

Abstract
Targeted ablation of the surfactant protein D (SP-D) gene caused chronic inflammation, emphysema, and fibrosis in the lungs of SP-D (−/−) mice. Although lung morphology was unperturbed during the first 2 weeks of life, airspace enlargement was observed by 3 weeks and progressed with advancing age. Inflammation consisted of hypertrophic alveolar macrophages and peribronchiolar-perivascular monocytic infiltrates. These abnormalities were associated with increased activity of the matrix metalloproteinases, MMP2 and MMP9, and immunostaining for MMP9 and MMP12 in alveolar macrophages. Hydrogen peroxide production by isolated alveolar macrophages also was increased significantly (10-fold). SP-D plays a critical role in the suppression of alveolar macrophage activation, which may contribute to the pathogenesis of chronic inflammation and emphysema.
Surfactant protein D (SP-D) is a 43-kDa member of a group of collagenous, lectin domain-containing polypeptides, termed the collectins. SP-D shares considerable structural homology with other C-type lectins, including surfactant protein A (SP-A), conglutinin, bovine collectin-43, and mannose binding protein (refs. 1–3 for review). SP-D is expressed at relatively high concentrations in nonciliated bronchiolar cells and in alveolar type II cells (4); however, SP-D mRNA also is expressed in other tissues, including the stomach, heart, kidney, and mesentery cells (5–7). Shared structural motifs with other collectins support the potential role of SP-D in innate host defense (3). In vitro, SP-D binds to various viral, fungal, and bacterial components and to both alveolar macrophages and polymorphonuclear cells (1–3). Despite numerous in vitro studies supporting its role in host defense, its function in vivo remains poorly understood. Targeted gene inactivation of the SP-D gene in mice caused increased alveolar surfactant phospholipid pools, associated with increased numbers of lipid-laden macrophages (8, 9) and enlarged distal airspaces (8). Surfactant protein B (SP-B) and surfactant protein C (SP-C) expression was unaltered, but SP-A mRNA was reduced by 40% in lung homogenates, whereas the concentration of SP-A in bronchoalveolar lavage fluid (BALF) was reduced by 25% (8). The present work describes the temporal progression of postnatal airspace enlargement and spontaneous inflammatory changes in the lungs of SP-D (−/−) mice. SP-D (−/−) mice developed progressive pulmonary emphysema and subpleural fibrosis, associated with chronic inflammation and increased matrix metalloproteinase (MMP) activity and oxidant production by alveolar macrophages.
Experimental Procedures
Animals.
SP-D (−/−) mice were generated by targeted gene inactivation as recently described (8). SP-D (−/−) mice survive and breed normally in the vivarium under barrier containment facilities at Children's Hospital Medical Center, Cincinnati. Experimental procedures were reviewed and approved by the Children's Hospital Institutional Animal Care and Use Committee. Sentinel mice kept in the rooms with SP-D (−/−) mice have been viral free as assessed by serology. In addition, no serological evidence of viral infection or histological evidence of bacterial infection was detected in SP-D (−/−) mice at necropsy. Swiss black SP-D (−/−) and age-matched Swiss black SP-D (+/+) mice were mated separately to generate animals for this study. At selected ages, body, lung, and heart weights were obtained by direct measurement, and lung and heart volumes were obtained by fluid displacement. Lung protein and DNA content were assessed by using BSA and salmon sperm DNA, respectively, as standards (10, 11).
Histology.
Animals were weighed, anesthetized with a 4:1:1 mixture of ketamine, acepromazine, and xylazine, and exsanguinated by severing the inferior vena cava and descending aorta. The trachea was cannulated, and the lungs were collapsed by piercing the diaphragm. The lungs were inflation-fixed at 25 cm of water pressure with 4% paraformaldehyde in PBS for 1 min. The trachea was ligated, and the excised lungs and heart were allowed to equilibrate in cold fixative. Lung and heart volumes were determined by fluid displacement (12). Each lobe then was measured along its longest axis, bisected perpendicularly to the long axis, and processed into paraffin blocks. Five-micrometer sections were cut in series throughout the length of each lobe, loaded onto polysine-coated slides, and stained with hematoxylin and eosin, Masson's trichrome stain for collagen (13), or orcein for elastin (13).
Morphometry.
