

Abstract
The defects in lymphocyte apoptosis that underlie the autoimmune lymphoproliferative syndrome (ALPS) are usually attributable to inherited mutations of the CD95 (Fas) gene. In this report, we present the histopathological and immunophenotypic features seen in the lymph nodes (n = 16), peripheral blood (n = 10), bone marrow (n = 2), spleen (n = 3), and liver (n = 2) from 10 patients with ALPS. Lymph nodes showed marked paracortical hyperplasia. Interfollicular areas were expanded and populated by T cell receptor-αβ CD3+ CD4−CD8− (double-negative, DN) T cells that were negative for CD45RO. CD45RA+ T cells were increased in all cases studied. The paracortical infiltrate was a result of both reduced apoptosis and increased proliferation, as measured by in situ detection of DNA fragmentation and staining with MIB-1, respectively. The paracortical proliferation may be extensive enough to suggest a diagnosis of malignant lymphoma. Many of the paracortical lymphocytes expressed markers associated with cytotoxicity, such as perforin, TIA-1, and CD57. CD25 was negative. In addition, most lymph nodes exhibited florid follicular hyperplasia, often with focal progressive transformation of germinal centers; in some cases, follicular involution was seen. A polyclonal plasmacytosis also was present. The spleens were markedly enlarged, more than 10 times normal size. There was expansion of both white pulp and red pulp, with increased DN T cells. DN T cells also were observed in liver biopsies exhibiting portal triaditis. In the peripheral blood, the T cells showed increased expression of HLA-DR and CD57 but not CD25. CD45RA+ T cells were increased in the four cases studied. Polyclonal B cell lymphocytosis with expansion of CD5+ B cells was a characteristic finding. Taken together, the histopathological and immunophenotypic findings, particularly in lymph nodes and peripheral blood, are sufficiently distinctive to suggest a diagnosis of ALPS. Of note, two affected family members of one proband developed lymphoma (T-cell-rich B-cell lymphoma and nodular lymphocyte predominance Hodgkin’s disease, respectively).
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder characterized by generalized, nonmalignant lymphadenopathy, hypergammaglobulinemia, lymphocytosis, splenomegaly, and autoimmune phenomena. A distinct feature of ALPS, and an early clue to its nature, is the occurrence of markedly increased numbers and percentage of T cell receptor (TCR)-αβ CD4−CD8−, double-negative (DN) T cells in the circulation and lymphoid tissues. Sneller et al 1 first reported a detailed clinical and immunological study of two patients, recognizing similarities to the MRL and C3H/Hej strains of mice possessing the lpr and gld mutations, respectively. Genetic elucidation of the lpr and gld loci as representing recessive mutations in the genes encoding CD95 (Fas/Apo-1) and CD95L (FasL), respectively, 2-4 led to the discovery of functional CD95 mutations in ALPS patients. 5-8 With the ability to diagnose specific CD95 mutations in ALPS cases, it was appreciated that patients affected with this disorder had been included in published series by a number of investigators of undefined chronic lymphoproliferation or splenomegaly with associated autoimmune phenomena. 9,10
CD95 is a 48-kd type I transmembrane protein belonging to the tumor necrosis factor receptor family. Interaction of CD95 and its ligand plays a critical role in controlling the homeostasis of peripheral lymphocytes by inducing signals leading to cellular apoptosis; thus, aberrations in this interaction would be expected to impair the apoptosis of normal and autoreactive lymphocytes. Indeed, mice bearing the lpr or gld mutations, who thus have defective expression of CD95 or its ligand, respectively, develop massive lymphoid hyperplasia and autoimmunity. 11-13
In vitro studies of B and T cells from ALPS patients and their family members verify that in humans, as well as in mice, CD95 mutations lead to defective lymphocyte apoptosis. The precise role of CD95 in lymphocyte homeostasis in these individuals is not completely understood. This fact is illustrated by the wide spectrum of clinical and immunological findings in ALPS patients and the occurrence of family members who possess identical mutations and defective in vitro apoptosis but few or no clinical signs of ALPS. 14
In this study, we report the spectrum of histological and immunophenotypic features of 10 patients with ALPS. The potential functional properties of expanded DN T cells characteristic of this syndrome are also discussed.
Materials and Methods
Patient Selection
Ten patients referred to the National Institutes of Health for evaluation of generalized lymphadenopathy, splenomegaly, peripheral lymphocytosis, and autoimmune phenomena, along with available relatives, were evaluated. The studies were performed under approved research protocols of the National Institute of Allergy and Infectious Diseases and the National Human Genome Research Institute and with the patients’ written consent. Peripheral blood, lymph nodes, spleen, bone marrow, and liver were available for examination. ALPS was defined as the presence of chronic, nonmalignant lymphoproliferation, defective lymphocyte apoptosis in vitro, and greater than or equal to 1% TCR-αβ CD3+CD4−CD8− T cells. 15 Two family members of one proband (patient 3) had lymphoma; these tissues also were obtained for histological analysis.
Flow Cytometry
Anticoagulated peripheral blood specimens were stained for flow cytometry using a whole-blood lysis method and analyzed with a FACScan and Cell Quest software (Becton Dickinson, San Jose, CA). In selected cases (three lymph nodes and one spleen), cell suspensions were prepared from freshly resected tissues and stained with fluorescein-isothiocyanate-conjugated or phycoerythrin-conjugated murine monoclonal antibodies (listed in Table 1 ▶ ) and analyzed by flow cytometry using a FACScan and Cell Quest software.
Table 1.
