is designated as a gray line.
Homologous Recombination
In addition to repair of double-strand DNA breaks by nonhomologous DNA end joining, some double-strand DNA breaks are repaired by homologous recombination (HR). The precise balance of homologous and nonhomologous recombination in cells is not yet fully determined. 4,66 It appears that during G0, G1, and early S phases of the cell cycle in vertebrate cells (and those of most multicellular eukaryotes), NHEJ is the dominant pathway. During late S and G2 phases of the cell cycle, HR plays a significant role. 67 The enzymes involved in HR include RAD51, -52, -54, -55, and -57. 68 BRCA1, BRCA2, and ATM may also play roles in this process. HR may be ideally suited for late S and G2 when the chromatids are aligned. Teleologically, it may be that as eukaryotic genomes became rich with repetitive DNA (which is 40% in humans), HR became less appropriate as a method for repairing double-strand breaks; otherwise, the number of potential, but not ideal, donors for breaks within the repetitive 40% of the genome may have made the donor search process extremely long. Moreover, some HR events involving the wrong donor repeat might lead to chromosomal translocations. NHEJ may be the safer alternative in multicellular eukaryotes during phases of the cell cycle when chromatids are not aligned. This is despite the fact that NHEJ is almost certain to result in the loss of some genetic information between the two DNA ends.
Some protein complexes, such as RAD50-Mre11-XRS(p95) complex, are involved in double-strand break repair, but it is not yet clear whether they are involved in NHEJ or HR or both. 69
NHEJ and Aging
As mentioned earlier, defects in the cleavage phase of V(D)J recombination can result in human SCID. Defects in the synapsis phase of V(D)J recombination may result in chromosomal translocations that yield follicular lymphomas, and similar events may occur for switch recombination in the case of sporadic Burkitt’s lymphoma.
However, a broader issue in human disease is the extent to which double-strand breaks generate irreversible changes in the genome over time. Any irreversible change in any nondegradable macromolecule may be contributory to the aging process. It is currently unclear as to the extent of the contribution of changes in DNA to aging. However, DNA is the one macromolecule in the cell whose information content cannot be regenerated once it is lost.
Single-strand DNA damage is not as relevant because the other strand provides for the information content. However, double-strand breaks typically result in the loss of the information between the two ends during NHEJ. Hence, double-strand breaks are the likely candidate for aging of DNA.
The one tangible observation thus far is that cells lacking the ability to carry out NHEJ appear to senesce in culture more quickly 41,70 (P. Hasty, personal communication). Additional studies will be needed to define whether the basis for this is actual DNA damage.
A key pending question related to this issue is how frequently double-strand breaks are created in the genome from free radicals generated by oxidative respiration and from ionizing radiation. If they are rare, they represent no significant contributory cause of aging. At this point, we do not yet know the rate at which double-strand breaks occur. This could be high and yet our NHEJ (and HR) may be sufficiently effective so as to repair the vast majority of these to restore the integrity of the chromosome. Nevertheless, restoration of chromosomal integrity would still be accompanied by loss of genetic information at the nucleotide level, and this loss is irreversible. There may soon be methods to measure the background level of double-strand breaks caused by oxidative free radicals arising from respiration and from ionizing radiation. Only at that point will we be able to evaluate this issue in a meaningful way.
Concluding Remarks
Biochemical definition of the steps and details of physiological and pathologic double-strand DNA breaks is still a young field. Yet there have been many rewards for this field concerning human disease. This may continue as we examine the relationship to broader issues, such as the time-dependent DNA changes that may occur with aging.
Figure 10.

Relationships between physiological and pathological DNA breaks and the pathways for their joining. The physiological forms of double-strand breakage occur in V(D)J recombination (mediated by RAG1 and RAG2) and in class switch recombination (the nuclease for this is not yet identified). One source of pathological breaks is free radicals generated from oxidative respiration. Pathological breaks are also caused by ionizing radiation. Ionizing radiation arises constantly as cosmic rays (hydrogen and helium nuclei) from solar sources generate showers of secondary cosmic rays (some of which are γ-rays). Ionizing radiation also arises from naturally occurring radionuclides in the earth. 72 During G0, G1, and early S phases of the cell cycle, pathological breaks are primarily repaired by NHEJ.
