

Abstract
The R1-R6 subclass of photoreceptor neurons (R cells) in the Drosophila compound eye form specific connections with targets in the optic ganglia. In this paper, we report the identification of a gene, brakeless (bks), that is essential for R1-R6 growth cone targeting. In brakeless mutants, R1-R6 growth cones frequently fail to terminate migration in their normal target, the lamina, and instead project through it and terminate in the second optic ganglion, the medulla. Genetic mosaic analysis and transgene rescue experiments indicate that bks functions in R cells and not within the lamina target region. bks encodes a nuclear protein. We propose that it participates in a gene expression pathway regulating one or more growth cone components controlling R1-R6 targeting.
Neurons form precise patterns of synaptic connections during development. Differentiating neurons extend axons that migrate along defined pathways toward their targets (1). The growth cone, a sensorimotor structure at the leading edge of the axon, plays a central role in axon guidance. It detects and integrates multiple signals (see, e.g., ref. 2) and translates them into directed motility (see, e.g., ref. 3). On reaching the target region, growth cone migration ceases and specific synapses are established. How growth cones select specific targets once they have reached the target region remains a central issue in developmental neurobiology.
Considerable progress has been made in defining the molecular basis of targeting. In the chick visual system, for instance, the formation of a precise topographic map between retinal ganglion cells and tectal cells relies on the graded expression of the Ephrin ligands in the tectum and their receptors, the Ephs, on retinal ganglion cell growth cones (4). Antibody disruption experiments suggest that cadherins play an important role in subsequent steps in which retinal ganglion cells form specific connections in distinct laminae within the tectum (5). A large family of odorant receptors play an essential role in determining the precise patterns of connections made by olfactory neurons with their postsynaptic target cells (6). That targeting may rely on multiple signals, both attractive and repulsive in nature, is supported by the genetic analysis of motoneuron target specificity in the Drosophila embryo (2).
Although targeting requires interactions between extracellular signals and receptors on the growth cone, the formation of neuronal connections also relies on the genetic programs that specify the precise spatial and temporal expression of these signaling molecules. Several transcription factors have been implicated in regulating axon guidance and target specificity (7–14). For instance, in Caenorhabditis elegans, a homeobox-containing protein UNC30 is required for normal pathfinding of a subset of axons suggesting that it may control the expression of specific guidance receptors; UNC-30 also controls genes necessary for other aspects of neuronal differentiation, including enzymes necessary for neurotransmitter metabolism. In both the vertebrate spinal cord and Drosophila ventral nervous system, LIM homeodomain proteins have been proposed to play key roles in determining patterns of connections elaborated by different classes of motoneurons (12, 13). Specific transcription factors also may act in targets to specify synaptic inputs. In C. elegans, for instance, the homeodomain-containing protein UNC-4 regulates the expression of genes in motoneurons that specify presynaptic input (9, 14). In principle, other nuclear factors, such as those regulating splicing, could also control connection specificity. In this paper, we describe the isolation, phenotypic characterization, and molecular analysis of a nuclear protein required for targeting of Drosophila photoreceptor neurons.
The Drosophila visual system comprises the compound eye and the optic lobe (see ref. 15). The compound eye contains ≈800 ommatidia, each containing eight different R cells. Different R-cell classes connect to target cells in distinct ganglia within the optic lobe. R1-R6 axons innervate the first optic ganglion, the lamina, whereas the R7 and R8 axons project through the lamina and terminate in the medulla. Targeting of R-cell axons to different ganglia occurs during the third and final stage of larval development. R cells differentiate in a defined sequence, with R8 differentiating first, followed by R1-R6 and then R7; the sequence of R-cell innervation appears to reflect the order of R-cell differentiation. R8 grows through the optic stalk, traverses the developing lamina, and terminates in the medulla neuropil. R1-R6 neurons from the same developing ommatidiun grow along the surface of the R8 axon into the developing lamina. On encountering a distinct layer of lamina glial cells, called marginal glia, these growth cones cease migration and elaborate a large expanded growth cone in the target region. In the absence of the lamina glia, the R1-R6 neurons fail to terminate and project into the medulla neuropil (I. Salecker and S.L.Z., unpublished data), supporting the view that these glia provide a stop signal specifically recognized by R1-R6 neurons (16). The last R cell in each cluster to differentiate is R7; it projects an axon along the bundle containing R1-R6 and R8. R7 is refractory to the lamina “stop” signal projecting through the lamina and into the medulla neuropil. Synaptic connections between R1-R6 and lamina target neurons are established during pupal development.