Morphometric measurements were performed on mice at 5 days (0.5 weeks), 14 days (2 weeks), 17 days (2.5 weeks), 3 and 6 weeks, and 6–7 months of age. Three to seven mice of both genotypes were studied at each age. The overall proportion (% fractional area) of respiratory parenchyma and airspace was determined by using a point counting method (14). Measurements were performed on sections taken at intervals throughout the left, right upper, or right lower lobes. Slides were viewed by using a 20× objective, and the images (fields) were transferred by video camera to a computer screen using metamorph imaging software (Universal Imaging, West Chester, PA). A computer-generated, 121-point lattice grid was superimposed on each field, and the number of intersections (points) falling over respiratory parenchyma (alveoli and alveolar ducts) or airspace was counted. Points falling over bronchioles, large vessels, and smaller arterioles and venules were excluded from the study. Fractional areas (% Fx area) were calculated by dividing the number of points for each compartment (n) by the total number of points contained within the field (N), and then multiplying by 100: % Fx area = n/N × 100.
Ten fields per section were analyzed to gather the data. The x and y coordinates for each field measured were selected by using a random number generator.
Pressure-Volume Curves.
Twelve-week-old mice were injected with sodium pentobarbital and placed in a chamber containing 100% oxygen to ensure complete collapse of alveoli by oxygen absorption. Mice were killed by exsanguination, and the trachea was cannulated and connected to a syringe linked to a pressure sensor via a three-way connector (Mouse Pulmonary Testing System, TSS, Cincinnati, OH). After opening the diaphragm, lungs were inflated in 75-μl increments every 10 sec to a maximum pressure of 28 cm of water and then deflated. Pressure-volume curves were generated for each animal, determining lung volumes (ml/kg) at 10, 5, and 0 cm of water during the deflation curve (n = 5–6 mice per genotype).
Cytokine Measurements.
Cytokine content was measured in BALF or lung homogenates after lavage from 3- and 9-week-old mice. Seven to nine animals of each genotype were used for each determination. Tumor necrosis factor α, IL-1β, IL-6, and macrophage inflammatory protein 2 were quantitated by using murine sandwich ELISA kits (R & D Systems) according to the manufacturer's directions. Levels of cytokine detection ranged from 1.5 to 5.1 pg/ml in the cytokine assays. All plates were read on a microplate reader (Molecular Devices) and analyzed with the use of a computer-assisted analysis program (softmax, Molecular Devices). Only assays having standard curves with a calculated regression line value of >0.95 were accepted for analysis. Data were expressed as pg per g of body weight (BW).
Hydrogen Peroxide Production.
Alveolar macrophages were collected from 8- to 10-week-old mice by bronchoalveolar lavage with 1 ml of dye-free RPMI media (GIBCO) times three. BALF from 8–10 mice was pooled to provide sufficient numbers of macrophages for each determination. The assay was repeated four times; 36–40 animals per genotype were used for these measurements. The lavage was centrifuged at 1,200 rpm for 10 min, and 1 × 106 macrophages were resuspended in PBS. Hydrogen peroxide production by macrophages was measured by using a commercially available assay (Bioxytech H2O2-560 assay, OXIS International, Portland, OR), based on the oxidation of ferrous ions (Fe2+) to ferric ions (Fe3+) by hydrogen peroxide under acidic conditions. Hydrogen peroxide production was determined after activation with 100 ng/ml of phorbol myristate acetate (PMA) or without stimulation.
MMP Activity and Immunohistochemistry.
MMP activity was measured in macrophage-conditioned media and BALF isolated from 8- to 10-week-old SP-D (+/+) and SP-D (−/−) mice. For analysis in BALF, samples from four mice of each genotype were centrifuged (100,000 × g, 1 h) in a SW-28 rotor (Beckman) and concentrated by using Centricon-30 filtration units (Amicon). The samples (200 μg protein) then were electrophoresed under nonreducing conditions (Laemmli) into 10% Zymogram gelatin gels (NOVEX, San Diego). After electrophoresis, gels were washed twice with 2.5% Triton X-100 (37°C, 15 min) and incubated for 16 h with 40 mM Tris⋅HCl (pH 7.5), 10 mM CaCl2, and 1 μM ZnCl2 (15). Gels were stained with 0.5% (wt/vol) Coomassie blue in 50% methanol and 10% acetic acid for 1 h, then destained. MMPs were detected as clear bands against a blue background. MMP2 and MMP9 mRNA was quantitated by Northern blot analyses of total lung mRNA from wild-type and SP-D (−/−) mice by using 32P-labeled cDNA probes (Chemicon).
MMP activity was measured in macrophage-conditioned media by using alveolar macrophages isolated from eight SP-D (+/+) or four SP-D (−/−) mice. The macrophages were pooled and placed in culture at a concentration of 1 × 105 cells per well for 24 h in serum-free RPMI medium. Proteinases in the conditioned media (100 μl per sample) were concentrated by incubation with gelatin-agarose beads (Amersham Pharmacia) for 2 h at 4°C (16). The beads then were pelleted and washed, and the proteinases were eluted by incubation in sample buffer for 45 min at 37°C. The samples then were assayed by zymography as described above for BALF.