Acknowledgments
I thank Drs. H. T. Blumenthal, T. Steck, M. Gellert, and K. Mizuuchi.
Footnotes
Address reprint requests to Dr. Michael R. Lieber, Department of Pathology, Norris Comprehensive Cancer Center, University of Southern California School of Medicine, 1441 Eastlake Avenue, Los Angeles, CA 91007. E-mail: lieber_m@froggy.hsc.usc.edu.
Supported by NIH grants and by the Rita and Edward Polusky Basic Cancer Research Professorship. The author is also a Leukemia Society of America Scholar.
References
- 1.Friedberg EC, Walker GC, Siede W: DNA Repair and Mutagenesis. 1995. ASM Press, Washington, DC,
- 2.Ramsden DA, Gellert M: Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J 1998, 17:609-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pang D, Yoo S, Dynan WS, Jung M, Dritschilo A: Ku proteins join DNA fragments as shown by atomic force microscopy. Cancer Res 1997, 57:1412-1415 [PubMed] [Google Scholar]
- 4.Roth D, Wilson J: Illegitimate recombination in mammalian cells. Kucherlapapti R Smith GR eds. Genetic Recombination. 1988, :pp 621-653 American Society for Microbiology, Washington, DC, [Google Scholar]
- 5.Tonegawa S: Somatic generation of antibody diversity. Nature 1983, 302:575-581 [DOI] [PubMed] [Google Scholar]
- 6.Schatz DG, Oettinger MA, Baltimore D: The V(D)J recombination activating gene, RAG-1. Cell 1989, 59:1035-1048 [DOI] [PubMed] [Google Scholar]
- 7.Oettinger MA, Schatz DG, Gorka C, Baltimore D: Rag-1 and Rag-2, adjacent genes that synergistically activate V(D)J recombination. Science 1990, 248:1517-1523 [DOI] [PubMed] [Google Scholar]
- 8.vanGent DC, McBlane JF, Ramsden DA, Sadofsky MJ, Hesse JE, Gellert M: Initiation of V(D)J recombination in a cell-free system. Cell 1995, 81:925-934 [DOI] [PubMed] [Google Scholar]
- 9.McBlane JF, vanGent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA: Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 1995, 83:387-395 [DOI] [PubMed] [Google Scholar]
- 10.vanGent DC, Mizuuchi K, Gellert M: Similarities between initiation of V(D)J recombination and retroviral integration. Science 1996, 271:1592-1594 [DOI] [PubMed] [Google Scholar]
- 11.Craig NL: V(D)J recombination and transposition: closer than expected. Science 1996, 271:1512-1513 [DOI] [PubMed] [Google Scholar]
- 12.Hoim K, Gellert M: A stable RAG1-RAG2-DNA complex that is active in V(D)J cleavage. Cell 1997, 88:65-72 [DOI] [PubMed] [Google Scholar]
- 13.Grawunder U, Leiber MR: A complex of RAG-1 and RAG-2 persists on the DNA after single-strand cleavae at V(D)J recombination signal sequences. Nucleic Acids Res 1997, 25:1375-1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.vanGent DC, Ramsden DA, Gellert M: The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell 1996, 85:107-113 [DOI] [PubMed] [Google Scholar]
- 15.Eastman QM, Leu TMJ, Schatz DG: Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature 1996, 380:85-88 [DOI] [PubMed] [Google Scholar]
- 16.Sawchuk D, Weis-Garcia F, Malik S, Besmer E, Bustin M, Nussenzweig M, Cortes P: V(D)J recombination: modulation of RAG1 and RAG2 cleavage activity on 12/23 substrates by whole cell extract and DNA-bending proteins. J Exp Med 1997, 185:2025-2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.vanGent D, Hoim K, Paull T, Gellert M: Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J 1997, 16:2665-2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Difilippantonio M, McMahan C, Eastman Q, Spanopoulou E, Schatz DG: RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell 1996, 87:253-262 [DOI] [PubMed] [Google Scholar]