Monoclonal Antibodies Used in Immunophenotypic Analysis
| Antigen | Name | Source |
|---|---|---|
| CD1a | OKT6 | Ortho Diagnostics, Raritan, ND |
| CD2 | Leu5b | Becton Dickinson, Mountain View, CA |
| CD3 | Leu-4 | Becton Dickinson |
| CD4 | Leu-3a, 1F6* | Becton Dickinson; Novocastra Labs,* Newcastle-on-Tyne, UK |
| CD5 | Leu-1 | Becton Dickinson |
| CD7 | Leu-9 | Becton Dickinson |
| CD8 | Leu-2a, 144* | Becton Dickinson; K. Gatter,* Oxford University, Oxford, UK |
| CD11c | Leu-M5 | Becton Dickinson, |
| CD16 | Leu-11 | Becton Dickinson, |
| CD25 | Anti-Tac | T. Waldmann, National Cancer Institute |
| CD56 | Leu-19 | Becton Dickinson |
| CD57 | Leu-7 | Becton Dickinson |
| CD20 | Leu-16, L26* | Becton Dickinson; Dako,* Carpinteria, CA |
| CD19 | Leu-12 | Becton Dickinson |
| κ light chain | Bethesda Research Laboratories, Bethesda, MD | |
| λ light chain | Bethesda Research Laboratories | |
| CD30 | BerH2 | Dako |
| CD38 | Leu-17 | Dako |
| HLA-DR | ||
| MIB-1* | Ki-67 | Dako |
| TCR γδ | TCRδ1 | Becton Dickinson; T-Cell Sciences, Cambridge, MA |
| TCR αβ | βF1 | Becton Dickinson; T-Cell Sciences |
| Perforin* | P1-8 | H. Yagita, Juntendo University, Tokyo, Japan |
| CGP* | TIA-1 | Coulter, Hialeah, FL |
| Polyclonal CD3* | Dako | |
| CD43* | Leu-22 | Becton Dickinson |
| CD45RA† | Leu 18 | Becton Dickinson |
| CD45RO* | A6 | Immunotech, Westbrook, ME |
CGP, cytotoxic granule-associated protein.
*Used in paraffin sections only.
†Used in flow cytometry only.
Histological Examination
Lymph nodes (n = 16), spleens (n = 3), livers (n = 2), and bone marrows (n = 2) were examined in formalin-fixed, paraffin-embedded hematoxylin and eosin (H&E)-stained sections. Immunophenotypic studies were performed on paraffin sections using the ABC immunoperoxidase technique as previously described. 16 In seven cases yielding five lymph nodes and two spleens, snap-frozen material was studied using cryoimmunohistochemical techniques. The antibodies used for paraffin-section and frozen-section immunohistochemical studies are listed in Table 1 ▶ . In one patient (17), no paraffin or frozen sections were available for immunohistochemical studies. Similarly, unstained sections were not available for the spleen from patient 3.
In Situ Detection of Apoptosis
In situ detection of cells undergoing apoptosis was determined in histological sections using the terminal deoxynucleotide transferase (Tdt)-mediated dUTP-biotin nick-end labeling (TUNEL) method according to the Apoptag kit (Oncor, Gaithersburg, MD).
Molecular Analyses
TCR γ-chain gene and immunoglobulin heavy chain (IgH) gene rearrangement analyses were performed using polymerase chain reaction (PCR) amplification techniques on high molecular weight DNA extracted from peripheral blood mononuclear cells or paraffin-embedded tissue sections as previously described. 16,17 Because of early suspicions that chronic Epstein-Barr virus infection might play a role in the chronic lymphadenopathy of these patients, 1 lymph node sections from five patients were examined for the expression of the Epstein-Barr viral RNA by in situ hybridization using the EBER-1 probe. 18 Fas (CD95) gene mutations were identified, as previously described. 5
Results
Patient Selection
Ten patients originally came to medical attention 2 months to 5 years after birth because of persistent lymph node and spleen enlargement. Detailed clinical data for 9 of these 10 patients have been presented previously by Sneller et al. 15 Other clinical manifestations included recurrent bouts of hemolytic anemia, neutropenia, idiopathic thrombocytopenia, and glomerulonephritis. 15 Nearly all patients exhibited pathological autoantibodies, most of which were directed at platelets or red blood cells. Important early clinical clues as to the difference between these patients and patients with defined lymphoproliferative malignancies or viral infections were the prolonged stable clinical course and the lack of opportunistic infections.
In the course of their diagnostic evaluations, the patients had undergone lymph node, bone marrow, and liver biopsies, often more than once. Splenectomy was performed on 7 of the 10 patients. Indications for splenectomy were hypersplenism, refractory hemolytic anemia or idiopathic thrombocytopenia, or suspicion for lymphoma.
Subsequently, all 10 patients (4 males and 6 females) and their relatives provided informed consent and were evaluated at the National Institutes of Health. All patients showed defective lymphocyte apoptosis in vitro, and all were analyzed for the presence of CD95 gene mutation using either peripheral blood mononuclear cells or paraffin-embedded tissue. 5 Eight of ten patients demonstrated a specific mutation in various parts of the CD95 gene. Investigation of family members demonstrated that all mutations were inherited with one exception; patient 6 had a confirmed de novo mutation. The two remaining patients (patients 11 and 14) did not have an identifiable mutation in the CD95 gene but were diagnosed as ALPS based on the clinical and laboratory findings as well as demonstrable apoptotic defects, as defined by Sneller et al. 15,19
Lymph Nodes
As detailed in Table 2 ▶ , 16 lymph nodes from 10 patients with ALPS were subjected to histological analysis. The lymph node architecture was intact. The most prominent and consistent finding was the marked paracortical expansion by lymphocytes in varying stages of immunoblastic transformation. The immunoblasts were intermingled with mature lymphocytes and polyclonal plasma cells (Figure 1) ▶ . The paracortical lymphocytes were intermediate to large in size and round to oval in shape and contained moderate amounts of clear to more deeply staining eosinophilic cytoplasm. The paracortical lymphoid cells demonstrated a high proliferative index as determined by MIB-1 positivity and frequent mitoses. In several cases, the cellular proliferation was so marked that a diagnosis of malignant lymphoma was suspected by the referring pathologist.
Table 2.