The formation of connections between R cells and lamina neurons requires a complex dialogue between R cells and cells in the lamina target region. R-cell growth cones release Hh protein, which drives lamina neuronal precursor cells through their final division (17, 18). Lamina neuronal differentiation depends on a second signal released from these growth cones, the EGF-like ligand Spitz (19). Although glial cell proliferation does not require R-cell innervation, glial cell differentiation depends on additional signals (16, 20, 21).
Little is known about the mechanisms regulating targeting of R1-R6 growth cones to the lamina. R1-R6 targeting requires the activity of several proteins localized to the growth cone, including the receptor tyrosine phosphatase DPTP69D (22), the SH3/SH2 adapter protein Dock (23), two serine/threonine kinases, Pak (24) and Msn (25), and a component of the COP9 signalosome, JAB1 (G. Suh and S.L.Z., unpublished data). The relationship of these proteins to specific target molecules on the lamina glia and receptors on growth cones is not known. In this paper, we report the characterization of a gene called brakeless (bks) encoding a nuclear protein required for R1-R6 growth cones to terminate in the lamina target.
Materials and Methods
Drosophila Stocks and Genetics.
Lethal P-element inserts, l (2)k06903, l (2)k0702 (bksP2), and l (2)k0444 (bksP3) (26, 27) were provided by the Berkeley Drosophila Genome Project. The deficiency stock, Df(2R)PC4, was provided by the Bloomington Drosophila Stock Center. w; P[neo, Rh1-LacZ] was provided by G. Rubin (University of California, Berkeley). bks alleles were maintained over In(2LR)GlaBcElp for larval phenotypic analysis. Excision of the bksP1 P-element was performed as described (28).
To generate transgenic flies for rescue experiments, the bks cDNA was subcloned into the pGMR and CaspeR-hs transformation vectors. These DNA constructs were introduced into flies by using standard methods (29). Heat shock rescue was performed as described (23). Three independent GMR-bks and CaspeR-hs-bks transgenic lines were used in rescue experiments.
Genetic Mosaic Analysis.
To generate genetic mosaics, w; bksP1/+; P[neo, Rh1-LacZ]/+ individuals were X-irradiated as first instar larvae (30). Homozygous bksP1 eye clones, carrying two copies of P[lac-W], were easily distinguished from heterozygous cells (containing only one copy of P[lac-W]) or wild-type cells by the increased amount of pigment. The R1-R6 termination pattern in mosaic heads was visualized with anti-LacZ staining as described (23). Plastic sections of bksP1 mosaic eyes were performed as described (31).
Molecular Biology.
Genomic sequence flanking the bksP1 P element insertion site was isolated by plasmid rescue. PCR was used to determine the P-element insertion site in bksP2 and the deleted sequence in bks4. The expressed sequence tag (EST) clone LD14810 containing the partial sequence of the brakeless cDNA was obtained from Research Genetics (Huntsville, AL). Additional 5′ sequence was isolated from a Drosophila embryonic plasmid cDNA library (32) by PCR, based on the sequence of the EST clone LD13770. The blast program (33) was used to search for homologous sequences in public databases.
Immunohistochemistry.
R-cell axons in third-instar larvae were stained with mAb 24B10 as described (31). The expression pattern and the subcellular localization of Brakeless in third-instar larvae were determined by using anti-Bks antibody at 1:100 dilution. Third-instar eye-brain complexes were also stained with antibodies to Elav (34), Dachshund (35), RK2 (36), Bar (37), Boss (31), and Prospero (38).
Western Blot Analysis.
GST-Bks fusion protein was prepared as described (39) and was used to raise a rabbit anti-serum using standard methods. Anti-Bks antibody was purified on Affi-Gel-10 beads (Bio-Rad) cross-linked with GST-Bks fusion protein (40). Western blot analysis was performed as described (41). Anti-Bks antibody and anti-Dock antibody (42) were used at 1:2,000 dilution.
Results and Discussion
Isolation of brakeless Mutations.