- 19.Spanopoulou E, Zaitseva F, Wang FH, Santagata S, Baltimore D, Panayotou G: The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell 1996, 87:263-276 [DOI] [PubMed] [Google Scholar]
- 20.Hsieh C, McCloskey RP, Lieber MR: V(D)J recombination on minichromosomes is not affected by transcription. J Biol Chem 1992, 267:5613-5619 [PubMed] [Google Scholar]
- 21.Schlissel M, Voronova A, Baltimore D: Helix-loop-helix transcription factor E47 activates germ-line immunoglobulin heavy-chain gene transcripton and rearrangement in a pre-T-cell line. Genes Dev 1991, 5:1367-1376 [DOI] [PubMed] [Google Scholar]
- 22.Alvarez JD, Anderson SJ, Loh DY: V(D)J recombination and allelic exclusion of a TCR beta chain minilocus occurs in the absence of a functional promoter. J Immunol 1995, 155:1191-1202 [PubMed] [Google Scholar]
- 23.Engler P, Roth P, Kim JY, Storb U: Factors affecting the rearrangement efficiency of an Ig test gene. J Immunol 1991, 146:2826-2835 [PubMed] [Google Scholar]
- 24.Ferrier P, Krippl B, Blackwell TK, Furley AJW, Suh H, Winoto A, Cook WD, Hood L, Constantini F, Alt FW: Separate elements control DJ and VDJ rearrangement in a transgenic recombination substrate. EMBO J 1990, 9:117-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capone M, Watrin F, Fernex C, Horvat B, Krippl B, Wu L, Scollay R, Ferrier P: TCRβ and TCRα gene enhancers confer tissue- and stage-specificity to V(D)J recombination events. EMBO J 1993, 12:4335-4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernex C, Caillol D, Capone M, Krippl B, Ferrier P: Sequences affecting the V(D)J recombinational activity of the IgH intronic enhancer in a transgenic substrate. Nucleic Acids Res 1994, 22:792-798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lichtenstein M, Keini G, Cedar H, Bergman Y: B cell-specific demethylation: a novel role for the intronic κ chain enhancer sequence. Cell 1994, 76:913-923 [DOI] [PubMed] [Google Scholar]
- 28.Engler P, Weng A, Storb U: Influence of CpG methylation and target spacing on V(D)J recombination in a transgenic substrate. Mol Cell Biol 1993, 13:571-577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsieh C, Lieber MR: CpG methylated minichromosomes become inaccessible for V(D)J recombination after undergoing replication. EMBO J 1992, 11:315-325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh C-L: The dependence of transcriptional repression on CpG methylation density. Mol Cell Biol 1994, 14:5487-5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Constantinescu A, Schlissel MS: Changes in locus-specific V(D)J recombinase activity induced by immunoglobulin gene products during B cell development. J Exp Med 1996, 185:609-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanhope P, Hudson KM, Shaffer AL, Constantinescu A, Schlissel MS: Cell type-specific chromatin structure determines the targeting of V(D)J recombinase activity in vitro. Cell 1996, 85:887-897 [DOI] [PubMed] [Google Scholar]
- 33.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W, Seger RA, Hansen HT, Desiderio S, Lieber MR, Bartram CR: RAG mutations in human B cell-negative SCID. Science 1996, 274:97-99 [DOI] [PubMed] [Google Scholar]
- 34.Roth DB, Menetski JP, Nakajima P, Bosma MJ, Gellert M: V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in SCID mouse thymocytes. Cell 1992, 70:983-991 [DOI] [PubMed] [Google Scholar]