Lymph Node Findings in ALPS Patients
| Patient | Age at Presentation | Gender | CD95 Mutation | Paracortical expansion | Follicular hyperplasia | Follicular involution | PTGC |
|---|---|---|---|---|---|---|---|
| 1 | 4 months | M | + | + | + | − | + |
| 2 | 18 months | F | + | + | + | + | − |
| 3 | 5 years | M | + | + | + | − | + |
| 4 | 2 years | M | + | + | + | − | − |
| 5 | 2 months | M | + | + | + | − | + |
| 6 | 4 months | F | + | + | + | + | + |
| 11 | 9 months | F | − | + | + | + | + |
| 14 | 9 months | F | − | + | + | − | + |
| 17 | 4 months | F | + | + | + | − | − |
| 20 | 3 years | F | + | + | + | + | − |
M, male; F, female.
Figure 1.
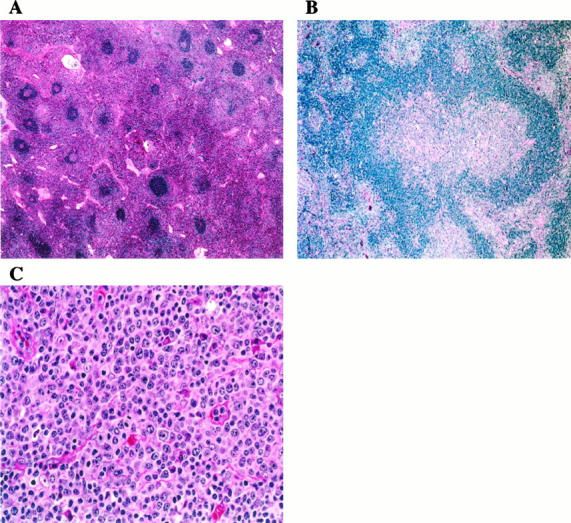
Histopathology of lymph nodes. A: The paracortex is markedly expanded by lymphocytes with pale pink cytoplasm. Primary and secondary follicles are numerous. Some follicles show regressive changes (upper right ). H&E; original magnification, ×100. B: Progressive transformation of germinal centers was a focal but relatively frequent finding. H&E; original magnification, ×200. C: The paracortex is populated by lymphocytes, plasma cells, and immunoblasts. Note frequent mitotic figures. H&E; original magnification, ×600.
There was a conspicuous absence of histiocytes containing apoptotic bodies within the paracortex, in contrast to what is characteristically seen in other reactive conditions. The status of apoptosis was determined by TUNEL assay, which demonstrated few cells undergoing programmed cell death in the interfollicular areas but readily identified numerous positive cells within the germinal centers (data not shown).
An additional histological finding of note was the presence of a spectrum of reactive germinal center changes, including florid follicular hyperplasia. Progressive transformation of germinal centers (PTGC) was observed focally in involved lymph nodes (6/10 cases). A minority of lymph nodes showed areas of atrophic follicles with regressive changes as seen in Castleman’s disease (4/10). Prominent postcapillary venules were seen in the interfollicular region of some cases.
Immunophenotypically, the majority of the paracortical cells were CD3+ T cells (Figure 2A) ▶ , of which only a small proportion showed staining with CD4 (Figure 2B) ▶ or CD8 (Figure 2C) ▶ . An increase in DN T cells was also documented by flow cytometry. DN T cells ranged from 27% to 54% of mononuclear cells, representing 51% to 78% of αβ T cells. In lymph nodes, CD4+ cells were more frequent than CD8+ cells (the CD4:CD8 ratio was approximately 2:1), in contrast to peripheral blood (see below), in which CD4+ and CD8+ cells tended to be present in more equal numbers. Normally the CD4:CD8 ratio in lymph nodes is 3:1 to 4:1, in contrast to the 2:1 to 3:1 ratio seen in peripheral blood. Therefore, the observed differences between lymph nodes and blood may be a reflection of a relatively higher proportion of CD4+ T cells normally found in lymph nodes. The majority of the CD4+ T cells were identified within germinal centers, and not in the expanded paracortical regions by immunohistochemistry (Figure 2B) ▶ .
Figure 2.
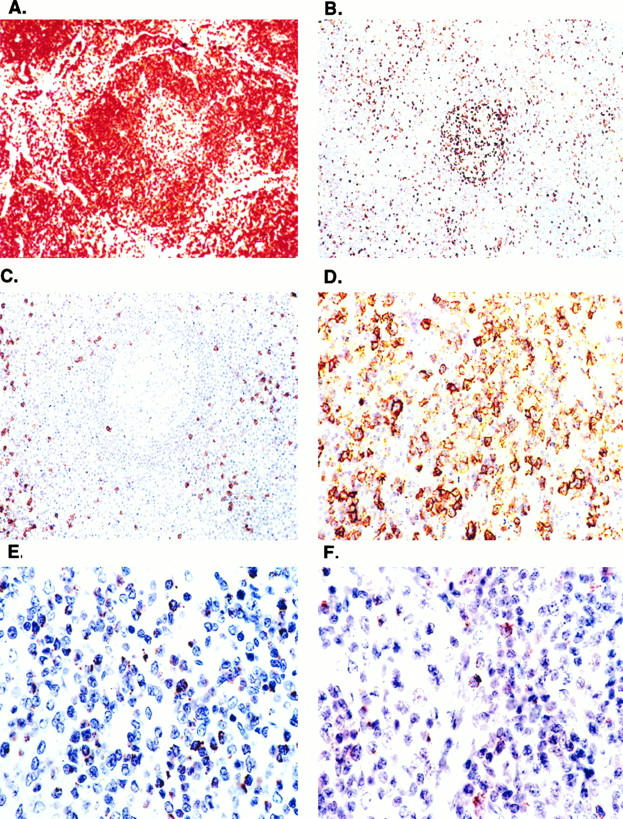
Lymph node demonstrates paracortical expansion by CD3+ DN T cells. Immunoperoxidase stains (ABC immunoperoxidase technique, hematoxylin counterstain; original magnification, ×200) performed in paraffin sections demonstrates expansion of interfollicular regions by CD3+ T cells (A) that are largely CD4− (B) and CD8− (C). Most CD4+ cells are within germinal centers. The paracortex contains numerous lymphocytes positive for CD57 (D) and TIA-1 (E) with weaker and patchy staining for perforin (F).