Immunohistochemistry for MMP9 was performed at dilutions of 1:500 and 1:1,000 by using a rabbit polyclonal antibody generated to murine MMP9 and provided by Robert Senior (Washington University, St. Louis, MO). Immunohistochemistry for MMP12 was performed at dilutions of 1:250 and 1:500 by using a rabbit polyclonal antibody generated to murine MMP12 (macrophage metalloelastase, aka MME), and provided by Steve Shapiro (Washington University). The antigen/antibody complexes were detected by using an avidin-biotin-peroxidase technique (Vectastain Elite ABC kit, Vector Laboratories). Pseudomonas-infected, wild-type mouse lung infiltrated with activated macrophages and neutrophils was used as a positive control for both antibodies. Elimination of the primary antibody from the reaction served as a negative control for nonspecific binding of the other kit components.
Statistical Analysis.
Statistically significant differences were determined by using either (i) ANOVA for fractional areas followed by the Student-Newman-Keuls procedure, (ii) the Student's t test for comparison of BWs, lung and heart volumes, volume/BW ratios, pressure-volume curves, total protein, DNA, and cytokine content, or (iii) the median nonparametric test for oxidant production. Differences of P < 0.05 were considered significant. Values are given as mean ± SE.
Results
BWs of SP-D (−/−) mice were slightly smaller before weaning, but were not significantly different from SP-D (+/+) mice after 3 weeks of age (Table 1). Although lung volumes were not significantly different, lung volume-to-BW ratios were increased in SP-D (−/−) mice at 3 and 6 weeks of age (Table 1). No significant differences were observed in heart volumes or heart volume-to-BW ratios (data not shown). At maturity (5 months), no changes in wet lung weight, total lung DNA, or protein content were noted (data not shown).
Table 1.
Comparison of BWs, lung volumes, and volume-to-BW ratios, mean ± SE
| Age | BW, g
|
Lung volumes, ml
|
LV∶BW, ml/g × 10−2
|
|||
|---|---|---|---|---|---|---|
| SP-D(−/−) | SP-D(+/+) | SP-D(−/−) | SP-D(+/+) | SP-D(−/−) | SP-D(+/+) | |
| 2 days | 1.8 ± 0.1* | 3.4 ± 0.1 | ND | ND | ND | ND |
| 5 days | 3.7 ± 0.3 | 4.6 ± 0.2 | ND | ND | ND | ND |
| 7 days | 3.9 ± 0.2* | 5.3 ± 0.2 | ND | ND | ND | ND |
| 14 days | 6.6 ± 0.2* | 7.7 ± 0.2 | ND | ND | ND | ND |
| 17 days | 10.9 ± 0.5 | 10.6 ± 0.7 | 0.36 ± 0.02 | 0.36 ± 0.03 | 3.25 ± 0.05 | 3.36 ± 0.03 |
| 3 wk | 10.9 ± 0.5* | 14.1 ± 1.2 | 0.36 ± 0.01 | 0.37 ± 0.03 | 3.43 ± 0.21† | 2.50 ± 0.18 |
| 6 wk | 23.2 ± 0.6 | 24.7 ± 0.5 | 0.63 ± 0.03 | 0.58 ± 0.02 | 2.71 ± 0.13† | 2.25 ± 0.01 |
| 9 wk | 25.2 ± 1.2 | 27.8 ± 1.3 | 0.55 ± 0.03 | 0.61 ± 0.02 | 2.10 ± 0.16 | 2.20 ± 0.09 |
| 28 wk | 36.9 ± 4.3 | 31.2 ± 1.6 | 0.67 ± 0.09 | 0.58 ± 0.06 | 2.03 ± 0.51 | 1.86 ± 0.10 |
LV∶BW, lung volume-to-BW ratio; ND, not determined.
*Significant statistical differences were observed in BWs, at 2 days, P = 0.0001; 7 days, P = 0.0002; 2 wk, P = 0.007; and 3 wk, P = 0.04.
† Significant statistical differences in LV∶BW ratios were observed at 3 wk (P = 0.02), due to differences in BW, and at 6 wk (P = 0.03), although BWs and lung volumes were not statistically different at the latter time point. n = 3–7 animals per group.
Morphology and Histochemical Analysis.
Histologic assessment of lungs from SP-D (−/−) mice at the light microscopic level demonstrated no detectable abnormalities in lung morphology within the first 2 weeks of life, although increased numbers of normal-appearing alveolar macrophages were noted in the alveoli of SP-D (−/−) mice at 2 weeks of age (Fig. 1). Abnormalities in lung histology were readily observed in SP-D (−/−) mice at 3, 6, and 9 weeks of age consisting of enlarged airspaces and infiltration with atypical, foamy, alveolar macrophages (Fig. 1). Enlarged airspaces associated with the accumulation of hypertrophic, foamy, alveolar macrophages and perivascular/peribronchiolar monocytic infiltrates were observed by 6–7 months of age, although the extent of airspace enlargement in individual SP-D (−/−) mice varied from moderate to severe in this age group (Fig. 2).