- 35.Danska JS, Holland DP, Mariathasan S, Williams KM, Guidos CJ: Biochemical and genetic defects in DNA-dependent protein kinase in murine scid lymphocytes. Mol Cell Biol 1996, 16:5507-5517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blunt T, Gell D, Fox M, Taccioli G, Lehman A, Jackson S, Jeggo PA: Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA 1996, 93:10285-10290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramsden D, Paull T, Gellert M: Cell-free V(D)J recombination. Nature 1997, 388:488-491 [DOI] [PubMed] [Google Scholar]
- 38.Leu TM, Eastman QM, Schatz DG: Coding joint formation in a cell-free V(D)J recombination system. Immunity 1997, 7:303-314 [DOI] [PubMed] [Google Scholar]
- 39.Gottlieb T, Jackson SP: The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 1993, 72:131-142 [DOI] [PubMed] [Google Scholar]
- 40.Zhu C, Bogue MA, Lim D-S, Hasty P, Roth DB: Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 1996, 86:379-389 [DOI] [PubMed] [Google Scholar]
- 41.Nussenzweig A, Chen C, Sores VdC, Sanchez M, Sokol K, Nussenzweig MC, Li GC: Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382:551-555 [DOI] [PubMed] [Google Scholar]
- 42.Ouyang H, Nussenzweig A, Kurimasa A, Soares V, Li X, Cordon-Cardo C, Li W, Cheong N, Nussenzweig M, Iliakis G, Chen DJ, Li GC: Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J Exp Med 1997, 186:921-929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu Y, Jin S, Gao Y, Weaver DT, Alt FW: Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci USA 1997, 8076–8081 [DOI] [PMC free article] [PubMed]
- 44.deVries E, Driel WV, Bergsma WG, Arnberg AC, Vliet PC: HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA-multimeric protein complex. J Mol Biol 1989, 208:65-78 [DOI] [PubMed] [Google Scholar]
- 45.Brush G, Anderson CW, Kelly TJ: The DNA-activated protein kinase is required for the phosphorylation of replication protein A during SV40 DNA replication. Proc Natl Acad Sci USA 1994, 91:12520-12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yaneva M, Kowaleski T, Lieber MR: Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy. EMBO J 1997, 16:5098-5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan Z, Amin AA, Gibbs E, Niu H, Hurwitz J: Phosphorylation of the p34 subunit of human single-stranded DNA-binding protein in cyclin A-activated G1 extracts is catalyzed by cdk-cyclin A complex and DNA-dependent protein kinase. Proc Natl Acad Sci USA 1994, 91:8343-8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dvir A, Peterson SR, Knuth MW, Lu H, Dynan WS: Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci USA 1992, 89:11920-11924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dvir A, Stein LY, Calore BL, Dynan WS: Purification and characterization of a template-associated protein kinase that phosphorylates RNA polymerase II. J Biol Chem 1993, 268:10440-10447 [PubMed] [Google Scholar]
- 50.Lieber MR, Hesse JE, Lewis S, Bosma GC, Rosenberg N, Mizuuchi K, Bosma MJ, Gellert M: The defect in murine severe combined immune deficiency: joining of signal sequences but not coding segments in V(D)J recombination. Cell 1988, 55:7-16 [DOI] [PubMed] [Google Scholar]
- 51.Levchenko I, Luo L, Baker TA: Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes Dev 1995, 9:2399-2408 [DOI] [PubMed] [Google Scholar]
- 52.Lieber MR, Hesse JE, Mizuuchi K, Gellert M: Lymphoid V(D)J recombination: nucleotide insertion at signal joints as well as coding joints. Proc Natl Acad Sci USA 1988, 85:8588-8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis SM: The mechanism of V(D)J joining: lessons from molecular immunological and comparative analyses. Adv Immunol 1994, 56:27-150 [DOI] [PubMed] [Google Scholar]
- 54.Agrawal A, Schatz DG: RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell 1997, 89:43-53 [DOI] [PubMed] [Google Scholar]