The paracortical lymphocytes did not show expression of the α-chain of the interleukin (IL)-2 receptor CD25. Similarly, a low percentage of T cells expressed CD25 by flow cytometry (<5% in all cases studied). The paracortical T cells stained strongly with CD3 and CD43 (Leu 22) but were negative for CD45RO and thus bore markers consistent with a naive, or virgin, phenotype. 20 Most CD45RO+ lymphocytes were within germinal centers. In the cases studied by flow cytometry, CD45RA+ T cells were markedly increased (79% to 90%).
A large number of the paracortical cells stained with CD57, TIA-1 (Figure 2, D and E) ▶ , and perforin (Figure 2F) ▶ , the latter two being granular serine proteases characteristically associated with cytotoxic T cells. Markers for natural killer (NK) cells (CD56 and CD16) were negative in both frozen sections and by flow cytometry (<3% of mononuclear cells). Notably, the lymph node histology in patients with and without CD95 mutations demonstrated similar histological features.
In situ hybridization for EBV performed on lymph nodes from 5/10 patients was negative. There was no evidence of T or B cell clonal expansion as demonstrated by TCR-γ gene or IgH-PCR (data not shown). PCR assays for antigen receptor gene rearrangement will detect clonality in 50% to 75% of cases of B-cell or T-cell lymphomas with these methods. 16,17
Spleen
Three spleens were examined by routine histology, and in two cases immunophenotypic studies were performed. Patient 3 underwent splenectomy at the age of 6 years because of suspicion for lymphoma. Patient 5 underwent splenectomy at 5 years because of refractory idiopathic thrombocytopenia. The spleens weighed 620 and 856 g, respectively. (Normal pediatric splenic weights are 47 g (5 years) and 58 g (6 years).) Patient 11 underwent splenectomy at 3 years because of severe hypersplenism. The splenic weight was not available in this case.
In each case, the splenic white pulp was moderately expanded with follicular hyperplasia and a prominent marginal zone (Figure 3) ▶ . The red pulp was greatly expanded and contained numerous polyclonal plasma cells and increased lymphocytes and immunoblasts, which were predominantly CD3+. The lymphoid cells in the red pulp were similar in morphology to the lymphoid cells of the paracortical reaction seen in lymph nodes and, like the latter, were generally CD4− and CD8−. DN T cells were present but less conspicuous in the white pulp than they were in lymph nodes. Splenic T cells were similar to lymph node T cells in other respects, being negative for CD25.
Figure 3.
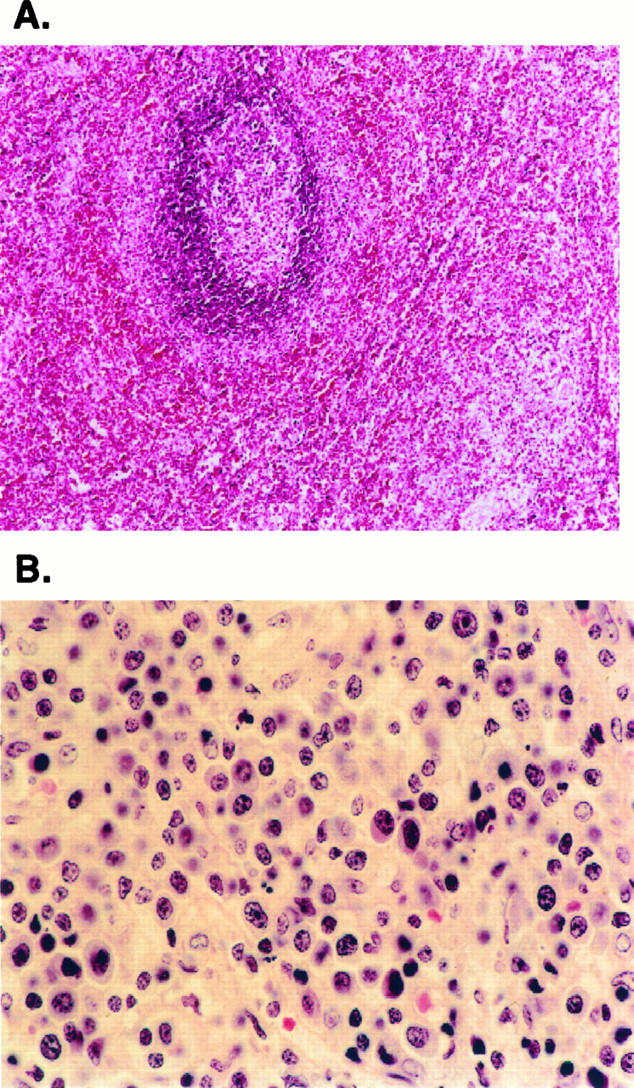
Spleen. A: The spleen shows follicular hyperplasia of white pulp, with an expanded marginal zone. The red pulp is greatly expanded. H&E; original magnification, ×200. B: The cytological composition of the red pulp resembles that of the lymph node paracortex and contains numerous lymphocytes, plasma cells, and immunoblasts. H&E; original magnification, ×400.
Liver
Liver biopsies were available for examination from patients 1 and 2. They demonstrated mild lymphocytic infiltrates composed of predominantly CD3+ T cells, of which greater than 50% were CD4−CD8−. Mild periportal fibrosis and extramedullary hematopoiesis was present in patient 1. Patient 2 developed chronic active hepatitis consistent with a form of autoimmune hepatitis.
Bone Marrow
Aspirate smears from patients 2 and 5 demonstrated scattered interstitial aggregates of large atypical lymphocytes. The cells showed clumped chromatin and prominent nucleoli with increased mitoses.