We screened a collection of lethal P element insertions on the second chromosome (26, 27) for mutations that disrupt the R-cell projection pattern (23). One insertion line, l (2)k06903, caused defects in both R-cell projections and R-cell patterning in the eye, as assessed in preparations of third instar eye-brain complexes stained with an antibody recognizing R-cell bodies and their axons. l (2)k06903 carries two P-element insertions at polytene chromosome bands 47D1-D2 and 55C1-C5. Genetic experiments demonstrated that the connectivity and patterning defects were genetically separable and that the connectivity defects resulted from the insertion at 55C1-C5. First, the projection, but not the eye, phenotype was uncovered by a deficiency [Df(2R)PC4] that selectively removes 55C1-C5. Second, the two P-element insertions were separated by meiotic recombination; larvae homozygous for the 55C1-C5 insertion exhibited the projection, but not the patterning, defect. And third, excision of the P element at 55C1-C5 restored R-cell projection pattern in 88 of 90 excision lines analyzed. Because many R1-R6 growth cones fail to stop in the lamina (see below), we named the mutation brakeless (bks) and the initial P allele bksP1.Two other lethal P-element insertions, l (2)k0702 and l (2)04440, were shown to be allelic to bksP1 and are designated bksP2 and bksp3, respectively. Two imprecise excision mutant alleles are designated bks4 and bks5. Mutations in bks were independently isolated by Dickson and coworkers (43).
bks Mutations Cause Defects in R-Cell Targeting.
The R-cell axon projection patterns in wild-type and mutant third-instar larval eye-brain complexes were visualized by using mAb24B10 (44). In the wild type (Fig. 1A), bundles of R-cell axons converge at the posterior end of the eye disk, where they enter the optic stalk. On exiting the stalk, axon bundles elaborate a topographic map in the developing lamina and medulla. The R1–6 growth cones terminate in the lamina with their expanded growth cones, forming a dense continuous layer of immunoreactivity in a region referred to as the lamina plexus. R7 and R8 growth cones project through the lamina and terminate in an array in the medulla neuropil.
Figure 1.

brakeless is required for R-cell projections. (A and B) R-cell projections were visualized with mAb 24B10 staining. In the wild type (A), R-cell axons project from the developing eye disk through the optic stalk (os) into the optic lobe. The dense staining layer in the lamina (la) consists mainly of expanded R1-R6 growth cones. R7 and R8 axons pass through the lamina into the medulla (me), where they form a highly ordered innervation pattern. In bks mutants (B), many R1-R6 growth cones fail to stop at the lamina and migrate into the medulla instead (also see Fig. 3B), leaving many uninnervated regions in the lamina. Consequently, abnormal thicker bundles and excessive growth cones appear in the medulla. The arrowhead in B indicates the terminus of Bolwig's nerve. (C and D) Wild-type (C) and bksP1 (D) third-instar larval optic lobes stained with anti-Dachshund. Dachshund is a nuclear protein expressed in the developing lamina neurons. Lamina neurons differentiate normally in bks mutants (D). (E and F) Optic lobes stained with the RK2 antibody recognizing Repo, a nuclear protein expressed in glial cells. In the wild type (E), R1–6 growth cones are surrounded by rows of RK2-positive glial cells. Although these cells migrate correctly into the target region and differentiate in a bks4 optic lobe (F), they appear less organized than the wild type, likely because of the abnormal R-cell innervations. (Bar = 20 μm.)
In bks mutant larvae, R-cell axons project through the optic stalk and into the optic lobe, largely as they do in the wild type. Projection defects in both the lamina and medulla were observed (Fig. 1B). In contrast to the wild type, the lamina plexus was discontinuous, with regions of dense staining separated by weakly stained regions or gaps. Thick bundles of axons were frequently observed between the lamina and medulla, in striking contrast to the lightly stained thin bundles in the wild type (Fig.1B). In the most severe cases, (Fig. 1B), the lamina plexus was largely missing and the medulla neuropil was hyperinnervated. In addition, disruptions in the normally smooth topographic map in the medulla were observed. However, R-cell projections did not extend through the medulla into deeper layers of the optic lobe, suggesting that R8 neurons terminate, as in the wild type, within the medulla neuropil. As R7 axons project into the optic lobe only later, and no early R7-specific axon markers are available, we were unable to assess the initial projections of these neurons to their medulla targets. These data are consistent with a strong mistargeting defect, where R1-R6 neurons fail to terminate in the lamina and, instead, project into the medulla neuropil (see below). These phenotypes are completely penetrant in bks4, bksP1, bksP2, and bks5 and partially penetrant in bksP3. Individuals homozygous for bks4 and bksP1 show the most severe phenotype. These phenotypes were not enhanced in trans to a deficiency for the region, Df(2R)PC4, suggesting that bks4 and bksP1 are strong loss-of-function, if not null, mutations. The relative strength of the connectivity defects is bks4 ≥ bksP1 > bks5 = bksP2 > bksP3. Stronger alleles not only increase penetrance, they also increase the severity of the mistargeting phenotype. Increasing strength of the alleles also correlated with progressive decrease in larval motility.
