

Abstract
Mitochondrial (mt)DNA defects, both deletions and tRNA point mutations, have been associated with cardiomyopathies. The aim of the study was to determine the prevalence of pathological mtDNA mutations and to assess associated defects of mitochondrial enzyme activity in dilated cardiomyopathy (DCM) patients with ultrastructural abnormalities of cardiac mitochondria. In a large cohort of 601 DCM patients we performed conventional light and electron microscopy on endomyocardial biopsy samples. Cases with giant organelles, angulated, tubular, and concentric cristae, and crystalloid or osmiophilic inclusion bodies were selected for mtDNA analysis. Mutation screening techniques, automated DNA sequencing, restriction enzyme digestion, and densitometric assays were performed to identify mtDNA mutations, assess heteroplasmy, and quantify the amount of mutant in myocardial and blood DNA. Of 601 patients (16 to 63 years; mean, 43.5 ± 12.7 years), 85 had ultrastructural evidence of giant organelles, with abnormal cristae and inclusion bodies; 19 of 85 (22.35%) had heteroplasmic mtDNA mutations (9 tRNA, 5 rRNA, and 4 missense, one in two patients) that were not found in 111 normal controls and in 32 DCM patients without the above ultrastructural mitochondrial abnormalities. In all cases, the amount of mutant was higher in heart than in blood. In hearts of patients that later underwent transplantation, cytochrome c oxidase (Cox) activity was significantly lower in cases with mutations than in those without or controls (P = 0.0008). NADH dehydrogenase activity was only slightly reduced in cases with mutations (P = 0.0388), whereas succinic dehydrogenase activity did not significantly differ between DCM patients with mtDNA mutations and those without or controls. The present study represents the first attempt to detect a morphological, easily identifiable marker to guide mtDNA mutation screening. Pathological mtDNA mutations are associated with ultrastructurally abnormal mitochondria, and reduced Cox activity in a small subgroup of non-otherwise-defined, idiopathic DCMs, in which mtDNA defects may constitute the basis for, or contribute to, the development of congestive heart failure.
In the last few years mitochondrial (mt)DNA deletions 1 or transfer (t)RNA point mutations 2-10 have been identified in cardiomyopathies phenotypically characterized by hypertrophy, atrio-ventricular block, or congestive heart failure. 11 Although some cardiomyopathies associated with mtDNA defects have been reported as clinically isolated disorders, 2-4 they may also be associated with peripheral myopathies 5-7 or with multisystem syndromes; 9-11 in these latter cases, cardiac involvement may be the major determinant of poor outcomes. 10,11
Mitochondrial tRNA gene mutations variably affect activity of the respiratory chain complexes. Activity of complex II is not affected as it is entirely encoded by nuclear DNA, whereas cytochrome C oxidase (Cox) activity (or complex IV), which is partially encoded by mtDNA genes and constitutes the electron transport chain along with complex I, 12 is frequently reduced in patients with mtDNA tRNA mutations. Pathological tRNA mutations are characteristically heteroplasmic and absent from controls and affect highly conserved regions; 12,13 tRNA mutations have been reported in patients with hypertrophic cardiomyopathy, particularly in those who develop late congestive heart failure 14 or with not otherwise defined cardiomyopathies and congestive heart failure. 5,6,9 Rarely, missense mutations in polypeptide-coding genes have been linked to multisystem syndromes. 15 Ultrastructural mitochondrial changes and enzyme defects characterize affected tissue in mtDNA-related disorders. 7,11,12,16 Similar ultrastructural mitochondrial changes and enzyme defects may be found in part of the cardiomyopathies. 2,4,9,17,18
Although molecular mitochondrial abnormalities constitute an attractive basis of development of dilated cardiomyopathy (DCM), the prevalence and characteristics of mtDNA mutations have not been extensively studied. In the present study, we attempted to determine the prevalence of mitochondrial abnormalities associated with mtDNA mutations in a large cohort of patients with idiopathic DCM and to assess whether mtDNA mutations were unique, heteroplasmic, localized to conserved regions, and absent in healthy controls, thus fulfilling the requisites for being accepted as having a pathological role. The genetic analysis was performed to identify mutations in tRNA, ribosomal (r)RNA, and flanking regions of mtDNA. The selection of cases for molecular analyses was based on mitochondrial ultrastructural changes in the endomyocardial biopsies (EMBs) of the patients. The study also evaluated the activity of oxidative enzyme complex (Cox, succinate dehydrogenase, (SDH), and the reduced form of nicotinamide-adenine dinucleotide (NADH) dehydrogenase).
Materials and Methods
DCM Patients and Control Subjects
A 10-year series of 601 consecutive DCM patients was evaluated in the Department of Cardiology at the IRCCS Policlinico San Matteo, Pavia, Italy. All patients underwent clinical examination, biochemical investigations (creatine-phosphokinase (CPK) and lactic dehydrogenase (LDH)), electrocardiography, chest x-rays, two-dimensional and Doppler echocardiography, right and left heart catheterization, coronary angiography, and right ventricular EMB.