Figure 1.
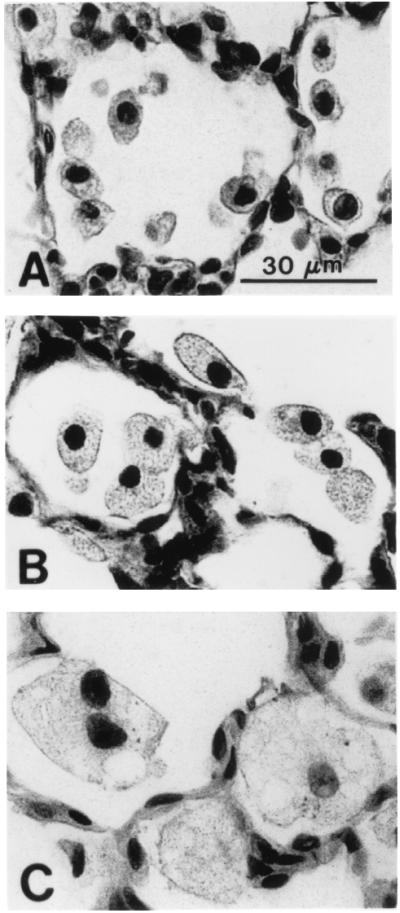
Progressive enlargement of alveolar macrophages from SP-D (−/−) mice. (A) Increased numbers of normal-appearing macrophages were observed by 2 weeks of age in SP-D (−/−) mice. (B) Enlarged, foamy, alveolar macrophages were readily observed by 3 weeks of age in SP-D (−/−) mice. (C) By 6 weeks of age, multiple foci of large, foamy, alveolar macrophages were observed throughout the lungs of SP-D (−/−) mice. (Scale bar = 30 μm.)
Figure 2.

Lung tissue from SP-D (+/+) (A) and SP-D (−/−) (B–D) mice killed at 7 months of age. Moderate (B) to severe (C) emphysematous changes were observed in SP-D (−/−) mice compared with age-matched SP-D (+/+) controls (A). Focal areas of macrophage accumulation and perivascular/peribronchiolar monocytic infiltrates also were observed in the lungs of 7-month-old SP-D (−/−) mice (D). (Scale bar = 100 μm.)
In 7-month-old SP-D (−/−) mice, subpleural fibrotic lesions that stained intensely for collagen were observed (Fig. 3). Abnormalities in elastin deposition also were observed in the parenchyma of lungs from SP-D (−/−) mice at this time (Fig. 3). These abnormalities consisted of regions of lung parenchyma with short, thick, and more highly coiled elastic fibers, as well as regions of inflammation where elastin staining was decreased in adjacent alveolar septa.
Figure 3.

Collagen (A–C) and elastin (D–F) staining of lung tissue from 7-month-old SP-D (−/−) mice. Increased collagen deposition was observed at the pleural surface (A), in focal areas of macrophage accumulation associated with localized fibrosis (B), and in alveolar septa (C) of SP-D (−/−) mice. Elastin staining was increased in alveolar septa of SP-D (−/−) mice (E) compared with age-matched SP-D (+/+) controls (D), the elastic fibers appearing shorter, thicker, and more highly coiled in the SP-D (−/−) mice. Elastin staining was decreased, however, in alveolar septa (arrows) adjacent to foci of macrophage accumulation and fibrosis (F). [Scale bars = 50 μm (A, B, D, and E) and 20 μm (C and F).]
Morphometric Analysis and Lung Volumes.
Although no differences in the relative proportion (% fractional area) of airspace and respiratory parenchyma were observed at 5 days (0.5 weeks), 14 days (2 weeks), or 17 days (2.5 weeks) of age, the % fractional area of airspace was increased significantly (P = 0.013) in SP-D (−/−) mice by 3 weeks of age (Fig. 4). Likewise, at this time, the % fractional area of respiratory parenchyma was decreased in SP-D (−/−) mice compared with age-matched SP-D (+/+) controls (34% parenchyma/66% airspace compared with 42.5% parenchyma/57.5% airspace, respectively) (Fig. 4). Relative proportions of airspace and respiratory parenchyma continued to diverge significantly from controls at later time points (Fig. 4). Increased lung volumes were readily apparent in SP-D (−/−) mice at 12 weeks of age, consistent with histologic and morphometric studies demonstrating emphysema (Fig. 5).
