

Abstract
Chemokines interact with specific G-protein-coupled receptors to activate and direct recruitment of immune cells. Some chemokines are up-regulated in pathological conditions of the central nervous system, and recently several chemokine receptors, including CCR5, were identified in the brain. However, little is known about the regulation of expression of chemokine receptors in the brain. Direct intracerebral injection of N-methyl-d-aspartate (NMDA), an excitatory amino acid agonist, elicits reproducible focal excitotoxic brain injury; in neonatal rats, intrahippocampal NMDA injection stimulates expression of pro-inflammatory cytokines and elicits a robust microglia/monocyte response. We hypothesized that NMDA-induced neurotoxicity would also stimulate expression of CCR5 in the neonatal rat brain. We evaluated the impact of intrahippocampal injections of NMDA on CCR5 expression in postnatal day 7 rats. Reverse transcription polymerase chain reaction revealed an increase in hippocampal CCR5 mRNA expression 24 hours after lesioning, and in situ hybridization analysis demonstrated that CCR5 mRNA was expressed in the lesioned hippocampus and adjacent regions. Western blot analysis demonstrated increased CCR5 protein in hippocampal tissue extracts 32 hours after lesioning. Complementary immunocytochemistry studies identified both infiltrating microglia/monocytes and injured neurons as the principal CCR5-immunoreactive cells. These results provide the first evidence that acute excitotoxic injury regulates CCR5 expression in the developing rat brain.
Recent experimental data indicate that inflammatory mediators contribute substantially to the pathogenesis of neonatal brain injury. 1 Heightened interest in the pathogenetic role of inflammatory mediators in the immature nervous system stems from clinical observations linking detection of pro-inflammatory cytokines in the amniotic fluid and in the neonatal brain with adverse neurodevelopmental outcome. 2,3 Studies in experimental models of hypoxic-ischemic and excitotoxic neonatal brain injury provide direct evidence for associated activation of pro-inflammatory mechanisms. 4-9
The maturational stage of postnatal day (P)7 rat brain corresponds roughly with late-gestation human brain maturation. A well characterized model of acute excitotoxic brain injury, elicited by direct intracerebral (i.c.) administration of the glutamate agonist N-methyl-d-aspartate (NMDA) into P7 rat brain 10 facilitates analysis of the molecular and cellular responses elicited by NMDA receptor over-activation in vivo. In the rat, NMDA-induced brain injury is dose dependent and reproducible; susceptibility to NMDA-induced neurotoxicity peaks at P7. 10 Over-activation of the NMDA-subtype glutamate receptor leads to increased intracellular calcium accumulation and increased neuronal nitric oxide production; a complex cascade of downstream molecular mechanisms, including generation of soluble injury mediators, determine the ultimate extent of tissue damage. 11 Direct i.c. administration of NMDA into P7 rat brain elicits rapid stimulation of interleukin (IL)-1β production, 9 and pharmacological antagonism of IL-1 markedly attenuates injury, 6 indicating the potential of inflammatory cytokines to exacerbate damage.
Recent observations suggest that chemokines, a family of small proteins involved in the activation and directed migration of leukocytes, 12 may be potential mediators of brain injury. Currently, four distinct subfamilies of chemokines have been identified; CC, CXC, C, and CX3C subfamilies are distinguished by the positioning of conserved cysteine residues. These structural distinctions correlate with functional differences in target specificity. For example, CC chemokines primarily recruit and activate monocytes and lymphocytes, 13-15 CXC chemokines recruit neutrophils, 16-18 lymphotactin (C subfamily) has chemotactic activity for lymphocytes, 19 and the newly described CX3C chemokine is chemotactic for lymphocytes and monocytes. 20 Most chemokines elicit their effects through interactions with seven-transmembrane-domain, G-protein-coupled receptors. There has been rapid progress in characterization of multiple distinct human and rodent chemokine receptors. 12,21,22
Interest in the potential pathophysiological role of chemokines and their receptors in the developing central nervous system (CNS) stems from several recent findings. First, excitotoxic injury in neonatal rats is characterized by an increase in activated microglia (macrophage-like cells in the CNS) in injured areas; 5 these activated microglia may contribute to the progression of neuronal injury through the release of inflammatory mediators. 23,24 In addition, acute excitotoxic injury in neonatal rats stimulates gene and protein expression of the chemokine monocyte chemoattractant protein (MCP)-1, a potent regulator of monocytes, in areas where activated microglia/monocytes subsequently accumulate. 7,8
Chemokine receptors, including CCR5, have been implicated in the pathogenesis of HIV-1 infection. 25,26 Recent evidence also suggests that HIV-1 infection of microglia, the major target cells of HIV-1 in the brain, is in part mediated by CCR5 27 and that HIV-1-infected microglia secrete neurotoxic factors that may contribute to neuronal death. 23,24 In experimental models of HIV-1-associated CNS injury, the HIV-1 envelope glycoprotein 120 (gp120) influences susceptibility to NMDA neurotoxicity in P7 hippocampus, 28 and microglia have been implicated as mediators of gp120 neurotoxicity. 24,29 Together, these observations prompted us to evaluate whether acute NMDA-mediated brain injury regulates CCR5 expression in the neonatal rat brain.
Materials and Methods
Animals and Reagents
P7 Sprague-Dawley rats were obtained from Charles River (Wilmington, MA). The following reagents were purchased: NMDA (Sigma Chemical Co., St. Louis, MO); Tri-Reagent (Molecular Research Center, Cincinnati, OH); MuLV RT, random hexamers, dNTPs, and RNAse inhibitor (Perkin Elmer, Foster City, CA); DNAse and in vitro transcription kit (Ambion, Austin TX); restriction endonucleases (Boehringer Mannheim, Indianapolis, IN); and [35S]UTP (NEN Dupont, Boston, MA). Protease inhibitors (aprotinin, phenylmethylsulfonyl fluoride, and sodium orthovanadate) and teleostean (cold-water fish skin) gelatin were from Sigma. BCA protein assay kit and enhanced luminol were obtained from Pierce (Rockford, IL). The following antibodies and related reagents were also purchased: goat anti-CCR5 antibody (Ab), CCR5 blocking peptide, normal goat IgG, and horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA); ED-1 monoclonal antibody (MAb; Serotec, Oxford, UK); glial fibrillary acidic protein (GFAP) polyclonal Ab (Dako, Carpinteria, CA); all biotinylated secondary Abs, ABC Elite kit, normal sera, and normal mouse and rabbit IgG (Vector Laboratories, Burlingame, CA); stable diaminobenzidine (DAB; Research Genetics, Huntsville, AL).