Biopsy specimens from all patients, in addition to routine histological study, were evaluated ultrastructurally and immunohistochemically. Of the 601 patients, 85 (ages 16 to 63 years; mean age, 43.5 ± 12.7; 67 males and 18 females) had a combination of the following peculiar mitochondrial ultrastructural abnormalities: mitochondrial proliferation and variable matrix density associated with giant ring-shaped mitochondria, concentric, angulated, and tubular cristae, crystalloid and osmiophilic inclusion bodies, and glycogen or lipid inclusions. Cristolysis, swelling, and variable size of mitochondria were considered as nonspecific changes. In the 67 male patients, defects of dystrophin immunolabeling with specific anti-COOH-terminus, NH-terminus, and rod domain antibodies (Novocastra, Newcastle, UK) were not identified. The biopsy specimens from these 85 patients were subjected to molecular analysis for mutations in mtDNA, tRNA, rRNA, and flanking regions. Of the 516 DCM patients without the ultrastructural changes considered for screening, 32 random cases (25 males; mean age, 41.3 ± 10.7 years) were selected as affected controls. Controls for mtDNA analysis were enrolled from the blood donor pool (see below). For comparative enzyme activity tests, normal myocardial tissue samples were obtained as right ventricular biopsies from in-hospital donor hearts before transplantation (EKG, echo, clinical, and biochemical evaluation; n = 6; 5 males; mean age, 38 ± 11 years) when total ischemic time was ≤60 minutes.
Endomyocardial Biopsy and Pathological Study
Five to seven right ventricular EMB samples were obtained for each patient and processed for light microscopy and ultrastructural study. 19-22 Sections were examined with a Zeiss 902 electron microscope. One frozen sample was used for nucleic acid extraction and genetic analysis. In 17 DCM cases (5 with mtDNA mutations and 12 without mtDNA mutations) that underwent cardiac transplantation later on in the course of the disease, we evaluated the mitochondrial Cox, NADH dehydrogenase, and SDH activity in myocardial tissue. 23-25 Pretransplant biopsies of six normal donor hearts were used as controls.
Mitochondrial DNA Study
Molecular analysis of mitochondrial DNA was performed both in the EMB samples as well as in peripheral blood of the 85 patients with ultrastructural mitochondrial abnormalities. Control blood samples were obtained from 111 healthy, age- and sex-matched blood donors (ages, 18 to 60 years; mean age, 39 ± 8 years; 83 males and 28 females). The control subjects were ensured to have normal electrocardiogram and biochemical parameters including CPK and LDH. Whenever a mtDNA mutation was identified, all available maternal relatives of the probands were also tested for the given mutation (n = 49 for the 85 patients).
For molecular analysis, total genomic DNA was extracted from EMBs and from peripheral blood samples. Specific oligonucleotide primers were synthesized. The selected regions were amplified by polymerase chain reaction (PCR) and subjected to denaturing gradient gel electrophoresis (DGGE) for tRNA and rRNA or to single-strand conformational polymorphism (SSCP) for coding regions 26 for mutation screening. The abnormal conformers underwent direct DNA sequencing for identification of mutation sites, followed by restriction enzyme digestion to assess heteroplasmy of each given mutation and by repeated densitometric analysis to quantify the amount of mutant in the heart and peripheral blood DNA (Tables 1 and 2) ▶ ▶ .
Table 1.
Heteroplasmic Mutations in DCM Patients with Ultrastructural Abnormalities of Mitochondria*
| Base change | S/F | Gene | RE +/− | Mismatch primer† | RE digestion‡ | Mutant % | n | ||
|---|---|---|---|---|---|---|---|---|---|
| Mutant | Wild type | Blood | Heart | ||||||
| A3260G | F | tRNALeuUUR | XmnI + | R: AAA3267-9TTC | 93 bp+ 50 bp | 143 bp | 55 | 75 | 1 |
| T4314A | S | tRNAIle | MseI− | 292 bp+ 61 bp | 134 bp+ 158 bp+ 61 bp | 60 | 85 | 1 | |
| A4315G | S | tRNAIle | MseI− | 292 bp+ 61 bp | 134 bp+ 158 bp+ 61 bp | 30 | 65 | 1 | |
| A5600T | S | tRNAAla | HinfI+ | F: A5597G | 24 bp+ 234 bp | 258 bp | 74 | 90 | 1 |
| T7581C | S | tRNAAsp | MnlI+ | R: TA7584-5CC | 125 bp+ 82 bp+ 27 bp | 125 bp+ 109 bp | 52 | 75 | 1 |
| C14684T | S | tRNAGlu | NlaIII + | 118 bp+ 147 bp | 265 bp | 75 | 85 | 1 | |
| T15889C | S | tRNAThr | AvaII− | F: T15887G | 25 bp+ 227 bp | 252 bp | 50 | 70 | 1 |
| A15902G | S | tRNAThr | MaeI+ | F: A15899C | 25 bp+ 215 bp | 240 bp | 55 | 75 | 1 |
| A15935G | F | tRNAThr | MboII+ | F: TG15932-3GA | 23 bp+ 182 bp | 205 bp | 54 | 92 | 1 |
| T1571G | S | 12 S rRNA | MspI+ | F: TG1568-9CC | 31 bp+ 182 bp | 213 bp | 62 | 82 | 1 |
| A1692T | S | 16 S rRNA | MnlI+ | 29 bp+ 141 bp + 60 bp | 29 bp+ 201 bp | 75 | 88 | 1 | |
| C1703T | S | 16 S rRNA | PflMI+ | R: TA1704-5GG | 175 bp+ 38 bp | 213 bp | 60 | 72 | 1 |
| T3197C§ | S | 16 S rRNA | MaeII− | F: TA3194-5AC | 27 bp+ 195 bp | 222 bp | 52 | 90 | 1 |
| T3228G | S | 16 S rRNA | NlaIII + | F: GG3225-6CA | 22 bp+ 87 bp+ 77 bp | 109 bp+ 77 bp | 75 | 88 | 1 |
| G3316A-Ala Thr¶ | F | ND1 | EaeI− | 222 bp | 147 bp+ 75 bp | 61 | 77 | 1 | |
| G3337A-Val Ile | S | ND1 | RsaI− | 222 bp | 170 bp+ 52 bp | 56 | 81 | 1 | |
| G3357A-Met Ile | S | ND1 | EaeI − | R: ATT3360-2CAA | 189 bp+ 32 bp | 147 bp+ 42 bp+ 32 bp | 70 | 85 | 1 |
| BalI− | |||||||||
| A5510C-Leu Phe | S | ND2 | MboII+ | 57 bp+ 280 bp+ 45 bp | 377 bp+ 45 bp | 85/78 | 95/90 | 2 |
* All of these mutations were absent in the control series. S/F, sporadic/familial; RE +/−, restriction enzyme gain or loss.