Although bks mutations lead to severe defects in neuronal connectivity, several markers indicated that development of cells in the target region occurs normally. As in the wild type, lamina neuronal precursor cells are driven through their final division, as assessed by BrdUrd incorporation (data not shown), and differentiate as assessed by the expression of the lamina neuronal markers, Dachshund (Fig. 1 C and D) and Elav (data not shown). Lamina glia, the intermediate targets for R-cell growth cones, migrate into the target region and differentiate largely as in the wild type (Fig. 1 E and F).
bks Encodes a Novel Protein.
Genomic DNA flanking the P-element insertion site in bksP1 was isolated by plasmid rescue. Database searches using the flanking sequence identified a Drosophila genomic fragment (GenBank accession no. AA004295) from a P1 clone, DS08374 (D180), which had been completely sequenced by the Berkeley Drosophila Genome Project. To look for transcripts close to the P-element insertion point, flanking genomic sequence was used to search a public database of expressed sequence tags (ESTs). A transcription unit corresponding to overlapping EST clones (LD35038, LD13770, LD14810, LD22820, and LD47245) was identified. The intron/exon structure was determined by comparing the sequence of the largest EST clone to the genomic sequence (Fig. 2A). Sequence analysis revealed that the bks P elements were inserted into either the first intron (bksP1 and bksP3) or the 5′-promoter region immediately adjacent to the first exon (bksP2). In the imprecise excision allele bks4, 436 base pairs were deleted from the third exon. To test whether disruption of this transcription unit alone accounts for the bks R-cell connectivity phenotype, a cDNA transgene rescue experiment was performed. An hsp-70 full length EST (LD13770) cDNA transgene was introduced into a homozygous bks mutant. Reiterative heat shocks completely rescued the connectivity defect (data not shown; see below for additional transgene rescue data).
Figure 2.

Molecular characterization of the brakeless gene. (A) Genomic structure of the brakeless gene. P element insertion sites for bksP1 and bksP3 are located within the first intron, 14 bp and ≈140 bp (based on the Berkeley Drosophila Genome Project database of transposon insertions) downstream of the exon/intron boundary, respectively. The P element insertion site for bksP2 is located 11 bp upstream of the first exon. The sequence deleted in bks4 is indicated as the shaded box. Filled boxes indicate the putative translated sequences whereas open boxes indicate untranslated sequences. (B) The predicted protein sequence of Brakeless. The sequence (amino acids 623–818) used to raise antiserum in rabbit is underlined. (C) Alignment of the Brakeless protein and homologous vertebrate sequences (ESTs). Identical amino acids are stippled. (D) Western blot analysis of extracts made from five third-instar larval eye-brain complexes. Anti-Bks antibody detected a single band at ≈80 kDa in the wild type, but not in bks4 homozygous mutants.
Sequence analysis of the bks cDNA revealed a single long ORF corresponding to a protein of 929 amino acids (Fig. 2B). The predicted molecular weight is 88 kDa. Bks protein does not resemble any protein of known function, nor does it contain known protein domains, motifs, or any significant stretches of sequence homology to known proteins. Bks protein contains an unusually high content of Ser (23.7%) and Thr (7.6%); repeated stretches of Ser-rich sequences are found in the amino-terminal half of the polypeptide. Sequences related to Bks, however, were identified in public databases of ESTs from human (GenBank accession no. AI223051), mouse (accession nos. AI644380 and AI614389), and Xenopus (accession no. AI031376) (Fig.2C). These ESTs may represent either portions of bks homologs or proteins that contain an evolutionarily conserved domain with undefined function. Senti et al. (43) identified a second alternatively spliced form of bks that gives rise to a larger protein with an extended C-terminal tail.
bks Is Required in the Eye for R1-R6 Growth Cone Targeting.