Figure 4.

Comparison of changes in fractional areas (% fx area) of airspace (a) and respiratory parenchyma (b) with age in SP-D (−/−) mice and age-matched SP-D (+/+) controls. No significant differences were observed in the fractional area of airspace and parenchyma found in the lungs of SP-D (−/−) mice when compared with wild-type controls at 5 days (0.5 wk), 14 days (2 wk), or 17 days (2.5 wk) of age. The fractional area devoted to both airspace and parenchyma diverged significantly between the two different genotypes at 3 weeks (*, P = 0.013), 6 weeks (*, P = 0.0007), and 28 weeks (*, P = 0.004) of age. Data are expressed as % fractional area and represent the mean ± SE.
Figure 5.

Deflation limbs of pressure-volume curves from SP-D (+/+) and SP-D (−/−) mice. Pressure-volume curves were generated in 5–6 mice at 12 weeks of age. Lung volumes associated with the deflation limbs of pressure-volume curves were significantly greater for 12-week-old SP-D (−/−) mice compared with age-matched SP-D (+/+) mice at 10 cm H2O and at the maximum pressure of 28 cm H2O (*, P < 0.05). Data are expressed as ml/kg and represent the mean ± SE.
Cytokines, Hydrogen Peroxide Production, and Metalloproteinases.
At 3 and 9 weeks of age, neither lung homogenates nor BALF from SP-D (−/−) mice contained inflammatory levels of the acute phase proinflammatory cytokines, tumor necrosis factor α, IL-1β, IL-6 or macrophage inflammatory protein 2, although basal levels of IL-1β were increased approximately 2-fold in lung homogenates. The concentration of IL-1β in SP-D (−/−) mice at 3 weeks of age was 9.2 ± 1.9 vs. 4.1 ± 0.49 pg/g BW in SP-D (+/+) mice (P < 0.001), and 3.9 ± 0.6 vs 1.9 ± 0.19 pg/g BW, respectively, at 9 weeks of age (P < 0.05). In general, the concentrations of all four cytokines were at or below the level of sensitivity of the assay in BALF from both SP-D (+/+) and SP-D (−/−) mice.
In contrast, oxidant production, as assessed by measuring hydrogen peroxide production by alveolar macrophages isolated from SP-D (−/−) mice, was increased 10-fold (Fig. 6). Because oxidant production has been associated with activation of a number of MMPs and with emphysema in both human and animal studies, MMP activities were estimated by degradation of gelatin substrates after SDS/PAGE of BALF and conditioned medium from alveolar macrophages isolated from SP-D (−/−) and SP-D (+/+) mice. Gelatinase activity was markedly increased in conditioned medium from alveolar macrophages isolated from SP-D (−/−) mice and migrated at approximately 72 kDa and 105 kDa, consistent with increased murine MMP2 (gelatinase A) and MMP9 (gelatinase B) activity (17) (Fig. 7). Alveolar macrophages in adult SP-D (−/−) mice stained positive for MMP9 and MMP12 (Fig. 8). Increased MMP12 staining also was detected in thickened alveolar septa adjacent to focal sites of macrophage accumulation (Fig. 8). On the other hand, bands of activity consistent with MMP2 and MMP9 were detected with equal intensity in BALF from both genotypes (data not shown). Likewise, the abundance of MMP2 and MMP9 mRNA was similar in whole lung RNA samples from SP-D (−/−) and SP-D (+/+) mice as assessed by Northern blot analysis (data not shown).
Figure 6.

Increased hydrogen peroxide production in alveolar macrophages from SP-D (−/−) mice. Hydrogen peroxide production was assessed from 1 × 106 macrophages isolated from BALF as described in Experimental Procedures. Macrophages from SP-D (−/−) mice (solid bar) generated significantly more hydrogen peroxide than macrophages from SP-D (+/+) mice (hatched bar) with and without PMA stimulation. Data are expressed as μM of H2O2 and represent the mean ± SE with n = 4 determinations per group. A total of 8–10 mice was used for each determination, *, P < 0.05, compared with SP-D (+/+) mice.
Figure 7.

Production of MMP by alveolar macrophages in SP-D (−/−) mice. MMP activity was assessed in macrophage-conditioned media. Pooled alveolar macrophages (1 × 105 cells/well) were incubated in 100 μl of serum-free media for 24 h. The supernatant was collected, concentrated, and analyzed by gelatin zymography. Production of 72-kDa (MMP2) and 105-kDa (MMP9) gelatinases was significantly increased in SP-D (−/−) mice, whereas there was no detectable enzyme activity in SP-D (+/+) mice. Alveolar macrophages were pooled from four SP-D (−/−) mice and eight SP-D (+/+) mice. The assay was performed in three different sets of animals. Data shown are for one assay and are presented as the reverse image of the original zymogram.