Peripheral Blood
The degree of peripheral blood lymphocytosis varied considerably from patient to patient. Sequential analysis of peripheral blood counts demonstrated fluctuations in patient 2 in response to immunosuppressive treatment (data not shown). Representative values obtained at time of immunophenotypic analysis are presented in Table 3 ▶ . The peripheral lymphocytosis ranged from 810 to 12,328 cells/mm3. DN T cells, expanded by definition in patients with ALPS, were increased 2- to 67-fold over normal values in all of the patients.
Table 3.
Summary of Peripheral Blood Lymphocyte Phenotype, CD95 Mutations and Lymphocyte Apoptosis Defects
| Patient | Lymphocytes (cells/mm3) | T cells (%) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3 | CD3/CD4 | CD3/CD8 | CD3/CD4−CD8− | CD57 | CD57/CD8 | αβ DN T cells | γδ DN T cells | DN T/CD45RA | % of DN T cells | |||||||||||
| 1 | 11,700 | 66 | 24 | 23 | 18 | 34 | 14 | 14 | 1 | ND | ND | |||||||||
| 2 | 5,712 | 65 | 11 | 16 | 39 | 42 | 22 | 29 | 3 | 29 | 74 | |||||||||
| 3 | 7,740 | 68 | 31 | 27 | 10 | 23 | 12 | 6 | 1 | 8 | 80 | |||||||||
| 4 | 1,870 | 70 | 23 | 37 | 9 | 39 | 16 | 4 | 3 | ND | ND | |||||||||
| 5 | 1,680 | 55 | 15 | 20 | 20 | 26 | 15 | 5 | 3 | ND | ND | |||||||||
| 6 | 12,328 | 67 | 15 | 26 | 26 | 32 | 22 | 13 | 8 | ND | ND | |||||||||
| 11 | 3,105 | 73 | 27 | 13 | 33 | 52 | 16 | 27 | 1 | ND | ND | |||||||||
| 14 | 5,900 | 59 | 14 | 27 | 17 | 15 | 8 | 7 | 10 | ND | ND | |||||||||
| 17 | 2,950 | 74 | 19 | 25 | 29 | 53 | 23 | 24 | 1 | 23 | 79 | |||||||||
| 20 | 810 | 76 | 31 | 29 | 14 | 29 | 16 | 9 | 3 | 8 | 57 | |||||||||
| Normal range | 1,173–2,640 | 61–84 | 32–58 | 11–36 | 1–7 | 5–29 | 3–24 | <1 | <5 | <3 | NA |
| Activation markers (%) | B cells (%) | NK cells (%) | CD95 mutation | Lymphocyte apoptosis defect | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3/HLA-DR | CD3/CD25 | CD20 | CD20/CD5 | % of CD20 cells | CD16/CD56 | CD16/CD56/CD3 | ||||||||||||||
| 46 | 11 | 27 | 21 | 76 | 3 | 3 | + | + | ||||||||||||
| 44 | 18 | 31 | 25 | 81 | 4 | 8 | + | + | ||||||||||||
| 17 | 7 | 24 | 22 | 92 | 5 | 3 | + | + | ||||||||||||
| 15 | 8 | 26 | 18 | 69 | 3 | 10 | + | + | ||||||||||||
| ND | ND | 41 | 38 | 92 | 4 | 7 | + | + | ||||||||||||
| 33 | 3 | 14 | 7 | 50 | 17 | 3 | + | + | ||||||||||||
| 57 | 5 | 17 | 15 | 90 | 6 | 7 | − | + | ||||||||||||
| 22 | 10 | 43 | 38 | 90 | 3 | 11 | − | + | ||||||||||||
| 56 | 4 | 20 | 10 | 50 | 4 | 1 | + | + | ||||||||||||
| 35 | 11 | 19 | 14 | 74 | 4 | 13 | + | + | ||||||||||||
| <15 | <37 | 5–16 | 1–10 | NA | 6–30 | 1–15 | Negative | Negative |
ND: Not determined; NA, not available.
*Percentage of DN T cells that also express CD45RA.
†Percentage of CD20 cells that also express CD5.
Although the percentage of CD3+ T cells was within normal limits, a relative decrease in the number of CD4+ T cells, resulting in an alteration of the CD4:CD8 ratio was seen in all but one patient. In four patients, CD8+ T cells exceeded CD4+ cells, and in five patients the ratio of CD4:CD8 was approximately 1:1. T cells showed generally increased expression of HLA-DR, without a concomitant increase in CD25 expression.
Of note, polyclonal B cell (CD20+) lymphocytosis (ranging from 20% to 43% of total lymphocytes) was seen in 7/10 patients. Notably, a high percentage of the B cells (50% to 92%) co-expressed CD5. There was no correlation between the extent of B cell lymphocytosis and the number of DN T cells. The B- and DN T-cell lymphocytosis did not reflect a general increase in all lymphocyte subsets, as the numbers of CD16+ or CD56+CD3− NK cells was within the normal range.
In concert with the findings in lymph nodes, CD57+ cells were significantly increased, without an increase in NK cells. Markedly increased numbers of CD45RA+ T cells were identified in the peripheral blood of the four patients studied.
As shown in Table 3 ▶ , the two ALPS patients without CD95 gene mutations (patients 11 and 14) also showed increased numbers of DN T cells as well a B cell lymphocytosis (patient 14).