Animal Methods
All surgical protocols were approved by the University of Michigan Committee on Care and Use of Animals. All lesioning was performed in P7 Sprague-Dawley rats of both genders, using previously reported methods. 30 Animals were deeply anesthetized by methoxyfluorane inhalation and placed in a standardized holder; the scalp was incised, and skull surface landmarks were identified. At the injection site, the skull was penetrated with a 22-gauge needle, and a 1-μl Hamilton syringe attached to a 25-gauge needle was used to deliver the NMDA-containing solution (10 nmol of NMDA/0.5 μl) over 2 minutes; stereotaxic coordinates were targeted to the right dorsolateral hippocampus (relative to Bregma: antero-posterior, 2.0 mm; lateral, 2.5 mm; dorsal, 4.0 mm). Thirty minutes later, after full recovery from anesthesia, animals were returned to their dams. Animals were housed in a temperature-regulated incubator (maintained at 37°C) during the recovery period. All experiments included controls that underwent the same procedures, in which an equal volume of PBS was substituted for NMDA, as well as unlesioned littermate controls. Animals were killed either by decapitation or by administration of a lethal dosage of chloral hydrate (3 g/kg) followed by perfusion-fixation.
RNA Isolation
RNA samples were prepared from the left and right hippocampus of animals that received right intrahippocampal injections of 10 nmol of NMDA 8, 16, 24, 48, or 72 hours earlier and from animals that had received PBS injections (24 hours earlier). Normal P8 hippocampus was also collected. Brains were divided along the midline, and left and right hippocampus were microdissected on ice. Four hippocampi were pooled per sample. Three independent samples were prepared from normal P8 rats and from animals that received NMDA injections and were killed 24 hours later. Total RNA was isolated using Tri-Reagent (1.2 ml) according to the manufacturer’s directions and stored at −70°C. Concentration and purity of RNA samples were estimated by spectrophotometric analysis.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
RNA samples were pretreated with DNAse; 1 μg of total RNA was suspended in 10 μl of diethylpyrocarbonate-treated H2O containing 2 U of DNAse in 20 mmol/L Tris/HCl (pH 8.4), 50 mmol/L KCl, and 20 mmol/L MgCl2 (15 minutes at room temperature); the reaction was stopped by the addition of EDTA (final concentration, 2.5 mmol/L), and DNAse was inactivated by heating (65°C for 15 minutes). RT was performed as previously described 4 with minor modifications. Briefly, 1 μg of DNAse-treated RNA was incubated with 50 U of MuLV reverse transcriptase, 2 μmol/L random hexamers, 20 U of RNAse inhibitor, and 0.5 mmol/L of each dNTP under the following conditions: 10 minutes at room temperature, 15 minutes at 42°C, and 5 minutes at 99°C. The RT product was diluted to a final volume of 100 μl in sterile H2O. Two sets of oligonucleotide primers were used to co-amplify the RT product: 1) sense (5′-CACCCTGTTTCGCTGTAGGAATG-3′)and antisense (5′-GCAGTGTGTCATCCCAAGAGTCTC-3′) primers to amplify a 219-bp fragment of the rat CCR5 cDNA sequence and 2) primers (5′-TCCTGCACCACCAACTGCTTAG-3′ and 5′-CAGATCCACAACGGATACATTGG-3′) to amplify a 298-bp fragment of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a ubiquitously expressed gene that was used to normalize CCR5 mRNA. In preliminary experiments, optimal MgCl2 concentration (1.0 mmol/L), pH 9.0, and primer concentrations (0.2 μmol/L for CCR5 primers and 0.05 μmol/L GAPDH primers) were determined; amplification conditions to yield results within the linear range of amplification for CCR5 and GAPDH were established, using 20 μl of RT product (94°C for 90 seconds, 56°C for 40 seconds, and 72°C for 40 seconds for 33 cycles). All RT-PCR reaction products were visualized in ethidium-bromide-stained 2% agarose gels. Results were quantified by fluorometric scanning of the gels, and measurement of arbitrary optical density units (expressed in counts/mm) of each band was performed using the Molecular Analyst imaging system (Bio-Rad, Hercules, CA); values for CCR5 mRNA in each sample were normalized, based on GAPDH mRNA content/sample.
In three independent RT-PCR assays, CCR5 expression was compared in samples from normal P8 samples and in left and right hippocampal samples from animals that had been lesioned 24 hours earlier; values for left and right hippocampal CCR5 mRNA expression were expressed as a percentage of corresponding normal control values and were compared using the Mann-Whitney ranking test.
To evaluate the efficacy of the DNAse treatment, a right hippocampal RNA sample (24 hours after NMDA injection) was DNAse treated and amplified using conditions described above, but without RT; no GAPDH or CCR5 fragments were amplified (data not shown).
In Situ Hybridization
A 310-bp EcoRV/BglII fragment of the rat CCR5 cDNA sequence (GenBank accession U77350), cloned into a pGEM7(+) vector, was used as a template for in vitro transcription reactions to generate 35S-labeled sense and antisense riboprobes. Briefly, the plasmid construct was linearized with either HindIII or EcoRI for sense and antisense (c)RNA probes, respectively. [35S]UTP (specific activity, 1100 to 1400 Ci/mmol) was incorporated using an in vitro transcription kit according to manufacturer’s directions.
Based on results of RT-PCR assays that showed increased CCR5 mRNA expression at 24 hours after right intrahippocampal NMDA injection, this time point was selected for in situ hybridization analysis. Samples were prepared from animals that had received right intrahippocampal NMDA (10 nmol) injections (n = 5) or PBS (n = 2) and from unlesioned P8 animals (n = 2); preliminary experiments were conducted with two samples from each group to ensure that methods were appropriate for detection of CCR5 mRNA. Using assay conditions that enabled detection of CCR5 mRNA, three additional NMDA-injected brains were assayed; 14 sections/brain were hybridized with the antisense riboprobe, and 6 sections/brain were hybridized with the sense riboprobe.
Brains were removed rapidly and frozen in crushed dry ice. Frozen, 20-μm coronal sections were collected on poly-l-lysine-coated slides. Sections were fixed in 4% formaldehyde for 1 hour and washed in PBS. Sections were then treated with proteinase K (5 μg/ml) for 5 minutes at 37°C and acetylated in 0.25% acetic anhydride with rapid stirring for 10 minutes at room temperature. After a 5-minute wash in 2X SSC, sections were dehydrated in graded ethanols. Sections were incubated with riboprobes (10 6 cpm/slide) in 50% formamide, 10% dextran sulfate, 1 mmol/L EDTA, 10 mmol/L Tris, and 0.1 mmol/L dithiothreitol for 20 hours at 55°C. On the following day, sections were washed for 30 minutes in 2X SSC at room temperature and 50% formamide/2X SSC at 55°C for 30 minutes, treated in RNAse A (50 μg/ml), and dehydrated in graded ethanols. Slides were apposed to x-ray film for 28 days.