Figure 8.

Immunohistochemical detection of MMP12 and MMP9 in 6- to 7-month-old SP-D (−/−) mice. Immunostaining for both MMP12 (A and B) and MMP9 (C and D) was enhanced in the lungs of SP-D (−/−) mice (B and D) compared with age-matched SP-D (+/+) mice (A and C). MMP12 immunopositive material was detected in regions of parenchyma composed of thickened alveolar septa and in adjacent macrophages (C). MMP9 immunopositive material was detected in alveolar spaces containing large, foamy and/or disintegrating alveolar macrophages (D). (Scale bar = 50 μm.)
Discussion
Pulmonary emphysema, associated with inflammation and increased oxidant production and MMP activity in isolated alveolar macrophages, was observed in SP-D gene-targeted mice. The timing and progressive nature of these pulmonary abnormalities support the conclusion that alveolar remodeling in SP-D (−/−) mice is related to activation of alveolar macrophages and inflammation, rather than to developmental abnormalities in alveologenesis. The present findings are consistent with an important and unanticipated role of SP-D in the modulation of oxidant and MMP production by alveolar macrophages in vivo and suggest that changes in the regulation or function of SP-D may play a role in the pathologic processes leading to emphysema after chronic lung injury.
Histologic and morphometric analyses of lungs from SP-D (−/−) mice revealed no abnormalities in lung structure until 3 weeks of postnatal age, 1 week after alveologenesis is completed in the mouse. This finding was consistent with the observation that the relative proportions of respiratory parenchyma and airspace were similar in both SP-D (−/−) and SP-D (+/+) mice between postnatal days 5 and 17. After 2 weeks of age, increased parenchymal-to-airspace ratios were observed in SP-D (−/−) mice, consistent with ongoing remodeling of the parenchyma and alveolar spaces. Enlarged airspaces generally were associated with focal accumulation of large, foamy, alveolar macrophages, although there was some heterogeneity in both localization and extent of inflammatory infiltrates and remodeling in older mice. Although focal accumulation of alveolar macrophages in lungs of SP-D (−/−) mice was observed as early as 2 weeks of age, macrophage morphology remained normal at this time. Abnormal alveolar macrophage morphology, consisting of enlarged foamy cells, was noted by 3 weeks of age and was coincident with enlargement of alveolar structures thereafter. Previous studies demonstrated increased numbers of enlarged alveolar macrophages in SP-D (−/−) mice by 8 weeks of age (8, 9). Thus, the development of emphysema in SP-D (−/−) mice is consistent with the temporal and spatial accumulation of activated macrophages, suggesting their role in the remodeling process. These findings do not support a role for SP-D in normal lung morphogenesis and alveologenesis, a process generally completed by approximately 2 weeks of postnatal age in mice.
Our findings support an important role for SP-D in the modulation of alveolar macrophage activation, oxidant production, and MMP activity leading to emphysema and fibrosis. Macrophage infiltration and lung remodeling in SP-D (−/−) mice were not associated with neutrophil infiltration or with marked differences in inflammatory levels of the acute phase proinflammatory mediators, tumor necrosis factor α, macrophage inflammatory protein 2, IL-1β, and IL-6, but rather with increased hydrogen peroxide production and MMP activity in isolated alveolar macrophages. Although basal levels of IL-1β in lung homogenates were modestly increased in SP-D (−/−) mice, IL-1β was not increased to levels typically detected in severe inflammation (18). It is unlikely, therefore, that the pulmonary abnormalities observed in SP-D (−/−) mice were influenced or mediated by these relatively modest changes in IL-1β. Although SP-D has been proposed to play an important role in host defense, there was no histologic or serologic evidence of infection in the SP-D (−/−) colony.
Enhanced hydrogen peroxide production and increased numbers of alveolar macrophages found in the lungs of SP-D (−/−) mice support the concept that SP-D may play a critical antioxidant role in the lung, influencing hydrogen peroxide production by alveolar macrophages in vivo. Relationships between oxidant injury and the development of emphysema and pulmonary fibrosis are well established in numerous animal and genetic models (19). Because activation of MMPs also has been associated with oxidant injury (16) and emphysema (19), MMP activities were assessed in BALF and in conditioned media from isolated alveolar macrophages. Proteinase activity, consistent with the migration of MMP2 and MMP9, was markedly increased in conditioned media from SP-D (−/−) alveolar macrophages, whereas increased immunostaining for MMP9 and MMP12 was observed in alveolar macrophages in vivo. These data suggest that increased MMP production and/or activity may play a role in the development of emphysema and alveolar remodeling in SP-D (−/−) mice. On the other hand, changes in the expression of MMP2 and MMP9 mRNA in lung homogenates or in MMP2 and MMP9 concentrations in BALF were not detected. These latter results suggest that SP-D deficiency alters local concentrations of proteinases in focal regions of alveolar macrophage accumulation, which were not detectable in whole lung or BALF.