† Combined with the corresponding primer either forward (F) or reverse (R).
‡ Fragment size in mutant and wild-type DNA after restriction enzyme (RE) digestion.
§ Reported by Hess et al 39 associated with A3243G MELAS mutation in a patient with MELAS and ischemic colitis.
¶ Reported by Nakagawa et al 40 associated with non-insulin-dependent diabetes mellitus.
Table 2.
Heteroplasmic Polymorphisms in DCM Patients
| Base change | Gene | RE +/− | RE digestion* | Mutant % | DCM | Controls | ||
|---|---|---|---|---|---|---|---|---|
| Mutant | Wild Type | Blood | Heart | |||||
| T4336C† | tRNA GlnC | AvaII + | 153bp+200bp | 353 bp | 50 | 72 | 1 /85 | 0 /111 |
| T10034C‡ | tRNA Gly | AluI + | 110bp+117bp | 227 bp | 75 | 90 | 1 /85 | 0 /111 |
| A10398G‡-Thr Ala | ND3 | DdeI+ | 37bp+38bp+160bp | 37 bp+ 198 bp | 58 | 75 | 1 /85 | 0 /111 |
DNA Extraction and Oligoprimer Synthesis
Total genomic DNA was isolated from heart and blood samples with conventional methods. 27 Oligonucleotide primers encompassing the 22 tRNA, 2 rRNA, and coding genes were designed, 28 synthesized with a 392 DNA-RNA synthesizer (Applied Biosystems, ABI, Foster City, CA), and purified with oligonucleotide purification cartridges (Applied Biosystems). Primers with GC clamps were synthesized for DGGE.
PCR
Selected mtDNA regions were amplified by PCR from 0.1 μg of the total DNA in an automated thermal cycler (Perkin Elmer Cetus Gene Amp PCR system 9600). Amplifications were performed in 50 μl of reaction solution containing 100 ng of genomic DNA, 200 mmol/L each dNTP, 1 mmol/L each primer, 1.25 U of AmpliTaq Gold DNA polymerase (Perkin Elmer), and PCR buffer (10 mmol/L TrisHCl, pH 8.4, containing 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.01% gelatin) after denaturation for 10 minutes at 94°C. PCR conditions are available on request. Amplified fragments were separated by electrophoresis on 2% agarose gel and stained with ethidium bromide.
DGGE and SSCP
Screening for mutations was performed with DGGE and/or SSCP analysis. 29,30 Single-stranded DNA was visualized with standard silver nitrate staining. Cases with abnormal DGGE or SSCP conformers were selected for automated DNA sequencing.
Automated DNA Sequencing
The double-stranded DNA fragments were purified with Centri-sep columns (ABI Perkin Elmer) and used for cyclic sequencing with the Taq DyeDeoxy Terminator cycle sequencing kit in a 373A DNA sequencer (ABI Perkin Elmer) under conditions recommended by the manufacturer. Sequence comparison and analyses were performed with the Sequencher TM 3.0 software (Gene Codes Corp., Ann Arbor MI).
Heteroplasmy Assessment
Although heteroplasmy can be suspected on DGGE pattern, most heteroplasmic mutations with a high amount of mutated DNA appeared to be homoplasmic according to the sequencing pattern. Thus, to assess the heteroplasmy of any given mutation, we used restriction enzyme analysis; for mutations that gained or lost restriction enzyme sites, the PCR fragments were digested with the enzyme and electrophoresed through an 8% nondenaturing polyacrylamide gel. For mutations that did not modify the restriction enzyme map, we synthesized a forward or reverse mismatch primer that, in combination with any given mutation, gained or lost a restriction enzyme site. Then, both digestion and electrophoresis were performed as above. The relative amounts of the mutant mitochondrial genomes were quantitated with a Bio-profile Bio-1D blot analyzer (Vilber Lourmat Biotechnology Division, Marne La Vallee, France). The mismatch sites for pathological mutations found in the study are detailed in Tables 1 and 2 ▶ ▶ .
Results
Ultrastructural examination of a 601-EMB-sample series identified 85 cases in which the mitochondria not only were increased in number but also showed abnormal matrix, increased size (giant organelles), abnormal cristae pattern, and inclusion bodies (Figure 1) ▶ . Of the 85 patients, 19 (22.35%) had mtDNA mutations that were not found in normal and diseased controls (Table 1) ▶ . Cox enzyme activity (Table 3) ▶ was severely affected, with significantly lower mean levels in the myocardium of patients with mtDNA mutations compared both with that of nonmutated DCM patients without mitochondrial abnormalities and with that of normal controls (P = 0.0008). NADH dehydrogenase activity was slightly reduced in DCM with mtDNA mutations (P = 0.0388), whereas differences in SDH activity in patients and controls did not reach significant values.