- 55.Lewis S, Hesse JE, Mizuuchi K, Gellert M: Novel strand exchanges in V(D)J recombination. Cell 1988, 55:1099-1107 [DOI] [PubMed] [Google Scholar]
- 56.Rolink A, Melchers F, Andersson J: The SCID but not the RAG-2 gene product is required for Sμ-Sɛ heavy chain switching. Immunity 1996, 5:319-330 [DOI] [PubMed] [Google Scholar]
- 57.Daniels GA, Lieber MR: RNA: DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res 1995, 23:5006-5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daniels GA, Lieber MR: Strand-specificity in the transcriptional targeting of recombination at immunoglobulin class switch sequences. Proc Natl Acad Sci USA 1995, 92:5625-5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reaban ME, Griffin JA: Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature 1990, 348:342-344 [DOI] [PubMed] [Google Scholar]
- 60.Reaban ME, Lebowitz J, Griffin JA: Transcription induces the formation of a stable RNA: DNA hybrid in the immunoglobulin alpha switch region. J Biol Chem 1994, 269:21850-21857 [PubMed] [Google Scholar]
- 61.Stavnezer J: Immunoglobulin class switching. Curr Opin Immunol 1996, 8:199-205 [DOI] [PubMed] [Google Scholar]
- 62.Lieber MR: The role of site-directed recombinases in physiologic and pathologic chromosomal rearrangements. Kirsch I eds. The Causes and Consequences of Chromosomal Translocations. 1992, :pp 239-275 CRC Press, Boca Raton, FL, [Google Scholar]
- 63.Lewis SM, Agard E, Suh S, Czyzyk L: Cryptic signals and the fidelity of V(D)J joining. Mol Cell Biol 1997, 17:3125-3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taki S, Schwenk F, Rajewsky K: Rearrangement of upstream Dh and Vh genes to a rearranged immunoglobulin variable region gene inserted into the DQ52-Jh region of the immunoglobulin heavy chain locus. Eur J Immunol 1995, 25:1888-1896 [DOI] [PubMed] [Google Scholar]
- 65.Bakhshi A, Wright JJ, Graninger W, Seto M, Owens J, Cossman J, Jensen JP, Goldman P, Korsmeyer SJ: Mechanism of the t(14;18) chromosomal translocation: structural analysis of both derivative 14 and 18 reciprocal partners. Proc Natl Acad Sci USA 1987, 84:2396-2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang F, Han M, Romanienko PJ, Jasin M: Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci USA 1998, 95:5172-5177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S: Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998, 17:598-608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanaar R, Hoeijmakers JHJ: Recombination and joining: different means to the same ends. Genes Funct 1997, 1:165-174 [DOI] [PubMed] [Google Scholar]
- 69.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JHJ: In situ visualization of DNA double-strand break repair in human fibroblasts. Science 1998, 280:590-592 [DOI] [PubMed] [Google Scholar]
- 70.Gu Y, Seidl K, Rathbun GA, Zhu C, Manis JP, vanderStoep N, Davidson L, Cheng HL, Sekiguchi J, Frank K, Stanhope-Baker P, Schlissel MS, Roth DB, Alt FW: Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 1997, 7:653-665 [DOI] [PubMed] [Google Scholar]
- 71.Sheehan KM, Lieber MR: V(D)J recombination: signal and coding joint resolution are uncoupled and depend on parallel synapsis of the sites. Mol Cell Biol 1993, 13:1363-1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sutton HE: Human Genetics. 1989. Saunders, Philadelphia
- 73.Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, Qin X, Besmer E, Kenter A, Kajewsky K, Nussenzweig M: Ku80 is required for immunoglobulin isotype switching. EMBO J 1998, 17:2404-2411 [DOI] [PMC free article] [PubMed] [Google Scholar]