Lymphoproliferative Disorders in Relatives of ALPS Patients
Family members of ALPS patients were surveyed for lymphoproliferative disease and malignant lymphoma. Two family members of patient 3 had a diagnosis of lymphoma. Subsequent genetic studies have demonstrated identical CD95 mutations in both patient 3 and the family members with lymphoma. Slides and paraffin blocks of biopsy and autopsy materials were obtained for histological review and immunophenotypic studies. The father and paternal uncle of patient 3 had two different but related types of B-cell lymphoma: disseminated T-cell-rich B-cell lymphoma (TCRBCL) and nodular lymphocyte predominance Hodgkin’s disease (NLPHD), respectively. The father of patient 3 died at the age of 26 with stage IV TCRBCL involving spleen, liver, bone marrow, and lymph nodes. In the uncle, the NLPHD was seen in a partially involved node, other portions of which demonstrated reactive follicular hyperplasia, PTGC, paracortical expansion by DN T cells, and plasmacytosis, features similar to those seen in ALPS patients.
Discussion
Sneller and co-workers 1 described an association between lymphoproliferation and autoimmune disease in two children with lymph node enlargement, splenomegaly, hypergammaglobulinemia, and expansion of TCR-αβ+CD4−CD8− T lymphocytes. After review of four additional patients with similar clinical and immunological findings, the syndrome was designated as ALPS. 5 Among all ALPS patients with CD95 mutations identified thus far, only one was a homozygous for the defect and thus expressed totally abnormal CD95 product. 7 All of the rest were heterozygous for the defects and expressed an abnormal CD95 protein that dominantly inhibited the activity of the co-expressed normal CD95 protein. The clinical and immunological features of 15 ALPS patients were recently reported by three independent groups. 6,7,15 Here, we report the spectrum of pathological findings seen in 10 patients as well as a description of the lymphoproliferative processes seen in their family members.
The most consistent and notable pathological finding in ALPS was the paracortical T-zone expansion by proliferating double-negative T cells. The extent of such expansion was variable from patient to patient. Although morphometric measurements of lymph nodes were not performed, it was our impression that in each case the paracortical hyperplasia correlated with the numbers of DN T cells in the peripheral blood. Notably, although the T cells in the paracortex exhibited the abnormal phenotype, T cells within germinal centers displayed a normal phenotype and were usually CD4+. Using the TUNEL method, we showed that although apoptotic cells were present in expected frequency in the germinal centers, apoptotic cells were reduced in the paracortex. This finding is consistent with reduced apoptosis in T cells with an abnormal CD95 gene. In addition, using the MIB-1 antibody to Ki-67, we identified prominent proliferative activity in the paracortex. Therefore, the marked lymphadenopathy observed in these patients is due to both defective cellular apoptosis as well as increased cellular proliferation. However, the lymph node T cells did not show increased expression of the α-chain of the IL-2 receptor (CD25).
Other relatively consistent features seen in the ALPS lymph nodes were follicular hyperplasia, prominent vascularity of interfollicular areas, and florid plasmacytosis. The follicles exhibited a spectrum of changes, including follicular hyperplasia with numerous secondary follicles, often with focal PTGC and focal regression of germinal centers. Follicular hyperplasia and plasmacytosis are typical of the lymphadenopathy associated with other autoimmune diseases, such as rheumatoid arthritis. PTGC is often seen in association with NLPHD. Notably, one affected family member of a patient in our study had NLPHD and a second had a TCRBCL, a disease closely linked to NLPHD in morphology and immunophenotype. 21,22
The complexity of the molecular processes regulating lymphocyte apoptosis is demonstrated by the existence of ALPS patients who do not possess CD95 mutations. In our series we could demonstrate no differences in the histological finding in the two ALPS patients without such mutations, as compared with the patients with such mutations. The role of other genetic factors in the pathogenesis of ALPS also is suggested by the existence of relatives of affected patients who have identical CD95 mutations but do not exhibit the clinical features of the syndrome. 5,14 This finding suggests that subtle immunological disturbances in addition to CD95 defects (or other defects of the CD95 pathway) must be present for the clinical expression of ALPS. Various other conditions with associated lymphadenopathy and immunological disturbances, such as Felty’s syndrome, lupus erythematosus, and rheumatoid arthritis may also be associated with aberrant T-cell apoptosis. Indeed, a mutation in the extracellular domain of CD95L has been reported in a patient diagnosed as having atypical lupus erythematosus. 23
In humans, TCR-αβ DN T cells are normally found in the thymus, 24 skin, 25 and peripheral blood. 25,26 They represent a small subpopulation of peripheral blood lymphocytes in most healthy adults but are increased in some patients with systemic and cutaneous autoimmune disorders. 27,28 The contribution of αβ DN T cells to the pathophysiology of ALPS, to these other diseases, or to normal immunity is not completely understood. Shivakumar et al 28 reported an increase of DN T cells in patients with active systemic lupus erythematosus and suggested that these cells induced the production of pathogenic anti-DNA autoantibodies. In addition, Wirt et al 29 described a patient with immunodeficiency in whom massive proliferation of DN T cells developed. In this case, it was suggested that the DN T cells were responsible for reactions resembling those of graft-versus-host disease. In contrast, studies from Sneller et al 1 and Fuss et al 30 show that the DN T cells in ALPS patients respond poorly to mitogens or antigens and fail to produce cytokines on activation. This finding suggests that the DN T cells in ALPS do not play a role in the causation of autoimmunity in this disease and may in fact represent an epiphenomenon.
There is some debate as to the origin of DN T cells. However, the weight of the evidence suggests that DN lymphocytes are derived from cells that previously expressed CD8. 31,32 This possibility is supported by our finding that the expanded population of paracortical lymphocytes express TIA-1 and perforin, markers associated with CD8+ cytotoxic T cells.
We noted that a significant proportion of the paracortical lymphocytes expressed CD57. In the five cases studied by either frozen-section immunohistochemistry or flow cytometry, very few lymphocytes expressed CD16 or CD56, markers characteristic of NK cells. Moreover, an increase in NK cells was not observed in the peripheral blood of these patients. Although double staining was not performed in lymph node sections, the frequency of CD57+ cells suggests that most, if not all, of the CD57+ lymphocytes in the lymph nodes are DN T cells. A comparable increase in CD57+ T cells was observed in the peripheral blood. Finally, the DN T cells in ALPS usually express HLA-DR, a marker of previous cell activation. Taken together, these observations indicate that the phenotype of DN T cells in ALPS patients is compatible with that of chronically activated cells that were derived from CD8+ T cells.