R-cell projection defects in bks mutants may reflect a role for bks in R cells, in their targets, or both. To distinguish between these possibilities, we carried out genetic mosaic analysis. Mutant eye tissue, homozygous for bksP1, was generated in otherwise heterozygous eyes by x-ray-induced mitotic recombination. R1-R6 targeting from mutant tissue into the lamina was assessed using an R1-R6-specific axonal marker, Rh1-LacZ (45). Cryostat sections of adult heads prepared from genetic mosaics and wild-type controls were stained with a rabbit anti-β-galactosidase antibody. In the wild type (Fig. 3A), all R1-R6 axons terminate in the lamina; no staining was observed in the medulla. In contrast, in all mosaic individuals examined (n = 8), many R1-R6 axons fail to terminate in the lamina and, instead, project through the lamina and terminate in the medulla (Fig. 3B). In support of these findings, the bks mutant phenotype was largely rescued by expressing a bks cDNA under the control of an eye-specific promoter, GMR (Fig. 3D) (46). These data indicate that expression of bks in the eye is not only necessary for normal R1-R6 connectivity, it is sufficient.
Figure 3.

brakeless is required in the eye for R1-R6 growth cone targeting. (A and B) Cryostat sections of adult heads carrying the adult R1-R6 marker Rh1-LacZ were stained with anti-β-galactosidase antibody. In the wild type (A), anti-LacZ staining shows R1-R6 cell bodies in the retina and their axons in the lamina. Note that no staining is observed in the medulla. (B) A patch of bksP1 mutant eye tissue was generated in an otherwise heterozygous or wild-type eye by x-ray-induced mitotic recombination. Some R1-R6 axons migrate into the medulla (arrowheads). (C and D) Third-instar larval eye-brain complexes were stained with mAb 24B10. (C) R-cell projections in a bks5 homozygote. (D) R-cell projections in a bks5 homozygote carrying a copy of GMR-bks transgene. R-cell termination pattern is very similar to that in the wild type. (Bar = 20 μm.)
Bks Is a Nuclear Protein.
To determine the expression pattern and the subcellular localization of Bks, antibodies to it were generated. A glutathione S-transferase fusion protein containing a fragment of the Bks protein (amino acids 623–818) was used as an immunogen in rabbits. Western blot analysis of extracts made from wild-type third instar eye-brain complexes using the affinity-purified antibody detected a single band at ≈80 kDa (Fig. 2D), which is close to the predicted size of the Bks protein. That this band corresponds to Bks is supported by its loss in bks mutant extracts (Fig. 2D); the faint band remaining at approximately the same molecular weight is a cross-reacting species. The distribution of the Bks protein in the eye imaginal disk was determined in whole-mount preparations. Neuronal differentiation occurs posterior to a dorsoventral groove called the morphogenetic furrow. Bks was detected in the nuclei of developing R-cells (Fig. 4A) and undifferentiated precursor cells (data not shown) lying in the basal region of the disk. Weak nuclear staining was also seen in cells located anterior to the morphogenetic furrow. This nuclear staining was missing in bks mutants (Fig. 4B) and was greatly enhanced in larvae overexpressing bks (data not shown), confirming the specificity of the antibody. In addition to the staining in the developing eye disk, Brakeless protein was also detected in the nuclei of glial cells in the optic stalk (data not shown). High background staining precluded an assessment of Bks protein in the central nervous system. Because bks mutations are lethal, however, bks must be required for functioning outside the eye. Transgene rescue experiments described in the preceding section indicate that bks is not required in these glial cells because the GMR promoter does not drive expression in them.
Figure 4.
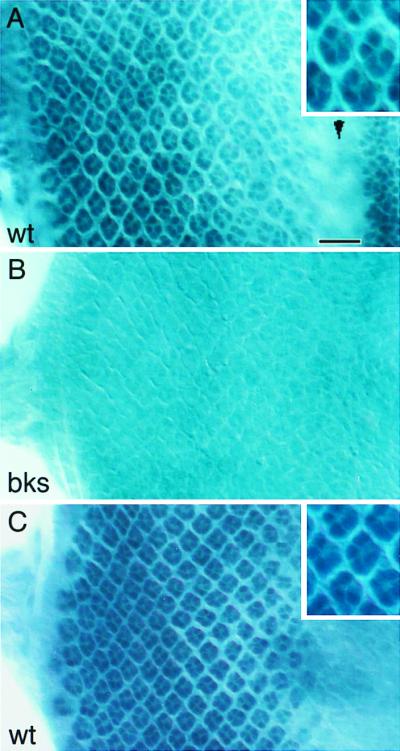
Expression pattern and subcellular localization of the Brakeless protein. Third-instar larval eye discs were stained with anti-Bks antibody (A and B) or anti-Elav antibody (C). Posterior is to the left. An apical focal plane is shown. (A) In the wild type, Bks was detected in the nuclei of developing R-cells behind the morphogenetic furrow (arrowhead). The intensity of staining increases gradually from anterior to posterior. Weak Bks staining also was seen in cells located anterior to the morphogenetic furrow. The inset is a high magnification view of the staining. (B) No Bks staining was seen in bks4 mutants. (C) The nuclei of differentiating R-cells in the wild type was visualized with anti-Elav staining. The inset is a high magnification view of the nuclear staining. Note that the Bks staining pattern in the inset of A is similar to that in C. (Bar = 15 μm.)