Western Blotting
A commercial polyclonal goat IgG Ab, directed against an epitope corresponding to an amino acid sequence mapping at the carboxy terminus of CCR5 of mouse origin (conserved in rat CCR5) was used; the Ab does not cross-react with other known C-C chemokine receptor gene-encoded proteins. To prepare hippocampal protein extracts, brains were rapidly removed and microdissected on ice; tissue from three animals was pooled for each sample. In addition, for preliminary experiments, samples were prepared from each cerebral hemisphere. Tissue extracts were prepared from left and right hippocampus or left and right hemispheres of animals that had received right intrahippocampal injections of NMDA (10 nmol) or PBS 32 hours earlier; samples were also collected from unlesioned P8 animals. Adult rat spleen extracts (a rich tissue source of chemokine receptors) were also prepared for use as positive controls. Samples were homogenized in 500 μl of buffer (PBS/0.1% Nonidet P-40/0.1% SDS/0.5% deoxycholic acid) containing aprotinin (5.7 μg/ml) and sodium orthovanadate (1 mmol/L); phenylmethylsulfonyl fluoride (100 μg/ml) was added after homogenization. Samples were then centrifuged (15,000 × g for 20 minutes), and supernatants were collected and stored at −20°C.
Sample protein content was measured using a BCA protein assay kit according to the manufacturer’s directions. Equal amounts of protein (20 μg) were resolved by SDS/10% polyacrylamide gel electrophoresis. Protein was electrotransferred to nitrocellulose in Tris/glycine buffer containing 20% methanol. Membranes were blocked overnight at 4°C in Tris-buffered saline (TBS) containing 5% gelatin and 0.2% Tween-20 and then incubated with goat anti-mouse CCR5 Ab (1:1000 in TBS/3% gelatin for 1 hour at room temperature). Membranes were washed (TBS and 0.05% Tween-20) and incubated with horseradish-peroxidase-conjugated donkey anti-goat IgG (1:15,000 in 3% gelatin in TBS), and signal was developed using enhanced luminol according to the manufacturer’s directions.
Immunocytochemistry
To prepare samples for immunocytochemistry, euthanized animals were perfused transcardially with 10 ml of PBS, followed by 10 ml of 2% paraformaldehyde in PBS. Brains were removed intact and cryoprotected in 20% sucrose as previously described; 5 14-μm frozen coronal sections were mounted onto gelatin-coated slides.
Samples were prepared from animals that had received right intrahippocampal injections of 10 nmol of NMDA 24 hours (n = 2), 32 hours (n = 5), 48 hours (n = 2), 72 hours (n = 2), or 5 days (n = 2) earlier or PBS (n = 2) 32 hours earlier; two samples from unlesioned P8 animals were also assayed. At least 12 sections/brain (all including the hippocampus) were assayed. The same anti-CCR5 Ab used for the Western blot assays was also used to detect CCR5 immunocytochemically. In addition, to facilitate identification of cells of the microglia or monocyte lineage that expressed CCR5, representative adjacent sections were probed with ED-1 Ab, a MAb that recognizes a cell surface antigen expressed by activated macrophages/monocytes. 5 In addition, to identify reactive astrocytes, GFAP immunocytochemistry was performed (using a polyclonal anti-bovine GFAP Ab that detects rat GFAP).
For CCR5, ED-1, and GFAP immunocytochemistry, sections were washed (in PBS for 5 minutes) and preincubated in normal horse serum (CCR5 and ED-1) or normal goat serum (GFAP) in PBS; all wash and dilution buffers for ED-1 and GFAP immunocytochemistry contained 0.1% Triton X-100. Sections were then incubated with the selected primary Ab (goat anti-CCR5, 1:100 dilution; mouse anti-ED-1, 1:500; rabbit anti-GFAP, 1:500) for 18 hours at 4°C. Equal amounts of isotype-matched IgG were substituted for the primary Ab in control samples (CCR5-goat IgG; ED-1-mouse IgG; GFAP-rabbit IgG). Sections were washed and incubated with the appropriate biotinylated secondary Ab (CCR5-horse anti-goat, 1:500; ED-1-rat adsorbed horse anti-mouse, 1:200; GFAP-goat anti-rabbit, 1:80). Sections were washed again, endogenous peroxidase activity was blocked (0.3% hydrogen peroxide in methanol for 10 minutes at room temperature), and a Vectastain ABC Elite kit was used to amplify the signal, followed by chromogenic detection with stable DAB. Sections were counterstained in 0.5% cresyl violet, dehydrated in graded ethanols, and coverslipped with Permount. To verify the specificity of CCR5 immunoreactivity, the CCR5 primary Ab was preincubated with a CCR5 blocking peptide; then the preadsorbed Ab was used under the same conditions as the CCR5 primary Ab.
Results
Intrahippocampal injection of 10 nmol of NMDA in P7 rats elicits reproducible focal neuronal loss and atrophy that is maximal in the CA3 subfield of the hippocampus; injury extends both in an antero-posterior plane from the injection site within the hippocampus and also extends to the adjacent thalamus and posterior striatum. 10 At 24 hours after injury, evidence of neuronal injury, eg, loss of Nissl staining, is detectable in the lesioned hippocampus and is most pronounced within the CA3 pyramidal cell layer; the contralateral hippocampus is intact (Figure 1) ▶ .
Figure 1.

NMDA-induced hippocampal injury in neonatal rat brain. Twenty-micron coronal frozen brain sections were prepared from a P8 animal that had received a right intrahippocampal injection of 10 nmol of NMDA 24 hours earlier; the section was stained with cresyl violet to evaluate tissue integrity. Left panel: Intact contralateral hippocampus; right panel: loss of Nissl staining, which is maximal in the Cornu Ammon (CA)3 subfield of the right hippocampus (arrow). CC, corpus callosum; DG, dentate gyrus. Scale bar, 0.5 mm.