The immunohistochemical studies showed most of the paracortical lymphocytes to be negative for CD45RO. This result is in parallel with the reported increase in CD45RA+ over CD45RO+ DN T cells in lpr/lpr mice. 33 This finding is puzzling because RA+ T cells are naive cells, and as indicated above, the DN T cells express HLA-DR, a marker of previous activation. One possible explanation is that the CD95 defect leads to disturbances in certain types of T-cell maturation.
Another important immunophenotypic feature of ALPS is the presence of a significant B-cell lymphocytosis, with a particular increase in CD5-co-expressing B cells observed in all patients but two. Although patients with B-cell chronic lymphocytic leukemia frequently exhibit autoimmune phenomena, recent studies suggest that the CD5+ B cell is itself not responsible for autoimmunity in this disease. 34 Thus, our finding of increased CD5+ B cells does not totally explain the presence of autoimmune phenomena in ALPS patients. However, the overall increase in B cells and plasma cells is in keeping with increased antibody production.
In previous studies we demonstrated that CD4+/DR+ T cells manifest a prominent Th-2 cytokine profile, characterized by a diminished secretion of IL-2 and interferon-γ but an increased secretion of IL-4 and IL-5 from stimulated cells. In addition, ALPS patients display a marked elevation in circulating IL-10 levels. 30 These Th-2 cytokines have been shown to play a role in polyclonal B cell activation and hypergammaglobulinemia, and may contribute to the extent of B-cell lymphocytosis seen in ALPS patients.
In summary, analysis of clinical, immunological, and pathological features in 10 unrelated patients with ALPS reveals a characteristic spectrum of histopathological features that correlates with disease severity. It is becoming more evident that ALPS is a consequence of several different genetic and immunological abnormalities. Indeed, abnormalities in peripheral blood apoptosis leading to accumulations of lymphocyte subsets may occur in the absence of CD95 gene mutations as evidenced by patient 11 and 14. 15,19
Despite possibly different etiological factors responsible for the disease, consistent histological findings are characteristic of the entity. We believe the observed histological findings in lymph nodes are sufficiently distinctive to warrant further evaluation for ALPS in patients exhibiting these changes. The role of CD95/CD95L interaction and/or DN T cells in the pathogenesis of other autoimmune diseases associated with generalized lymphadenopathy and abnormal T cell proliferations such as rheumatoid arthritis and systemic lupus erythematosus may also be a productive area for future study. Additional studies examining other genes in the apoptosis pathway may reveal other genetic aberrations associated with ALPS.
Acknowledgments
We gratefully acknowledge the assistance of Drs. Laszlo Krenacs and Douglas W. Kingma, Laboratory of Pathology, NCI, for assistance with immunohistochemical and in situ hybridization studies, respectively.
Footnotes
Address reprint requests to Dr. Elaine S. Jaffe, Hematopathology Section, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Building 10, Room 2N 202, 10 Center Drive MSC-1500, Bethesda, MD 20892. E-mail: elaine.jaffe@nih.gov.
M.S. Lim is a recipient of a clinical fellowship from the Medical Research Council of Canada, Alberta Heritage Foundation for Medical Research, and the Fogarty Visiting Fellowship from the National Institutes of Health.
References
- 1.Sneller MC, Straus SE, Jaffe ES, Jaffe JS, Fleisher TA, Stetler-Stevenson M, Strober W: A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest 1992, 90:334-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe-Fukunaga R, Brannan PE, Copeland NG, Jenkins N, Nagata S: Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992, 356:314-317 [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, Tanaka M, Rannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S: Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 1994, 76:969-977 [DOI] [PubMed] [Google Scholar]
- 4.Lynch DH, Watson ML, Alderson MR, Baum PR, Miller RE, Tough T, Gibson M, Davis-Smith T, Smith CA, Hunter K, Bhat D, Din W, Goodwin RG, Seldin MF: The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity 1994, 1:131-140 [DOI] [PubMed] [Google Scholar]
- 5.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM: Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81:935-946 [DOI] [PubMed] [Google Scholar]
- 6.Drappa J, Vaishnaw A, Sullivan KE, Chu J, Elkon KB: Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 1996, 335:1643-1649 [DOI] [PubMed] [Google Scholar]
- 7.Rieux-Laucat F, Le Diest F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP: Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995, 268:1347-1349 [DOI] [PubMed] [Google Scholar]
- 8.Le Deist F, Emile J-F, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, Fischer A: Clinical, immunological and pathological consequences of fas-deficient conditions. Lancet 1996, 348:719-723 [DOI] [PubMed] [Google Scholar]
- 9.Canale VC, Smith CH: Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr 1967, 70:891-899 [DOI] [PubMed] [Google Scholar]
- 10.Rao LM, Shahidi NT, Opitz JM: Hereditary splenomegaly with hypersplenism. Clin Genet 1974, 5:379-386 [DOI] [PubMed] [Google Scholar]
- 11.Gillette-Ferguson I, Sidman CL: A specific intercellular pathway of apoptotic cell death is defective in the mature peripheral T cells of autoimmune lpr and gld mice. Eur J Immunol 1994, 24:1181-1185 [DOI] [PubMed] [Google Scholar]