Figure 1.
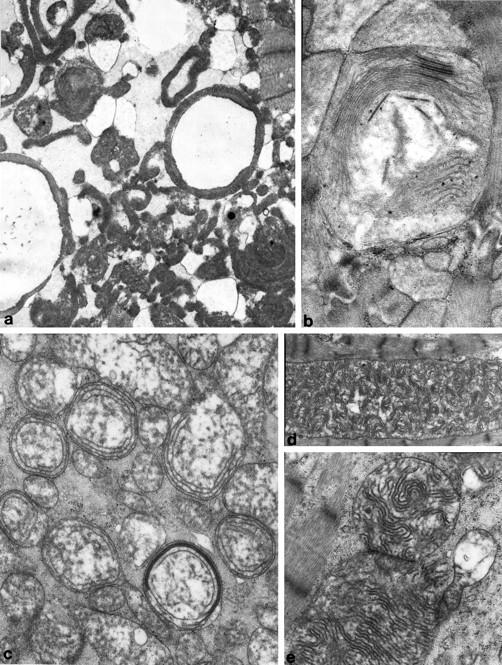
Electron micrographs showing characteristic ultrastructural changes of mitochondria in patients with pathological mtDNA mutations. a: Ring mitochondria; b: giant mitochondria with membrane fusion and circular cristae; c: concentric cristae; d and e: giant organelles with irregularly whorled and undulated cristae. Uranyl acetate, lead citrate; magnification, ×5600 and (a and d), ×16,000 (b) and ×9600 (c and e).
Table 3.
Mitochondrial Enzyme Activity in Normal Donor Heart Samples (n = 6) and in DCM Explanted Hearts from Patients without (n = 12) and with (n = 5) mtDNA Mutations
| Controls | DCM* without mtDNA mutations | DCM with mtDNA mutations | P values | |
|---|---|---|---|---|
| Cytochrome c oxidase | 170.450 | 143.917 | 86.117 | 0.0008 |
| NADH dehydrogenase | 1401.5 | 1475.083 | 1278.600 | 0.0388 |
| Succinic dehydrogenase | 49.217 | 42.730 | 49.750 | NS |
Statistical analysis was performed with the Student’s t-test. NS, not significant.
Pathological mtDNA Mutations from the 85 Patients (Table 1) ▶
We identified nine tRNA, five rRNA, and five missense mutations. In addition, polymorphic changes were seen both in controls (n = 111) and in DCM subjects with (n = 85) and without (n = 32) mitochondrial ultrastructural changes.
Transfer RNA Mutations
Of the 85 patients, 9 had heteroplasmic mutations in 6 of the 22 tRNA (Figures 2 and 3) ▶ ▶ . Five mutations affected tRNA stems, and four affected the loops. The tRNA mutations were present both in blood and in heart mtDNA, but the amount of mutated DNA was higher in heart than in blood and varied from case to case (Figures 2 and 3) ▶ ▶ . All nine patients had unique mutations. Two patients had familial cardiomyopathy. The seven remaining patients had sporadic forms. Most mutations occurred in bases that are highly or moderately conserved through evolution (alignments are available on request).
Figure 2.
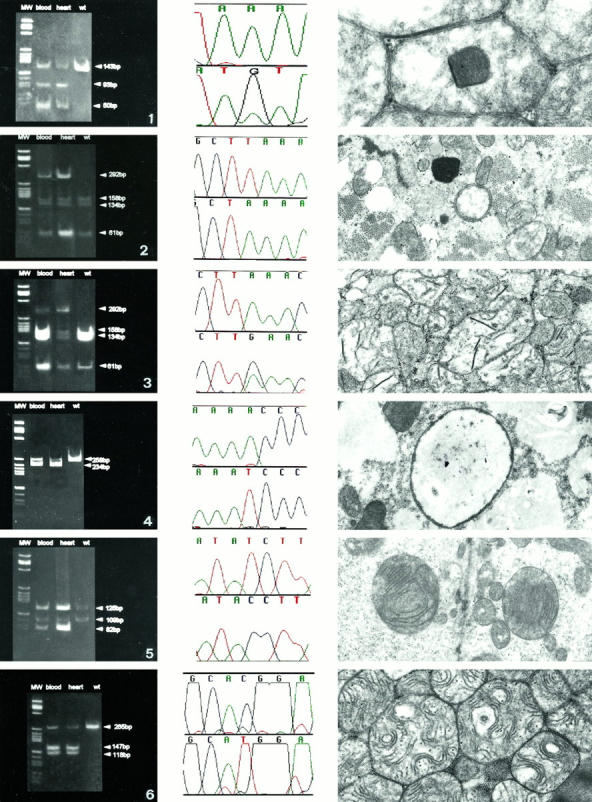
Enzyme cuts show higher amount of mutants in heart than in blood DNA. 1: Middle, A3260G, tRNALeuUUR; left, XmnI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with inclusion polygonal bodies. 2: Middle, T4314A, tRNAIle; left, MseI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with ring organelles. 3: Middle, A4315G, tRNAIle; left, MseI digestion in blood and inheart DNA; right, ultrastructural characteristics of the heart mitochondria with crystallized compacted cristae and osmiophilic bodies. 4: Middle, A5600T, tRNAAla; left, HinfI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with large organelles, devoid of cristae and often full of glycogen. 5: Middle, T7581C, tRNAAsp; left, MnlI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with giant organelles, peripheral cristolysis, and central compacted cristae. 6: Middle, C14684T, tRNAGlu; left, NlaIII digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with concentrically arranged and undulated cristae. Uranyl acetate, lead citrate; magnification, ×6000.