R-Cell Fate and Differentiation Occur Normally in bks.
As Bks is expressed in the nuclei of R cells, defects in R-cell targeting may be an indirect consequence of more general defect in R-cell fate determination or differentiation. To assess this possibility, mutant eye discs were stained with various markers, and adult mosaic eyes were analyzed by toluidine blue staining of plastic sections. The Prospero (R7 marker; n = 12 discs) (Fig. 5 A and E) and Boss (R8 marker; n = 14 discs) (Fig. 5 B and F) staining patterns in bks and the wild type were indistinguishable. Thus, targeting to the medulla, and the failure of R cells to terminate in the lamina, is unlikely to reflect transformation of R1-R6 into either R7 or R8 neurons. Furthermore, R1 and R6 cell-fate acquisition appears to occur normally because the spatiotemporal pattern of Bar antibody staining (n = 12 discs) in these cells is similar to the wild type (Fig. 5 C and G). PTP69D and Dock, two proteins necessary for R1-R6 targeting, are expressed in R cells in bks mutants at a level similar to that in the wild type.
Figure 5.

R-cell differentiation and patterning appear normal in bks mutants. Wild-type (A, B, C, and D) and bks4 mutant (E, F, G, and H) third-instar eye-imaginal discs stained with cell-type-specific markers: anti-Prospero (A and E); anti-Boss (B and F); anti-Bar (C and G); and anti-Elav (D and H). Apical surface is up and posterior to the left. (Bar = 15 μm.)
Neuronal differentiation occurs normally, as well. The onset and pattern of expression of the pan-neuronal nuclear marker Elav occurs in a sequential fashion, as it does in the wild type (Fig. 5 D and H). Furthermore, toluidine-blue stained plastic sections of homozygous bks mutant patches in the adult eye (n = 6 eyes; 180 ommatidia) revealed that the adult morphology of R cells and the composition of ommatidia were unaltered (data not shown). As in the wild type, bks mutant R cells are seen as unstained cell bodies in the periphery of the ommatidium with their densely stained photosensitive organelles, the rhabdomeres, projecting toward the center. In addition to the apparently normal morphology of R cells, the trapezoidal arrangement of these cells within in the ommatidium is also retained in bks mutants. Of 180 ommatidia, 3 showed abnormal polarity. This may reflect a weak role for bks in regulating the cellular movements underlying polarity establishment. In summary, these data indicate that the targeting defects in bks do not reflect a secondary effect of an earlier role for bks in R-cell development.
Concluding Remarks
In this paper, we demonstrate that bks encodes a nuclear protein essential for the establishment of specific neuronal connections in the fly visual system. In particular, in bks mutants, many R1-R6 neurons fail to terminate in their appropriate target region but, rather, project through it. As bks does not appear to regulate cell fate determination or cellular differentiation more generally, we propose that it regulates the expression, either at the transcriptional or posttranscriptional level, of genes necessary for targeting. Genes controlled by bks may encode targeting determinants on the surface of growth cones or proteins necessary for transducing signals to the motility apparatus of the growth cone.
Acknowledgments
We thank the Berkeley Drosophila Genome Project and Bloomington Stock Center for fly stocks; L. Ballard for technical assistance; R. Carthew for anti-Prospero antibody; J. Clemens and J. Dixon for anti-Dock antibody; G. Mardon for anti-Dachshund mAb; G. M. Rubin for Rh1-LacZ line; K. Saigo for anti-Bar antibody; K. Ronan for help with the manuscript; I. Salecker and G. Suh for assistance on figures; and A. Tomlinson for an anti-RK2 antiserum. We also acknowledge Barry Dickson and his colleagues for sharing their results before publication. This work was supported by an operating grant to Y.R. (MT-14688) and Scholarship (to Y.R.) from the Medical Research Council of Canada, and a McKnight Development Award (to S.L.Z.). S.L.Z. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- bks
brakeless
- EST
expressed sequence tag
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database [accession no. AF238858 (brakeless)].