Although surfactant phospholipid content was increased in SP-D (−/−) mice and was associated with increased numbers of large, foamy, alveolar macrophages (8, 9), increased phospholipid content alone is not likely to be sufficient to cause the alveolar remodeling observed in SP-D (−/−) mice. In fact, the overall general effect of surfactant phospholipids appears to be anti-inflammatory, decreasing phagocytosis, oxidant production, and cytokine release, and inhibiting lymphocyte proliferation, Ig production, and expression of adhesion molecules (20). Increased oxidant production in the SP-D (−/−) mice, however, may generate oxidized lipid or protein intermediates that could influence lung inflammation and/or tissue degradation.
The modest reduction of lung SP-A concentrations found in SP-D (−/−) mice also is not likely to have contributed to the changes in lung morphology observed in these mice, because neither SP-A (+/−) nor SP-A (−/−) mice develop emphysema (21). Furthermore, in contrast to SP-D deficiency, SP-A deficiency was associated with decreased hydrogen peroxide production by isolated alveolar macrophages (22).
Although concentrations of SP-D in the lung change during development, increasing with advancing age (4), SP-D levels also are influenced by various clinical conditions (1, 4). Recent studies demonstrated marked reduction of SP-D concentrations in BALF obtained from patients with cystic fibrosis (CF) (23), supporting a potential role for SP-D in the pathogenesis of the chronic inflammation associated with CF lung disease. SP-D levels also were reduced in BALF of smokers, suggesting that decreased levels of SP-D may contribute to later development of chronic obstructive pulmonary disease and emphysema in these patients (24). Although concentrations of SP-D in BALF were increased in patients with pulmonary alveolar proteinosis (PAP), patients with idiopathic pulmonary fibrosis (IPF) and interstitial pneumonia associated with collagen vascular disease (IPCD) had decreased BALF levels of SP-D (25). On the other hand, serum concentrations of SP-D were increased in patients with PAP, IPF, and IPCD, although serum levels of both SP-A and SP-D varied with the severity of IPF and during the course of anti-inflammatory therapies (25). These clinical findings, as well as the present study, demonstrate that SP-D is required for maintenance of normal lung architecture and support the concept that changes in SP-D concentrations may be involved in the pathogenesis of lung injury associated with various clinical conditions. In our previous studies, no abnormalities in alveolar macrophages or lung morphology were observed in the heterozygous SP-D (+/−) mice (8), demonstrating that a 50% reduction in SP-D concentration in BALF is not sufficient to cause these pulmonary abnormalities.
SP-D (−/−) mice developed progressive pulmonary emphysema and subpleural fibrosis in association with chronic inflammation. Alveolar remodeling and macrophage abnormalities were apparent as early as 3 weeks of age, whereas mild, focal, pulmonary fibrosis was observed at 6–7 months of age, demonstrating a role for SP-D in the regulation of inflammation and alveolar remodeling. The present study also demonstrated an unexpected role for SP-D in the modulation of oxidant and MMP production by alveolar macrophages, both of which may contribute to inflammation and emphysema in the lungs of SP-D (−/−) mice. Whether SP-D deficiency contributes to ongoing inflammation or to the development of emphysema and fibrosis found in various human chronic lung diseases, including those caused by smoking and other oxidants, remains to be determined.
Acknowledgments
We thank Paula Blair, Sherri Profitt, and Jodi Gwozdz for technical assistance; Ann Maher for secretarial assistance; and Jaymi Semona for assistance with mouse husbandry. This work was supported by National Institutes of Health Grants HL56387 (J.A.W., S.E.W., and M.I.), HL41320 (J.H.F.), HL58795 (T.R.K.), and HL63329–01 (M.I.), and the Cystic Fibrosis Foundation.