NMDA Stimulates CCR5 mRNA Expression
In preliminary RT-PCR assays, CCR5 mRNA was detected in P7 to P8 hippocampus. A semiquantitative RT-PCR method was used to evaluate whether NMDA stimulated CCR5 mRNA expression acutely. CCR5 and GAPDH mRNA expression were compared in the left and right hippocampus of animals injected with PBS 24 hours earlier, in animals that received right intrahippocampal NMDA injections 8, 16, 24, 48, and 72 hours earlier, and in normal P8 animals (Figure 2) ▶ . Increased CCR5 expression was detected as early as 16 hours, peaked at 24 hours after NMDA lesioning, and remained elevated at 72 hours after NMDA lesioning. To confirm that CCR5 mRNA content was increased in the NMDA-lesioned hippocampus, CCR5 expression was compared in samples from left and right hippocampal samples obtained at 24 hours after NMDA injection along with unlesioned P8 controls in three independent experiments, and values were expressed as a percentage of matched control values. There was a consistent, greater than twofold mean increase in CCR5 mRNA in the right hippocampus (P < 0.04, Mann Whitney ranking test, comparing values from the left and right hippocampus).
Figure 2.

Evaluation of NMDA-induced changes in CCR5 mRNA expression by RT-PCR. Samples from left (L) and right (R) hippocampus of PBS-injected controls (evaluated at 24 hours after injection), animals that received right intrahippocampal NMDA (10 nmol) injections (evaluated at 8, 16, 24, 48, and 72 hours after injection), and normal P8 samples were assayed concurrently. PCR products were electrophoresed through a 2% agarose gel, which was stained with ethidium bromide to reveal GAPDH (298 bp) and CCR5 (219 bp) bands (lower panel) in all samples. The histogram (upper panel) provides a quantitative estimate of CCR5 mRNA (values in arbitrary optical density units), based on normalization to GAPDH mRNA/sample. CCR5 gene expression peaked at 24 hours after injection in the right hippocampus; a consistent, greater than twofold mean increase in CCR5 mRNA was found in three independent samples assayed at this time interval (P < 0.04, Mann Whitney test, comparing CCR5 mRNA expression in the left and right hippocampus).
In preliminary in situ hybridization analysis using an antisense 35S-labeled riboprobe to detect CCR5 mRNA, a substantial hybridization signal was detected ipsilaterally in brain samples obtained at 24 hours after a right intrahippocampal NMDA (10 nmol) injection, whereas no hybridization signal was detected in samples from either normal unlesioned P8 or PBS-injected control animals (data not shown). Under the same assay conditions, brain samples from three additional NMDA-lesioned animals were evaluated, and representative autoradiograms are presented in Figure 3 ▶ . Consistent features of the distribution of CCR5 mRNA included an intense, focal hybridization signal in the pyramidal cell layer of the lesioned hippocampus and a more diffuse hybridization signal extending to adjacent ipsilateral structures, including the posterior striatum, thalamus, and cortex. Note that NMDA-mediated neuronal injury typically extends from the injection site, both within the lesioned hippocampus and also to these adjacent regions. There was a corresponding increase in CCR5 mRNA expression in these vulnerable regions. Of interest, no hybridization signal was evident in the corpus callosum, where physiologically activated microglia are concentrated at this developmental stage. 5 No hybridization signal was detected in sections assayed concurrently with sense strand control riboprobes (data not shown).
Figure 3.

In situ hybridization analysis of CCR5 mRNA in NMDA-lesioned neonatal rat brain. These dark-field autoradiograms were prepared from a P8 animal that had received a right intrahippocampal injection of 10 nmol of NMDA 24 hours earlier. A to C: Generated from sections at three sequential anatomical levels of the hippocampus. 35S-Labeled antisense riboprobe was used to detect CCR5 mRNA (see Materials and Methods). Increased hybridization signal was evident at all three anatomical levels, both in the NMDA-lesioned hippocampus and also in adjacent structures, including the ipsilateral posterior striatum (eg, in A), thalamus (eg, in B), and cortex; hybridization signal was consistently most intense in the right hippocampal pyramidal cell layer. Note the absence of hybridization signal in the corpus callosum (B), an area that has physiologically activated microglia at this developmental stage. Similar results were obtained in two additional NMDA-lesioned animals, and all control sections assayed with sense riboprobes revealed no hybridization signal (data not shown). C, cortex; H, hippocampus; S, striatum; T, thalamus; CC, corpus callosum.
NMDA Stimulates CCR5 Protein Expression
The only method that is currently feasible to estimate tissue content of CCR5 is a Western blot assay, which does not yield quantitative data. Western blot assay conditions detected a single CCR5 protein band (41 kd) in extracts of cerebral hemispheres of lesioned and normal P8 rats and normal adult spleen. CCR5 was detected in all samples assayed; the signal was more intense in extracts obtained from the lesioned side as compared with the contralateral or control cerebral hemisphere samples (data not shown). To directly evaluate changes in hippocampal CCR5 protein expression, extracts were prepared from pooled microdissected hippocampal samples of animals that had received right intrahippocampal injection of NMDA (10 nmol) or PBS 32 hours earlier and from unlesioned littermates. CCR5 was detected in all hippocampal samples (Figure 4) ▶ . A substantial increase in CCR5 protein in the lesioned hippocampus was evident in comparison with samples from either normal P8 or PBS-injected animals. In addition, a less pronounced increase was evident in the contralateral hippocampus of NMDA-lesioned animals. These trends were replicated in three independent assays using different tissue samples collected at 32 hours after lesioning (data not shown).
Figure 4.

Western blot analysis of CCR5 protein in normal and lesioned neonatal rat hippocampus. Samples were prepared from left (L) and right (R) hippocampus of animals that received a right intrahippocampal injection of 10 nmol of NMDA or PBS 32 hours earlier and from unlesioned P8 animals were analyzed, together with a sample from adult rat spleen (included as a positive control; see Materials and Methods). A single immunoreactive band of the expected size (approximately 41 kd) was detected in all samples tested. There was a robust increase in CCR5 content in the NMDA-lesioned hippocampus in comparison with samples from normal P8 and PBS-injected animal; a less pronounced increase was also evident in the left hippocampus of NMDA-lesioned animals.