Figure 3.
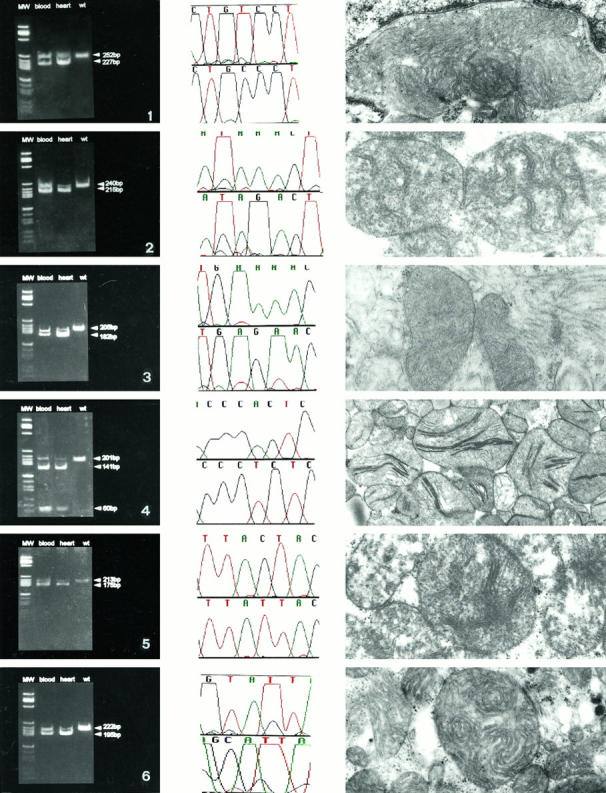
Left column shows the heteroplasmy and the higher amount of mutant in heart than in blood DNA. 1: Middle, T15889C, tRNAThr; left, AvaII + digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with concentric cristae and giant mitochondria. 2: Middle, A15902G, tRNAThr; left, MaeI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with giant organelles and undulated cristae. 3: Middle, A15935G, tRNAThr; left, MboII digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with undulated, compacted cristae. 4: Middle, A1692T, 16 S rRNA; left MnlI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with giant organelles and crystallized compacted cristae. 5: Middle, C1703T, 16 S rRNA; left, PflMI digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with giant organelles, peripheral cristolysis, and central compacted cristae. 6: Middle, T3197C, 16 S rRNA; left, MaeII digestion in blood and in heart DNA; right, ultrastructural characteristics of the heart mitochondria with giant organelles and undulate, swollen cristae. Uranyl acetate, lead citrate; magnification, ×6000.
Ribosomal RNA Mutations
Heteroplasmic mutations in the 12 S rRNA and in the 16 S rRNA were found in five unrelated patients with sporadic DCM (Figure 3) ▶ . Analogously to tRNA mutations, the amount of mutated DNA was higher in heart than in blood. All five mutations were not seen in control myocardial or blood samples.
Missense Mutations
Missense heteroplasmic mutations, with a higher amount of mutant DNA in heart than in blood, were identified in five cases. One (G3316A) was familial (sister and mother died of DCM). Two unrelated patients shared an identical mutation (A5510C) in the ND2 gene; these two patients also had similar clinical history, morphology, and outcome.
Polymorphic mtDNA Changes in DCM Patients and Controls
A series of known neutral polymorphisms, most of which were homoplasmic, was identified in both patients and controls (mutations and relative frequencies in cases and in controls are available on request). Three tRNA neutral polymorphisms in three different patients (T4336C, T10034C, and A10398G) were absent in controls and heteroplasmic at repeated restriction enzyme digestions (Table 2) ▶ . In addition, three other patients carried the T4216C, Tyr-Lys homoplasmic LHON mutation, which was also found in three controls. Three homoplasmic mutations, T14766C Ile-Thr (16.4% of patients versus 14.4% of controls), T14798C Phe-Leu (10.5% of patients versus 12.6% of controls), and G15884A Ala-Thr (1.1% of patients versus 0.9% of controls), were missense and were found in patients but also in healthy controls. All other mutations in polypeptide-coding genes were synonymous.
Discussion
In a large cohort of idiopathic DCM, we have identified a small subgroup of patients with either known or novel pathological mutations in mtDNA. These mutations were found in 22.35% of idiopathic DCM cases with ultrastructural mitochondrial changes consisting of giant organelles combined with abnormally arranged cristae and/or intraorganelle inclusions.
All pathological mutations were heteroplasmic, had a higher proportion of mutant in the affected heart than in the corresponding blood, and were not detected in controls. Some of the mutations occurred at nucleotide positions highly conserved through evolution (ie, A5600T tRNAAla), whereas other mutations affected moderately (such as A4315G tRNAIle) or less conserved (such as T7581C tRNAAsp, or A15902G tRNAThr) positions. Previously described mtDNA mutations with similar evolutionary conservation (eg, T3271C) have been proven to be involved in the pathogenesis of mitochondrial encephalomyopathies. 31 The nucleotide position of the mutation in any given tRNA may be related to the effects; in the tRNA stems, the mutations disrupt highly conserved base pairs, whereas in the tRNA loops, the mutations adversely affect the clover leaf structure of tRNAs. The functional relevance of rRNA mutations is less clear. As ribosomes are fundamental for translation mechanisms, rRNA mutations may cause major assembly defects or interfere with tRNA protection activity. 32 Additional studies are needed to elucidate the possible pathogenic role of rRNA mutations.