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.110135297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.110135297
References
- 1.Tessier-Lavigne M, Goodman C S. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 2.Winberg M L, Mitchell K J, Goodman C S. Cell. 1998;93:581–591. doi: 10.1016/s0092-8674(00)81187-3. [DOI] [PubMed] [Google Scholar]
- 3.Hong K, Hinck L, Nishiyama M, Poo M M, Tessier-Lavigne M, Stein E. Cell. 1999;97:927–941. doi: 10.1016/s0092-8674(00)80804-1. [DOI] [PubMed] [Google Scholar]
- 4.Flanagan J G, Vanderhaeghen Annu Rev Neurosci. 1998;21:309–345. doi: 10.1146/annurev.neuro.21.1.309. [DOI] [PubMed] [Google Scholar]
- 5.Inoue A, Sanes J R. Science. 1997;276:1428–1431. doi: 10.1126/science.276.5317.1428. [DOI] [PubMed] [Google Scholar]
- 6.Wang F, Nemes A, Mendelsohn M, Axel R. Cell. 1998;93:47–60. doi: 10.1016/s0092-8674(00)81145-9. [DOI] [PubMed] [Google Scholar]
- 7.Giniger E, Tietje K, Jan L Y, Jan Y N. Development (Cambridge, UK) 1994;120:1385–1398. doi: 10.1242/dev.120.6.1385. [DOI] [PubMed] [Google Scholar]
- 8.Jin Y, Hoskins R, Horvitz H R. Nature (London) 1994;372:780–783. doi: 10.1038/372780a0. [DOI] [PubMed] [Google Scholar]
- 9.Miller D M, Niemeyer C J. Development (Cambridge, UK) 1995;121:2877–2886. doi: 10.1242/dev.121.9.2877. [DOI] [PubMed] [Google Scholar]
- 10.Wightman B, Baran R, Garriga G. Development (Cambridge, UK) 1997;124:2571–2580. doi: 10.1242/dev.124.13.2571. [DOI] [PubMed] [Google Scholar]
- 11.Prasad B C, Ye B, Zackhary R, Schrader K, Seydoux G, Reed R R. Development (Cambridge, UK) 1998;125:1561–1568. doi: 10.1242/dev.125.8.1561. [DOI] [PubMed] [Google Scholar]
- 12.Sharma K, Sheng H Z, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff S L. Cell. 1998;95:817–828. doi: 10.1016/s0092-8674(00)81704-3. [DOI] [PubMed] [Google Scholar]
- 13.Thor S, Andersson S G, Tomlinson A, Thomas J B. Nature (London) 1999;397:76–80. doi: 10.1038/16275. [DOI] [PubMed] [Google Scholar]
- 14.Winnier A R, Meir J Y-J, Ross J M, Tavernarakis N, Driscoll M, Ishihara T, Katsura I, Miller D M., III Genes Dev. 1999;13:2774–2786. doi: 10.1101/gad.13.21.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolff T, Martin K A, Rubin G M, Zipursky S L. In: Molecular and Cellular Approaches to Neural Development. Cowan W M, Jessell T M, Zipursky S L, editors. New York: Oxford Univ. Press; 1997. pp. 474–508. [Google Scholar]
- 16.Winberg M L, Perez S E, Steller H. Development (Cambridge, UK) 1992;115:903–911. doi: 10.1242/dev.115.4.903. [DOI] [PubMed] [Google Scholar]
- 17.Selleck S B, Steller H. Neuron. 1991;6:83–99. doi: 10.1016/0896-6273(91)90124-i. [DOI] [PubMed] [Google Scholar]
- 18.Huang Z, Kunes S. Cell. 1996;86:411–422. doi: 10.1016/s0092-8674(00)80114-2. [DOI] [PubMed] [Google Scholar]
- 19.Huang Z, Shilo B-Z, Kunes S. Cell. 1998;95:693–703. doi: 10.1016/s0092-8674(00)81639-6. [DOI] [PubMed] [Google Scholar]
- 20.Perez S E, Steller H. J Neurobiol. 1996;30:359–373. doi: 10.1002/(SICI)1097-4695(199607)30:3<359::AID-NEU5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Gong Q, Rangarajan R, Seeger M, Gaul U. Development (Cambridge, UK) 1999;126:1451–1456. doi: 10.1242/dev.126.7.1451. [DOI] [PubMed] [Google Scholar]