To identify the cellular source(s) of CCR5, an immunocytochemistry assay was developed. To facilitate identification of CCR5-immunoreactive cells, representative adjacent sections were also processed for ED-1 and GFAP immunocytochemistry. In all samples evaluated, ED-1-immunoreactive activated microglia were identified within the corpus callosum bilaterally (Figure 5) ▶ , an expected finding at this developmental stage. In NMDA-lesioned brains, ED-1-immunoreactive cells were consistently detected throughout the right hippocampus; this was accompanied by an apparent relative depletion of ED-1-immunoreactive cells in the ipsilateral corpus callosum (as illustrated in Figure 5B ▶ ). A consistent feature of ED-1 immunostaining was that reactive cells infiltrated the pyramidal cell layer in the lesioned hippocampus. These ED-1-immunoreactive cells included both cells with morphological features of activated microglia (enlarged cell bodies with short thickened processes) and round cells (which could be either fully activated microglia and/or blood-derived monocytes). GFAP-immunoreactive astrocytes were also consistently detected in P8 rat brain; their concentration was highest in the corpus callosum and other white matter tracts (data not shown). Immunocytochemical analysis using isotype-matched mouse and rabbit IgG revealed no immunoreactivity (data not shown).
Figure 5.

ED-1 immunocytochemistry analysis to evaluate the microglial/monocyte response after NMDA lesioning. Coronal brain sections were prepared from a P8 animal that had received a right intrahippocampal injection of 10 nmol of NMDA 32 hours earlier and were processed for ED-1 immunocytochemistry (see Materials and Methods); sections were lightly counterstained with cresyl violet to facilitate identification of anatomical landmarks. In the left hippocampus, contralateral to the lesion, there are few ED-1-immunoreactive cells (A); however, immunoreactive cells are concentrated in the overlying corpus callosum (A, arrow). In contrast, ED-1-immunoreactive cells infiltrate the lesioned hippocampus, whereas fewer, more diffusely distributed immunoreactive cells are apparent in the adjacent corpus callosum (B, arrow). C and D: Enlargements of the boxed regions in A and B, respectively. Within the pyramidal cell layer of the unlesioned hippocampus no infiltrating microglia or monocytes are evident (C). In the lesioned hippocampus, there are many ED-1-immunoreactive cells both within the pyramidal cell layer (arrows) and adjacent to it; the morphology of these immunoreactive cells includes both cells with short, thick processes, indicative of activated microglia, and round cells (which could be fully activated microglia or blood-derived monocytes). Immunocytochemistry with isotype-matched mouse IgG revealed no positive staining (data not shown). Peroxidase staining protocol with DAB chromogenic detection and cresyl violet counterstain; scale bars, 0.5 mm (A and B) and 50 μm (C and D).
Although CCR5 protein was detected in tissue homogenates from normal P8 brain, no specific cellular staining was detected immunocytochemically in unlesioned and PBS-injected control P8 brain (not shown). The absence of CCR5 immunoreactivity in tissue sections likely reflects the lower sensitivity of the immunocytochemistry assay than of the Western blot (which assayed tissue extracts pooled from three animals). Similarly, although CCR5 was detected in protein extracts from the contralateral hippocampus of animals that had received right intrahippocampal injections of NMDA (10 nmol) 32 hours earlier (Figure 4) ▶ , in coronal brain sections prepared from comparably lesioned animals, few immunoreactive cells were detected in the contralateral hippocampus (Figure 6A) ▶ . In contrast, CCR5-immunoreactive cells were widely distributed in the lesioned hippocampus of animals that had received right intrahippocampal injections of NMDA (10 nmol) 32 hours earlier (Figure 6B) ▶ . Immunoreactive cells were concentrated in the pyramidal cell layers of the lesioned hippocampus. The distribution of CCR5 immunoreactivity was similar in the right hippocampus at 48 and 72 hours after NMDA lesioning, but CCR5 immunoreactivity was no longer detected at 5 days after lesioning (data not shown). In samples evaluated at an earlier time, 24 hours after lesioning, no CCR5-immunoreactive cells were seen within the pyramidal cell layer of the right hippocampus (Figure 6C) ▶ . Note that no CCR5-immunoreactive cells were detected in the corpus callosum, which has a high density of physiologically activated microglia in P8 rat brain. Immunocytochemistry assays performed with anti-CCR5 Ab that had been preadsorbed with the blocking peptide revealed no reactive cells (Figure 6D) ▶ ; similarly, there was no staining if isotype-matched goat IgG was substituted for the primary Ab (data not shown). In PBS-injected and unlesioned controls, CCR5-immunoreactive cells were sparsely distributed throughout the brain (data not shown).
Figure 6.
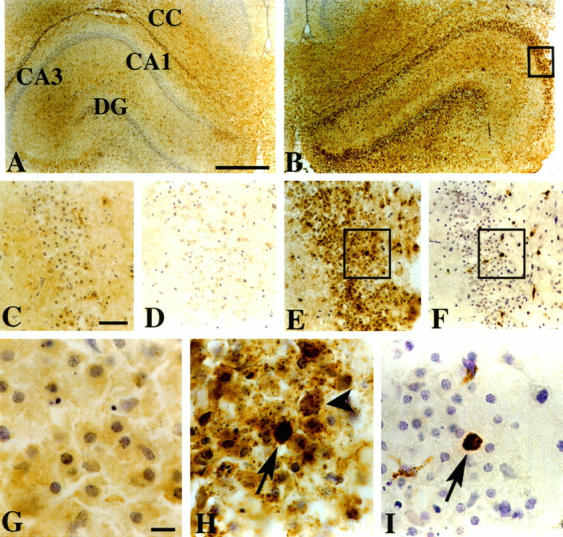
CCR5 immunocytochemistry in NMDA-lesioned brain. In coronal sections, analyzed at 32 hours after a right intrahippocampal injection of NMDA, CCR5 immunocytochemistry revealed no distinct cellular staining in the intact contralateral hippocampus (A); in the lesioned hippocampus, immunoreactivity was concentrated within the pyramidal cell layer (B, E, and H). E: Enlargement of boxed region in B. In all samples evaluated at an earlier time, 24 hours after lesioning, no immunoreactivity was evident in the right hippocampal CA3 subfield (C). D: Evidence of the specificity of immunostaining in the right hippocampus at 32 hours after injection of NMDA; preadsorption of the CCR5 Ab with a blocking peptide effectively eliminated immunoreactivity. In the pyramidal cell layer of the lesioned hippocampus at 32 hours, two distinct patterns of CCR5 immunoreactivity could be distinguished: in the majority of reactive cells (which had the distribution and morphology of injured neurons), there was a diffuse, speckled pattern of staining (H, arrowhead); in a minority of cells there was much more intense homogeneous staining (H, arrow). To evaluate the cellular identity of these intensely immunoreactive cells, adjacent tissue sections were processed for ED-1 immunocytochemistry (see Figure 5 ▶ ). ED-1-immunoreactive cells (both microglia with thickened processes and round cells) infiltrated the pyramidal cell layer (F). H and I: Enlargements of the boxed regions in E and F, respectively. Comparison of the distributions of CCR5-immunoreactive and ED-1-immunoreactive cells in adjacent sections revealed that the homogeneously, intensely stained CCR5-immunoreactive cells corresponded well with ED-1-immunoreactive cells (H and I, corresponding arrows). Immunocytochemistry with isotype-matched goat IgG revealed no positive staining (data not shown). Peroxidase staining protocol with DAB chromogenic detection and cresyl violet counterstain; scale bars, 0.5 mm (A and B), 50 μm (C to F), and 10 μm G to I). CA, Cornu Ammon; DG, dentate gyrus; CC, corpus callosum.