We attributed a pathological significance only to heteroplasmic mutations with a higher amount of mutated DNA in heart than in peripheral blood (Table 1) ▶ . In general, the threshold of mutated DNA in the affected organ varies from one mutation to the other and is influenced by the functional significance of a given base change. Myocardial samples from the maternal relatives of our patients were not available, and mutations could be evaluated only in their peripheral blood DNA. Due to the heteroplasmic characteristics of the mutations, the difference in mutant DNA in blood of patients and of unaffected relatives does not necessarily reflect identical gradients in myocardial tissue. Therefore, the evaluation of the amount of mutant DNA in blood of relatives does not provide any information about the likelihood of occurrence of cardiomyopathy. We are following up unaffected mutated relatives. If the cardiomyopathy phenotype indeed is related to the amount of mutated mtDNA in the affected organ as in encephalomyopathies, 13 clinical follow-up of mutated but healthy relatives should allow early detection of disease. A recent study done in 58 unrelated patients with DCM showed that mtDNA point mutations are significantly more frequent in patients than in controls. Of the 43 mutations identified, 4 were heteroplasmic and affected evolutionarily conserved regions. 33 None of these mutations, as well as none of the mutations previously reported in patients with cardiomyopathy, was identified in our series, supporting the uniqueness of missense mutations found to date in cardiomyopathies.
All pathological heteroplasmic mutations were absent from our normal and diseased control subjects. However, it would not be surprising if some of these mutations were subsequently considered as polymorphic changes. For instance, Lauber et al originally proposed that the A12308G transition in the tRNALeuCUN gene in four patients with mitochondrial encephalomyopathy was associated with the disease because it occurred at a site that had been highly conserved in various species during evolution and was not present in the general population. 34 A subsequent study showed that this mutation, with a frequency of 16%, was also present in the general population. 35 The same considerations applies to the LHON 4216 mutation, which seems to contribute to the disease only when interacting with other mutations; 15,35 such a mutation has not been considered, by itself, to be pathological in our patients.
Although the heart was the only clinically affected organ in our patients, we cannot exclude the presence of subclinical skeletal myopathies, particularly in the patient with the A3260G mutation. 5 Clinically overt peripheral myopathy did not occur in the other cases with family history of cardiomyopathy and congestive heart failure of the present series. Furthermore, none of our patients had diabetes mellitus or other disorders that may be associated with pathological mtDNA mutations. Compared with severe mitochondrial phenotypes affecting children (such as MELAS), our mutations were associated with a relatively milder phenotype. Most of our patients were young adults or adults, and most of them had a slowly evolving illness. It is not unlikely that in addition to the dose of mutant DNA in the affected myocardial tissue, the unique characteristics of the tissue itself could influence the phenotype. As such, post-mitotic or slowly dividing tissue cells that are highly dependent on mitochondrial oxidative function (such as myocardium) may be affected only when the proportion of the mutated DNA is considerably high. The resulting dysfunction in the overall mtDNA activity could impair oxidative phosphorylation processes and reduce the supply of the energy necessary for the contractile function of cardiac myocytes, thus leading to chronic heart failure. Although the mitochondrial cardiomyopathies may appear less severe than most encephalomyopathies, they often show inexorable progression of chronic heart failure and may be equally fatal.
mtDNA mutations fulfilling pathological criteria occurred in approximately one-fourth of our DCM patients with ultrastructural mitochondrial abnormalities. Furthermore, biochemical study showed a significant decrease in Cox activity and in NADH dehydrogenase activity in myocardial tissue obtained from patients who subsequently underwent heart transplantation. For the remaining three-fourths of cases with abnormal mitochondrial ultrastructure, either nuclear DNA mutations or signaling defects can be hypothesized, based on the mitochondrial and nuclear DNA interactions and on the nuclear DNA contribution to the synthesis of the respiratory chain enzymes. 36,37
A limit of the present study is that we cannot exclude a priori that DCM patients with nonspecific mitochondrial changes carry mtDNA mutations. However, no pathological mutation was identified in the 32 DCM cases with nonspecific ultrastructural mitochondrial changes.
Furthermore, it cannot be stated that mtDNA mutations were the only DNA defects in these patients. In a recent report we have described the association of mtDNA mutations plus β-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathies complicated by congestive heart failure evolution. 38 In these hypertrophic cardiomyopathy patients we also found a severe reduction in Cox activity in the heart and a wide spectrum of mitochondrial ultrastructural abnormalities. Therefore, we cannot exclude that mtDNA mutations may act as cofactors to unidentified pathological nuclear gene mutations.
The present study provides evidence that pathological mtDNA mutations may occur in a small proportion of otherwise idiopathic DCM cases. These mutations carry the same characteristics that define pathogenicity in mitochondrial encephalomyopathies and are associated with ultrastructurally abnormal mitochondria and decreased Cox activity. The findings support the hypothesis that in a small subset of patients, mitochondrial DNA defects may constitute the basis for, or contribute to, the development of dilated cardiomyopathy and congestive heart failure.
Footnotes
Address reprints requests to Dr. Eloisa Arbustini, Istituto di Anatomia Patologica, Via Forlanini 16, 27100 Pavia, Italy. E-mail: e.arbustini@smatteo.pv.it.
Supported by grants RF-IRCCS Policlinico San Matteo, Pavia 1992–1995.