- 22.Garrity P A, Lee C H, Salecker I, Robertson H C, Desai C J, Zinn K, Zipursky S L. Neuron. 1999;22:707–717. doi: 10.1016/s0896-6273(00)80730-8. [DOI] [PubMed] [Google Scholar]
- 23.Garrity P A, Rao Y, Salecker I, McGlade J, Pawson T, Zipursky S L. Cell. 1996;85:639–650. doi: 10.1016/s0092-8674(00)81231-3. [DOI] [PubMed] [Google Scholar]
- 24.Hing H, Xiao J, Harden N, Lim L, Zipursky S L. Cell. 1999;97:853–863. doi: 10.1016/s0092-8674(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 25.Ruan W, Pang P, Rao Y. Neuron. 1999;24:595–605. doi: 10.1016/s0896-6273(00)81115-0. [DOI] [PubMed] [Google Scholar]
- 26.Karpen G H, Spradling A C. Genetics. 1992;132:737–753. doi: 10.1093/genetics/132.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torok T, Tick G, Alvarado M, Kiss I. Genetics. 1993;135:71–80. doi: 10.1093/genetics/135.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebens A J, Garren H, Cheyette B N R, Zipursky S L. Cell. 1993;74:15–27. doi: 10.1016/0092-8674(93)90291-w. [DOI] [PubMed] [Google Scholar]
- 29.Spradling A C, Rubin G M. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner M. Drosophila: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Van Vactor D L, Cagan R L, Kramer H, Zipursky S L. Cell. 1991;67:1145–1155. doi: 10.1016/0092-8674(91)90291-6. [DOI] [PubMed] [Google Scholar]
- 32.Brown N H, Kafatos F C. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- 33.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Robinow S, White K. J Neurobiol. 1991;22:443–461. doi: 10.1002/neu.480220503. [DOI] [PubMed] [Google Scholar]
- 35.Mardon G, Solomon N, Rubin G M. Development (Cambridge, UK) 1994;120:3473–3486. doi: 10.1242/dev.120.12.3473. [DOI] [PubMed] [Google Scholar]
- 36.Campbell G, Goring H, Lin T, Spana E, Andersson S, Doe C Q, Tomlinson A. Development (Cambridge, UK) 1994;120:2957–2966. doi: 10.1242/dev.120.10.2957. [DOI] [PubMed] [Google Scholar]
- 37.Higashijima S, Kojima T, Michiue T, Ishimaru S, Emori Y, Saigo K. Genes Dev. 1992;6:50–60. doi: 10.1101/gad.6.1.50. [DOI] [PubMed] [Google Scholar]
- 38.Kauffmann R C, Li S, Gallagher P A, Zhang J, Carthew R W. Genes Dev. 1996;10:2167–2178. doi: 10.1101/gad.10.17.2167. [DOI] [PubMed] [Google Scholar]
- 39.Smith D B. Methods Mol Cell Biol. 1983;4:220–229. [Google Scholar]
- 40.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. p. 313. [Google Scholar]
- 41.Biggs W H, Zipursky S L. Proc Natl Acad Sci USA. 1992;89:6295–6299. doi: 10.1073/pnas.89.14.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clemens J C, Ursuliak Z, Clemens K K, Price J V, Dixon J E. J Biol Chem. 1996;271:17002–17005. doi: 10.1074/jbc.271.29.17002. [DOI] [PubMed] [Google Scholar]
- 43.Senti, K.-A., Keleman, K., Eisenhaber, F. & Dickson, B. J. (2000) Development (Cambridge, U.K.), in press. [DOI] [PubMed]
- 44.Zipursky S L, Venkatesh T R, Teplow D B, Benzer S. Cell. 1984;36:15–26. doi: 10.1016/0092-8674(84)90069-2. [DOI] [PubMed] [Google Scholar]
- 45.Mismer D, Rubin G M. Genetics. 1987;116:565–578. doi: 10.1093/genetics/116.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hay B A, Wolff T, Rubin G M. Development (Cambridge, UK) 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]