- 12.Adachi M, Suematsu S, Kondo T, Ogasawara J, Tanaka T, Yoshida N, Nagata S: Targeted mutation in the Fas gene causes hyperplasia in peripheral lymphoid organs and liver. Nature Genet 1995, 11:294-300 [DOI] [PubMed] [Google Scholar]
- 13.Adachi M, Suematsu S, Suda T, Watanabe D, Fukuyama H, Ogasawara J, Tanaka T, Yoshida N, Shigekazu N: Enhanced and accelerated lymphoproliferation in Fas-null mice. Biochem 1996, 93:2131-2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Infante AJ, Britton HA, DeNapoli T, Middelton LA, Lenardo MJ, Jackson CE, Wang J, Fleisher T, Straus SE, Puck JM: The clinical spectrum in a large kindred with autoimmune lymphoproliferative syndrome (ALPS) due to a fas mutation that impairs lymphocyte apoptosis. J Pediatr (in press) [DOI] [PubMed]
- 15.Sneller MC, Wang J, Dale JK, Strober W, Middleton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE: Clinical, immunologic and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood 1997, 89:1341-1348 [PubMed] [Google Scholar]
- 16.Elenitoba-Johnson KSJ, Kumar S, Lim MS, Kingma DW, Raffeld M, Jaffe E: Marginal zone B-cell lymphoma with monocytoid B-cell lymphocytes in pediatric patients without immunodeficiency: a report of two cases. Am J Clin Pathol 1997, 107:92-98 [DOI] [PubMed] [Google Scholar]
- 17.McCarthy KP, Sloane JP, Kabarowski JH, Matutes E, Wiedermann LM: A simplified method of detection of clonal rearrangement of the TCR-γ chain gene. Diagn Mol Pathol 1992, 1:173-179 [PubMed] [Google Scholar]
- 18.Natkunam Y, Elenitoba-Johnson KS, Kingma DW, Kamel O: Epstein-Barr virus strain type and latent membrane protein-1 gene deletions in lymphoma in patients with rheumatic disease. Arthritis Rheum 1997, 40:1152-1156 [DOI] [PubMed] [Google Scholar]
- 19.Dianzani U, Bragardo M, DiFranco D, Alliaudi C, Scagni P, Buonfiglio D, Redoglia V, Bonissoni S, Correra A, Dianzani I, Ramenghi U: Deficiency of the Fas apoptosis pathway without Fas gene mutations in pediatric patients with autoimmunity/lymphoproliferation. Blood 1997, 89:2871-2879 [PubMed] [Google Scholar]
- 20.Tedder TF, Cooper MD, Clement LT: Human lymphocyte differentiation antigens HB-10 and HB-11. II. Differential production of B cell growth and differentiation factors by distinct helper T cell subpopulations. J Immunol 1985, 134:2989-2994 [PubMed] [Google Scholar]
- 21.Chittal SM, Brousset P, Voigt J-J, Delsol G: Large B-cell lymphoma rich in T-cells and simulating Hodgkin’s disease. Histopathology 1992, 19:211-220 [DOI] [PubMed] [Google Scholar]
- 22.Delabie J, Vandenberghe E, Kennes C, Verhoef G, Foschini MP, Stul M, Cassiman JJ, De Wolf-Peeters C: Histiocyte-rich B-cell lymphoma: a distinct clinicopathologic entity possibly related to lymphocyte predominant Hodgkin’s disease, paragranuloma subtype. Am J Surg Pathol 1992, 16:32-48 [PubMed] [Google Scholar]
- 23.Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD: Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest 1996, 98:1107-1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De la Hara A, Toribio ML, Marquez C, Marcos MAR, Cabrero E, Martinez AC: Differentiation of human mature thymocytes: existence of a T3+4−8− intermediate stage. Eur J Immunol 1986, 16:653-658 [DOI] [PubMed] [Google Scholar]
- 25.Groh V, Fabbi M, Hochstenbach F, Maziarz R: Double negative (CD4−CD8−) lymphocytes bearing T cell receptor α and β chains in normal human skin. Proc Natl Acad Sci USA 1989, 86:5059-5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanier LL, Ruitenberg JJ, Phillips JH: Human CD3+ T lymphocytes that express neither CD4 nor CD8 antigens. J Exp Med 1986, 164:339-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenkei R, Andersson B: High correlations of anti-CMV titers with lymphocyte activation status and CD57 antibody-binding capacity as estimated with three-color, quantitative flow cytometry in blood donors. Clin Immunol 1995, 77:131-138 [DOI] [PubMed] [Google Scholar]
- 28.Shivakumar S, Tsokos GC, Datta SK: T cell receptor α/β expressing double negative and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol 1989, 143:103-112 [PubMed] [Google Scholar]
- 29.Wirt DP, Rooks EG, Vaidya S, Klimpel GR, Waldmann TA, Goldblum RM: Novel T-lymphocyte population in combined immunodeficiency with features of graft-versus-host disease. N Engl J Med 1989, 321:370-374 [DOI] [PubMed] [Google Scholar]
- 30.Fuss IJ, Strober W, Dale JK, Fritz S, Pearlstein GR, Puck JM, Lenardo MJ, Straus SE: Characteristic T helper 2 T cell cytokine abnormalities in autoimmune lymphoproliferative syndrome, a syndrome marked by defective apoptosis and humoral autoimmunity. J Immunol 1997, 158:1912-1918 [PubMed] [Google Scholar]
- 31.Giese T, Davidson WF: Chronic treatment of C3H-lpr/lpr and C3H-gld/gld mice with anti-CD8 monoclonal antibody prevents the accumulation of double negative T cells but not autoantibody production. J Immunol 1994, 152:2000-2010 [PubMed] [Google Scholar]
- 32.Giese T, Davidson WF: In CD8+ T cell-deficient lpr/lpr mice, CD4+B220+, and CD4+B220− T cells replace B220+ double-negative T cells as the predominant populations in enlarged lymph nodes. J Immunol 1995, 154:4986-4995 [PubMed] [Google Scholar]
- 33.Warren HS, Skipsey SM: Loss of activation-induced CD45RO with maintenance of CD45RA expressing prolonged culture of T cells and NK cells. Immunology 1991, 74:78. [PMC free article] [PubMed] [Google Scholar]
- 34.Pritsch O, Maloum K, Dighiero G: Basic biology of autoimmune phenomena in chronic lymphocytic leukemia. Semin Oncol 1998, 25:34-41 [PubMed] [Google Scholar]