References
- 1.Ozawa T, Tanaka M, Sugiyama S, Hattori K, Ito K, Ohno K, Takahashi A, Sato W, Takada G, Mayumi B: Multiple mitochondrial DNA deletions exist in cardiomyocytes of patients with hypertrophic or dilated cardiomyopathy. Biochem Biophys Res Commun 1990, 170:830-836 [DOI] [PubMed] [Google Scholar]
- 2.Merante F, Tein I, Benson L, Robinson BH: Maternally inherited hypertrophic cardiomyopathy due to a novel T-to-C transition at nucleotide 9997 in the mitochondrial tRNAGlycine gene. Am J Hum Genet 1994, 55:437-446 [PMC free article] [PubMed] [Google Scholar]
- 3.Casali C, Santorelli F, D’Amati G, Bernucci P, DeBiase L, DiMauro S: Novel mtDNA point mutation in maternally inherited cardiomyopathy. Biochem Biophys Res Commun 1995, 213:588-593 [DOI] [PubMed] [Google Scholar]
- 4.Merante F, Myint T, Tein I, Benson L, Robinson BH: An additional mitochondrial tRNAIle point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Hum Mutat 1996, 8:216-222 [DOI] [PubMed] [Google Scholar]
- 5.Zeviani M, Cellera C, Antozzi C, Rimoldi M, Morandi L, Villani F, Tiranti V, DiDonato S: Maternally inherited myopathy and cardiomyopathy: association with mutation in mitochondrial DNA tRNALeu(UUR). Lancet 1991, 338:143-147 [DOI] [PubMed] [Google Scholar]
- 6.Silvestri G, Santorelli FM, Shanske SB, Whitley CB, Schimmenti LA, Smith SA, DiMauro S: A new mitochondrial DNA mutation in the tRNALeu(UUR) gene associated with maternally inherited cardiomyopathy. Hum Mutat 1994, 3:37-43 [DOI] [PubMed] [Google Scholar]
- 7.Taniike M, Fukushima H, Yanagihara I, Tsukamoto H, Tanaka J, Fujimura H, Nagai T, Sano T, Yamaoka K, Inui I, Okada S: Mitochondrial tRNAIle mutation in fatal cardiomyopathy. Biochem Biophys Res Commun 1992, 186:47-53 [DOI] [PubMed] [Google Scholar]
- 8.Santorelli FM, Mak S, Vazquez-Aceredo M, Gonzalez-Astiazaran A, Ridaura-Sauz C, Gonzalez-Halphen D, DiMauro S: A novel mitochondrial DNA point mutation associated with mitochondrial encephalocardiomyopathy. Biochem Biophys Res Commun 1995, 216:835-840 [DOI] [PubMed] [Google Scholar]
- 9.Santorelli FM, Mak S, El-Schahawi M, Casali C, Shanske S, Baram TZ, Madrid RE, DiMauro S: Maternally inherited cardiomyopathy and hearing loss associated with a novel mutation in the mitochondrial tRNALys gene (G8363A). Am J Hum Genet 1996, 58:933-939 [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka M, Ino H, Ohno K, Hattori K, Sato W, Ozawa T, Tanaka T, Itoyama S: Mitochondrial mutation in fatal infantile cardiomyopathy. Lancet 1990, 2:1452. [DOI] [PubMed] [Google Scholar]
- 11.Anan R, Nakagawa M, Miyata M, Higuchi I, Nakao S, Suehara M, Osame M, Tanaka H: Cardiac involvement in mitochondrial disease: a study on 17 patients with documented mitochondrial DNA defects. Circulation 1995, 91:955-961 [DOI] [PubMed] [Google Scholar]
- 12.Wallace DC: Diseases of the mitochondrial DNA. Annu Rev Biochem 1992, 61:1175-1212 [DOI] [PubMed] [Google Scholar]
- 13.DiMauro S: Mitochondrial encephalomyopathies. What next? J Inher Metab Dis 1996, 19:489-503 [DOI] [PubMed] [Google Scholar]
- 14.Obayashi T, Hattori K, Sugiyama S, Tanaka M, Itoyama S, Deguchi H, Kawamura K, Koga Y, Toshima H, Takeda N, Nagano M, Ito T, Ozawa T: Point mutations in mitochondrial DNA in patients with hypertrophic cardiomyopathy. Am Heart J 1992, 124:1263-1269 [DOI] [PubMed] [Google Scholar]
- 15.Manfredi G, Schon EA, Moraes CT, Bonilla E, Berry GT, Sladky JT, DiMauro S: A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene. Neuromusc Disord 1995, 5:391-398 [DOI] [PubMed] [Google Scholar]
- 16.Goto Y, Tojo M, Horai S, Nonaka I: A novel point mutation in the mitochondrial tRNALeu(UUR) gene in a family with mitochondrial myopathy. Ann Neurol 1992, 31:672-675 [DOI] [PubMed] [Google Scholar]
- 17.Hug G, Schubert WK: Idiopathic cardiomyopathy: mitochondrial and cytoplasmic alterations in heart and liver. Lab Invest 1970, 22:541-552 [PubMed] [Google Scholar]
- 18.Ferrans VJ, Butany JW: Ultrastructural pathology of the heart. Trump BF Jones RT eds. Diagnostic Electron Microscopy. 1983, :pp 319-473 John Wiley and Sons, New York [Google Scholar]
- 19.Caves PK, Stinson EB, Billingham ME, Shumway NE: Percutaneous transvenous endomyocardial biopsy in human heart recipients. Ann Thorac Surg 1973, 16:325-336 [DOI] [PubMed] [Google Scholar]
- 20.Arbustini E, Grasso M, Diegoli M, Bramerio M, Scotti Foglieni A, Albertario M, Martinelli L, Gavazzi A, Goggi C, Campana C, Viganò M: Expression of tumor necrosis factor in human acute cardiac rejection. Am J Pathol 1991, 139:709-715 [PMC free article] [PubMed] [Google Scholar]