In the NMDA-lesioned hippocampus, within the pyramidal cell layer, two distinct patterns of CCR5 immunoreactivity could be distinguished (as illustrated in Figure 6H ▶ ): in the majority of reactive cells, there was a diffuse, speckled pattern of staining, whereas in a minority of cells there was much more intense homogeneous staining. The cells with punctate CCR5 immunostaining had the distribution and morphology of injured neurons. To evaluate the cellular identity of the intensely immunoreactive cells, the distributions of CCR5-immunoreactive and both ED-1- and GFAP-immunoreactive cells were compared in adjacent tissue sections; the homogeneous, intensely stained CCR5-immunoreactive cells corresponded well with ED-1-immunoreactive cells (Figure 6, H and I) ▶ . These results demonstrated that infiltrating microglia/monocytes expressed CCR5. There was no overlap between the distribution of CCR5-immunoreactive cells and the distribution of GFAP-immunoreactive astrocytes.
Discussion
Our data demonstrate that acute excitotoxic injury stimulates CCR5 mRNA and protein expression in the neonatal rat brain. Increased CCR5 mRNA expression was consistently noted at 24 hours after lesioning; increased CCR5 protein expression was first evident at 32 hours, persisted up to 72 hours, and was no longer detected at 5 days after lesioning. CCR5 expression increased selectively in regions vulnerable to irreversible tissue damage (ie, ipsilateral hippocampus and adjacent posterior striatum, cortex, and thalamus). The most surprising feature of CCR5 expression was its anatomical distribution; CCR5 was identified in two distinct cell populations. Scattered, intensely immunoreactive cells within the lesioned hippocampus were identified as infiltrating, activated microglia/monocytes, based on morphological features and expression of an activated macrophage-specific antigen (ED-1); in addition, many cells with the morphology and distribution of injured pyramidal neurons expressed CCR5. Immunocytochemical assays demonstrated distinct staining patterns in the two cell types: an intense, homogeneous staining pattern in microglia/monocytes and a punctate, speckled pattern (typical of membrane-bound receptors) in other immunoreactive cells.
Both excitotoxic and hypoxic-ischemic injury elicit a robust microglial response in neonatal rat brain. 31,5 NMDA lesioning elicited a relatively rapid microglia/monocyte reaction in P7 animals, as has been previously reported. 31 Based on morphological studies using lectin histochemistry, 31 in which the transition from resting to activated microglia can be visualized, it is evident that a substantial fraction of these infiltrating cells are activated microglia. The contribution of blood-derived monocytes to this inflammatory response is uncertain, as fully activated microglia and blood-derived monocytes cannot readily be distinguished by conventional methods using morphological or immunological criteria. 32
At this developmental stage, there is a dense population of physiologically activated microglia in the corpus callosum; our data suggest that there is a redistribution of activated microglia from the adjacent corpus callosum into the lesioned hippocampus (Figure 5) ▶ . Both resident microglia within the hippocampus and physiologically activated microglia in the adjacent corpus callosum could respond to acute excitotoxic injury. Based on comparison with the distribution of ED-1-immunoreactive cells within the lesioned hippocampus, our results also demonstrate that only a small fraction of activated microglia/monocytes expressed CCR5, even in the lesion core. In addition, neither in situ hybridization nor immunocytochemistry revealed any CCR5 expression in physiologically activated microglia in the corpus callosum. Together, these findings suggest that activated microglia/monocytes do not represent a homogeneous cell population; the features that distinguish the subset of CCR5-expressing cells are currently unknown.
Whether CCR5-expressing microglia/monocytes play a distinct pathophysiological role in contributing to progression of injury and/or intrinsic repair mechanisms remains to be determined. Although investigation of the known ligands for CCR5 was beyond the scope of this study, certain ligands for CCR5, such as macrophage inflammatory protein (MIP)-1α and RANTES, are up-regulated in acute and chronic models of brain injury. 33-36 These studies suggest that CCR5-specific ligands could play a role in immune-mediated responses in the brain.
Both RT-PCR and Western blot assays of tissue homogenates provided evidence that CCR5 is expressed in normal neonatal brain. However, levels of expression were below the limits of sensitivity of both our in situ hybridization and immunocytochemical analysis so that the anatomical distribution and cellular identity of CCR5-expressing cells in normal brain could not be determined. These results are consistent with previous reports that CCR5 is expressed in normal human brain tissue 37-39 and in cultured human fetal neurons. 40 These observations, along with our data, suggest that CCR5 may have a physiological role in the CNS.
Several recent studies have also reported constitutive and/or disease-related increases in expression of chemokine receptors in the brain. CCR5, 37,40 CXCR4, 38,40,41 CXCR2, 40,42 and CCR1 40 have been detected in human neurons. In addition, CCR5 has been detected in microglia, 27,38,43 and CXCR4 has been detected in both astrocytes and microglia. 38,41 Increased CCR5 expression has been detected in perivascular infiltrates in brains of primates infected with simian immunodeficiency virus and in children and adults infected with HIV; 38,39 of interest, multiple chemokine receptors, including CCR5, were expressed on hippocampal pyramidal neurons. 37,39 In cultured human fetal neurons, CD4-independent binding of the HIV-1 envelope protein gp120 to CXCR4 was recently reported. 40 Perhaps most surprisingly, these neurons had a chemotactic response to stromal-derived factor-1, the ligand for CXCR4. Together with our observations, these studies strongly suggest that chemokines may exert direct effects on neurons under both physiological and pathological conditions.