Abbreviations
- SP-D
surfactant protein D
- SP-A
surfactant protein A
- MMP
matrix metalloproteinase
- BALF
bronchoalveolar lavage fluid
- BW
body weight
- PMA
phorbol myristate acetate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.100448997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.100448997
References
- 1.Crouch E C. Biochim Biophys Acta. 1998;1408:278–289. doi: 10.1016/s0925-4439(98)00073-8. [DOI] [PubMed] [Google Scholar]
- 2.Reid K B M. Biochem Biophys Acta. 1998;1408:290–295. doi: 10.1016/s0925-4439(98)00074-x. [DOI] [PubMed] [Google Scholar]
- 3.Mason R J, Greene K, Voelker D R. Am J Physiol. 1998;275:L1–L13. doi: 10.1152/ajplung.1998.275.1.L1. [DOI] [PubMed] [Google Scholar]
- 4.Crouch E, Rust K, Marienchek W, Parghi D, Chang D, Persson A. Am J Respir Cell Mol Biol. 1991;5:13–18. doi: 10.1165/ajrcmb/5.1.13. [DOI] [PubMed] [Google Scholar]
- 5.Fisher J G, Mason R. Am J Respir Cell Mol Biol. 1995;12:13–18. doi: 10.1165/ajrcmb.12.1.7811466. [DOI] [PubMed] [Google Scholar]
- 6.Chailley-Heu B, Rubio S, Rougier J P, Ducroc R, Barlier-Mur A M, Ronco P, Bourbon J R. Biochem J. 1997;328:251–256. doi: 10.1042/bj3280251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motwani M, White R A, Guo N, Dowler L L, Tauber A I, Sastry K N. J Immunol. 1995;155:5671–5677. [PubMed] [Google Scholar]
- 8.Korfhagen T R, Sheftelyevich V, Burhans M S, Bruno M D, Ross G F, Wert S E, Stahlman M T, Jobe A H, Ikegami M, Whitsett J A, et al. J Biol Chem. 1998;273:28438–28443. doi: 10.1074/jbc.273.43.28438. [DOI] [PubMed] [Google Scholar]
- 9.Botas C, Poulain F, Akiyama J, Brown C, Allen L, Goerke J, Clements J, Carlson E, Gillespie A M, Epstein C, et al. Proc Natl Acad Sci USA. 1998;95:11869–11874. doi: 10.1073/pnas.95.20.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 11.Hill B T, Whatley S. FEBS Lett. 1975;56:20–23. doi: 10.1016/0014-5793(75)80102-5. [DOI] [PubMed] [Google Scholar]
- 12.Gehr P, Gliser M, Stone K C, Crapo J D. In: Toxicology of the Lung. Gardner D E, Crapo J D, McClellan R D, editors. New York: Raven; 1993. pp. 111–154. [Google Scholar]
- 13.Sheehan D C, Hrapchak B B. Theory and Practice of Histotechnology. Columbus, OH: Battelle; 1987. [Google Scholar]
- 14.Bolender R P, Hyde D M, deHoff R T. Am J Physiol. 1993;265:L521–L548. doi: 10.1152/ajplung.1993.265.6.L521. [DOI] [PubMed] [Google Scholar]
- 15.Woessner J F. Methods Enzymol. 1995;248:510–528. doi: 10.1016/0076-6879(95)48033-1. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan S, Meng X P, Ramasamy S, Harrison D G, Galis Z S. J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka H, Hojo K, Yoshida H, Yoshioka T, Sugita K. Biochem Biophys Res Commun. 1993;190:732–740. doi: 10.1006/bbrc.1993.1110. [DOI] [PubMed] [Google Scholar]
- 18.LeVine A M, Kurak K E, Bruno M D, Stark J M, Whitsett J A, Korfhagen T R. Am J Respir Cell Mol Biol. 1998;19:700–708. doi: 10.1165/ajrcmb.19.4.3254. [DOI] [PubMed] [Google Scholar]
- 19.Senior R M, Shapiro S D. In: Fishman's Pulmonary Diseases and Disorders. Fishman A, editor. New York: McGraw–Hill; 1998. pp. 659–682. [Google Scholar]
- 20.Wright J R. Physiol Rev. 1997;77:931–962. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 21.Korfhagen T R, Bruno M D, Ross G F, Huelsman K M, Ikegami M, Jobe A H, Wert S E, Stripp B R, Morris R E, Glasser S W, et al. Proc Natl Acad Sci USA. 1996;93:9594–9599. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeVine A M, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T. J Clin Invest. 1999;103:1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Postle A D, Mander A, Reid K B, Wang J Y, Wright S M, Moustaki M, Warner J O. Am J Respir Cell Mol Biol. 1999;20:90–98. doi: 10.1165/ajrcmb.20.1.3253. [DOI] [PubMed] [Google Scholar]
- 24.Honda Y, Takahashi H, Kuroki Y, Akino T, Abe S. Chest. 1996;109:1006–1009. doi: 10.1378/chest.109.4.1006. [DOI] [PubMed] [Google Scholar]
- 25.Kuroki Y, Takahashi H, Chiba H, Akino T. Biochim Biophys Acta. 1998;1408:334–345. doi: 10.1016/s0925-4439(98)00079-9. [DOI] [PubMed] [Google Scholar]