- 21.Patella V, Marino I, Lamparter B, Arbustini E, Adt M, Marone G: Human heart mast cells: isolation, purification, ultrastructure, and immunological characterization. J Immunol 1995, 154:2855-2865 [PubMed] [Google Scholar]
- 22.Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, Bellini O, Dal Bello B, Campana C, Piccolo G, Febo O, Opasich C, Gavazzi A, Ferrans VJ: Restrictive cardiomyopathy, atrio-ventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J Am Coll Cardiol 1998, 31:645-653 [DOI] [PubMed] [Google Scholar]
- 23.Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A: An electron transfer system associated with the outer membrane of the mitochondria. J Cell Biol 1967, 32:415-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King TE, Howard RL: Preparation and properties of soluble NADH dehydrogenase from cardiac muscle. Methods Enzymol 1967, 10:322-331 [Google Scholar]
- 25.King TE, Howard RL: Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. Methods Enzymol 1967, 10:322-331 [Google Scholar]
- 26.Sartore M, Grasso M, Piccolo G, Fasani R, Bergamaschi R, Malaspina A, Ceroni M, Kobayashi M, Semeraro A, Arbustini E, Surrey S, Fortina P: Leber’s hereditary optic neuropathy (LHON)-related mitochondrial DNA sequence changes in Italian patients presenting with sporadic bilateral optic neuritis. Biochem Mol Med 1995, 56:54-61 [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch SF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989:pp 9.14-9.21 Cold Spring Harbor Laboratory Press Cold Spring Harbor, NY
- 28.Anderson S, Bankier AT, Barrel BG, de Bruijin MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG: Sequence and organization of the human mitochondrial genome. Nature 1981, 290:457-465 [DOI] [PubMed] [Google Scholar]
- 29.Myers RM, Maniatis T, Lerman LS: Detection and localization of single base change by denaturing gradient gel electrophoresis. Methods Enzymol 1987, 155:501-527 [DOI] [PubMed] [Google Scholar]
- 30.Hayashi K: PCR-SSCP: a method for detection of mutations. Genet Anal Tech Appl 1992, 9:73-79 [DOI] [PubMed] [Google Scholar]
- 31.Hayashi JI, Otha S, Takai D, Miyabayashi S, Skuta R, Goto Y, Nonaka I: Accumulation of mtDNA with a mutation at position 3271 in tRNALeuUUR gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem Biophys Res Commun 1993, 197:1049-1055 [DOI] [PubMed] [Google Scholar]
- 32.Noller HF: Ribosomal RNA and translation. Annu Rev Biochem 1991, 60:191-227 [DOI] [PubMed] [Google Scholar]
- 33.Li YY, Maisch B, Rose ML, Hengstenberg C: Point mutations in mitochondrial DNA of patients with dilated cardiomyopathy. J Mol Cell Cardiol 1997, 29:2699-2709 [DOI] [PubMed] [Google Scholar]
- 34.Lauber J, Marsac C, Kadenbach B, Seibel P: Mutations in mitochondrial tRNA genes: a frequent cause of neuromuscular diseases. Nucleic Acids Res 1991, 19:1393-1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van den Ouweland JMW, Bruining GJ, Lindhout D, Wit JM, Veldhuyzen BFE, Maassen JA: Mutations in mitochondrial tRNA genes: non-linkage with syndromes of Wolfram and chronic progressive external ophthalmoplegia. Nucleic Acids Res 1992, 20:679-682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC: A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart muscle isoform of the adenine nucleotide translocation. Nature Genet 1997, 16:226-234 [DOI] [PubMed] [Google Scholar]
- 37.Zeviani M, Antozzi C: Nuclear DNA contribution to the synthesis of the respiratory chain enzymes. Mol Hum Reprod 1997, 3:133-148 [DOI] [PubMed] [Google Scholar]
- 38.Arbustini E, Fasani R, Morbini P, Diegoli M, Grasso M, Dal Bello B, Marangoni E, Banfi P, Banchieri N, Bellini O, Comi G, Narula J, Campana C, Gavazzi A, Danesino C, Viganò M: Coexistence of mitochondrial DNA and β-myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure. Heart 1998, 80: (in press) [DOI] [PMC free article] [PubMed]
- 39.Hess J, Burkhard P, Morris M, Lalioti M, Myers P, Hadengue A: Ischaemic colitis due to mitochondrial cytopathy. Lancet 1995, 346:189-190 [PubMed] [Google Scholar]
- 40.Nakagawa Y, Ikegami H, Yamato E, Takekawa K, Fujisawa T, Hamada Y, Ueda H, Uchigata Y, Miki T, Kumahara Y: A new mitochondrial DNA mutation associated with non-insulin-dependent diabetes mellitus. Biochem Biophys Res Commun 1995, 209:664-668 [DOI] [PubMed] [Google Scholar]
- 41.Leroy D, Norby S: A new human mtDNA polymorphism: tRNAGlu/4336 (T→C). Clin Genet 1994, 45:109-110 [PubMed] [Google Scholar]
- 42.Marzuki S, Noer AS, Letrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E: Normal variants of human mitochondrial DNA and translation products: the building of a reference data base. Hum Genet 1991, 88:139-145 [DOI] [PubMed] [Google Scholar]