Certain chemokine receptors, including CCR5, can act as co-receptors for HIV-1-mediated infection of CD4-positive lymphocytes 25,26,44 and microglia. 27 In addition, the ligands for CCR5 can inhibit infection by certain strains of HIV-1, 45 and decreased susceptibility to HIV-1 infection has been linked with mutations in the CCR5 gene. 46,47 Although the role of CCR5 in the CNS remains unknown, the identified human CCR5 mutations have no significant effect on CNS development. Despite the rapidly growing body of data suggesting that chemokines may be involved in a diverse array of CNS inflammatory processes, there is little information about the functional significance of chemokine receptor expression in neurons. Expression of certain CC chemokines, such as MCP-1 and MIP-1α, are up-regulated in experimental models of hypoxic-ischemic injury, 8,33,34,48 mechanical brain injury, 49,50 and experimental autoimmune encephalomyelitis; 35,36,50,51 MIP-1α has also been localized to reactive astrocytes 34 and macrophages. 33,36
There is currently no information about the regulatory mechanisms that could stimulate CCR5 expression in the brain. The timing of CCR5 induction by excitotoxic injury in the neonatal rat brain differs from that of other inflammatory mediators evaluated in this brain injury model. NMDA lesioning elicits a rapid increase in interleukin (IL)-1β and tumor necrosis factor (TNF)-α gene expression. 1 IL-1β expression peaks at 6 hours after NMDA lesioning and wanes rapidly; MCP-1 mRNA expression peaks at 8 to 16 hours and is absent at 24 hours after lesioning. 7 In addition, induction of mRNA expression of the related chemokine receptor CCR2 peaks at 16 hours and wanes substantially at 24 hours (J.M. Galasso and F.S. Silverstein, unpublished observations). The distinct temporal features of NMDA-stimulated CCR2 and CCR5 mRNA expression suggest that there are distinct molecular signals that regulate chemokine receptor expression after acute brain injury. IL-1β and TNF-α can induce expression of MIP-1α and MIP-1β in human fetal microglia. 52 Interestingly, the temporal pattern of cytokine-stimulated MIP-1 expression, observed in vitro, closely parallels the time course of CCR5 expression in our model of neonatal brain injury. Whether pro-inflammatory cytokines regulate CCR5 expression is unknown.
Excitatory amino acid receptor over-activation, by endogenous excitatory neurotransmitters such as glutamate, represents a pivotal mechanism contributing to neurodegeneration in neurological disorders such as cerebral ischemia and acute brain trauma. 10,11 Our data provide the first evidence that excitotoxic brain injury regulates CCR5 expression. Both our data and recent studies demonstrating neuronal CCR5 expression in HIV-1-infected brains suggest that CCR5 may play a pathophysiological role in the brain’s response to diverse acute and chronic insults.
Acknowledgments
We thank Drs. Rory M. Marks and Jeffrey S. Warren for their critical evaluations of this manuscript. Preliminary reports of these results were presented at the annual meetings of the Child Neurology Society (Phoenix, October 1997) and the Society for Neuroscience (New Orleans, October 1997).
Footnotes
Address reprint requests to Dr. Faye S. Silverstein, 8301 MSRB III, 1150 West Medical Center Drive, University of Michigan, Ann Arbor, MI 48109-0646. E-mail: fsilvers@umich.edu.
Supported by US Public Health Service grant NS 31054 (to F.S. Silverstein).
References
- 1.Silverstein FS, Barks JDE, Hagan P, Liu XH, Ivacko J, Szaflarski J: Cytokines and perinatal brain injury. Neurochem Int 1997, 30:375-383 [DOI] [PubMed] [Google Scholar]
- 2.Dammann O, Leviton A: Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 1997, 42:1-8 [DOI] [PubMed] [Google Scholar]
- 3.Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO: Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1β, and tumor necrosis factor-α), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol 1997, 177:19-26 [DOI] [PubMed] [Google Scholar]
- 4.Szaflarski J, Burtrum D, Silverstein FS: Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stroke 1995, 26:1093-1100 [DOI] [PubMed] [Google Scholar]
- 5.Ivacko JA, Sun R, Silverstein FS: Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res 1996, 39:1-9 [DOI] [PubMed] [Google Scholar]
- 6.Hagan P, Barks JDE, Yabut M, Davidson BL, Roessler B, Silverstein FS: Adenovirus-mediated over-expression of interleukin-1 receptor antagonist reduces susceptibility to excitotoxic brain injury in perinatal rats. Neuroscience 1996, 75:1033-1045 [DOI] [PubMed] [Google Scholar]
- 7.Szaflarski J, Ivacko J, Liu XH, Warren JS, Silverstein FS: Excitotoxic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. Mol Brain Res 1998, 55:306-314 [DOI] [PubMed] [Google Scholar]
- 8.Ivacko J, Szaflarski J, Malinak C, Flory C, Warren JS, Silverstein FS: Hypoxic-ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J Cereb Blood Flow Metab 1997, 17:759-770 [DOI] [PubMed] [Google Scholar]
- 9.Hagan P, Poole S, Bristow AF, Tilders F, Silverstein FS: Intracerebral NMDA injection stimulates production of interleukin-1β in perinatal rat brain. J Neurochem 1996, 67:2215-2218 [DOI] [PubMed] [Google Scholar]
- 10.McDonald JW, Silverstein FS, Johnston MV: Neurotoxicity of N-methyl-d-aspartate is markedly enhanced in developing rat central nervous system. Brain Res 1988, 459:200-203 [DOI] [PubMed] [Google Scholar]
- 11.Choi DW: Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci 1995, 18:58-60 [PubMed] [Google Scholar]
- 12.Premack BA, Schall TJ: Chemokine receptors: gateways to inflammation and infection. Nature Med 1996, 2:1174-1178 [DOI] [PubMed] [Google Scholar]
- 13.Schall TJ, Bacon K, Camp RDR, Kaspari JW, Goeddel DV: Human macrophage inflammatory protein-α (MIP-1α) and MIP-1β chemokines attract distinct populations of lymphocytes. J Exp Med 1993, 177:1821-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA: Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci USA 1994, 91:3652-3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell MD, Taub DD, Perry VH: Overriding the brain’s intrinsic resistance to leukocyte recruitment with intraparenchymal injections of recombinant chemokines. Neuroscience 1996, 74:283-292 [DOI] [PubMed] [Google Scholar]
- 16.Baggiolini M, Walz A, Kunkel SL: Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest 1989, 84:1045-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, Leonard EJ: Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci USA 1987, 84:9233-9237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walz A, Burgener R, Car B, Baggiolini M, Kunkel SL, Strieter RM: Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin-8. J Exp Med 1991, 174:1355-1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelner GS, Kennedy J, Bacon KB, Kleyensteuber S, Largaespada DA, Jenkins NA, Copeland NG, Bazan JF, Moore KW, Schall TJ, Zlotnik A: Lymphotactin: a cytokine that represents a new class of chemokine. Science 1994, 266:1395-1399 [DOI] [PubMed] [Google Scholar]
- 20.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ: A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385:640-644 [DOI] [PubMed] [Google Scholar]
- 21.Dunstan CN, Salafranca MN, Adhikari S, Xia Y, Feng L, Harrison JK: Identification of two rat genes orthologous to the human interleukin-8 receptors. J Biol Chem 1996, 271:32770-32776 [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Salafranca MN, Adhikari S, Xia Y, Feng L, Sonntag MK, deFiebre CM, Pennell NA, Streit WJ, Harrison JK: Chemokine receptor expression in cultured glia and rat experimental allergic encephalomyelitis. J Neuroimmunol 1998, 86:1-12 [DOI] [PubMed] [Google Scholar]
- 23.Giulian D, Vaca K, Noonan CA: Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990, 250:1593-1596 [DOI] [PubMed] [Google Scholar]
- 24.Giulian D, Wendt E, Vaca K, Noonan CA: The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci USA 1993, 90:2769-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA: CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272:1955-1958 [DOI] [PubMed] [Google Scholar]
- 26.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA: HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381:667-673 [DOI] [PubMed] [Google Scholar]
- 27.He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D: CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385:645-649 [DOI] [PubMed] [Google Scholar]
- 28.Barks JDE, Liu XH, Sun R, Silverstein FS: gp120, a human immunodeficiency virus-1 coat protein, augments excitotoxic hippocampal injury in perinatal rats. Neuroscience 1997, 76:397-409 [DOI] [PubMed] [Google Scholar]
- 29.Lipton SA: Requirements for macrophages in neuronal injury induced by HIV envelope protein gp120. NeuroReport 1992, 3:913-915 [DOI] [PubMed] [Google Scholar]
- 30.McDonald JW, Roeser NF, Silverstein FS, Johnston MV: Quantitative assessment of neuroprotection against NMDA-induced brain injury. Exp Neurol 1989, 106:289-296 [DOI] [PubMed] [Google Scholar]
- 31.Acarin L, Gonzalez B, Castellano B, Castro AJ: Microglial response to N-methyl-d-aspartate-mediated excitotoxicity in the immature rat brain. J Comp Neurol 1996, 367:361-374 [DOI] [PubMed] [Google Scholar]
- 32.Flaris NA, Densmore TL, Molleston MC, Hickey WF: Characterization of microglia and macrophages in the central nervous system of rats: definition of the differential expression of molecules using standard and novel monoclonal antibodies in normal CNS and in four models of parenchymal reaction. Glia 1993, 34–40 [DOI] [PubMed]
- 33.Takami S, Nishikawa H, Minami M, Nishiyori A, Sato M, Akaike A, Satoh M: Induction of macrophage inflammatory protein MIP-1α mRNA on glial cells after focal cerebral ischemia in the rat. Neurosci Lett 1997, 227:173-176 [DOI] [PubMed] [Google Scholar]
- 34.Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KMA: Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol 1995, 56:127-134 [DOI] [PubMed] [Google Scholar]
- 35.Godiska R, Chantry D, Dietsch GN, Gray PW: Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol 1995, 58:167-176 [DOI] [PubMed] [Google Scholar]
- 36.Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM: Synchronous synthesis of α- and β-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol 1997, 150:617-630 [PMC free article] [PubMed] [Google Scholar]
- 37.Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ: Cellular localization of the chemokine receptor CCR5. Am J Pathol 1997, 151:1341-1351 [PMC free article] [PubMed] [Google Scholar]
- 38.Vallat AV, De Girolami U, He J, Mhashilkar A, Marasco W, Shi B, Gray F, Bell J, Keohane C, Smith TW, Gabuzda D: Localization of HIV-1 co-receptors CCR5 and CXCR4 in the brain of children with AIDS. Am J Pathol 1998, 152:167-178 [PMC free article] [PubMed] [Google Scholar]
- 39.Westmoreland SV, Rottman JB, Williams KC, Lackner AA, Sasseville VG: Chemokine receptor expression on resident and inflammatory cells in the brain of macaques with simian immunodeficiency virus encephalitis. Am J Pathol 1998, 152:659-665 [PMC free article] [PubMed] [Google Scholar]
- 40.Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, Taub D, Horuk R: CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in human neurons. Curr Biol 1997, 7:112-121 [DOI] [PubMed] [Google Scholar]
- 41.Lavi E, Strizki JM, Ulrich AM, Zhang W, Fu L, Wang Q, O’Connor M, Hoxie JA, Gonzalez-Scarano F: CXCR-4 (fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons. Am J Pathol 1997, 151:1035-1042 [PMC free article] [PubMed] [Google Scholar]
- 42.Horuk R, Martin AW, Wang Z, Schweitzer L, Gerassimides A, Guo H, Lu Z, Hesselgesser J, Perez HD, Kim J, Parker J, Hadley TJ, Peiper SC: Expression of chemokine receptors by subsets of neurons in the central nervous system. J Immunol 1997, 158:2882-2890 [PubMed] [Google Scholar]
- 43.Xia MQ, Qin SX, Wu LJ, Mackay C, Hyman BT: Immunohistochemical study of chemokine receptors CCR3 and CCR5 and their ligands in Alzheimer’s disease and control brains. Soc Neurosci Abstr 1997, 23:563 [Google Scholar]
- 44.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW: A dual-tropic primary HIV-1 isolate that uses fusin and the β-chemokine receptors CKR-5, CKR-3, CKR-2b as fusion cofactors. Cell 1996, 85:1149-1158 [DOI] [PubMed] [Google Scholar]
- 45.Deng H, Liu R, Ellmeimer W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR: Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381:661-666 [DOI] [PubMed] [Google Scholar]
- 46.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86:367-377 [DOI] [PubMed] [Google Scholar]
- 47.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyidermans G, Verhofstede C, Burtonboy G, Georges M, lmai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M: Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382:722-725 [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Yue T, Barone FC, Feuerstein GZ: Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke 1995, 26:661-666 [DOI] [PubMed] [Google Scholar]
- 49.Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM: Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol 1996, 156:4363-4368 [PubMed] [Google Scholar]
- 50.Berman JW, Guida MP, Warren J, Amat J, Brosnan CF: Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J Immunol 1996, 156:3017-3023 [PubMed] [Google Scholar]
- 51.Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK: Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB 1993, 7:592-600 [DOI] [PubMed] [Google Scholar]
- 52.McManus CM, Brosnan CF, Berman JW: Cytokine induction of MIP-1α and MIP-1β in human fetal microglia. J Immunol 1998, 160:1449-1455 [PubMed] [Google Scholar]