

Abstract
Molecular dynamics simulations coupled with functional analyses of the major yeast phosphatidylinositol/phosphatidylcholine transfer protein Sec14p identify structural elements involved in regulating the ability of Sec14p to execute phospholipid exchange. The molecular dynamics simulations suggest large rigid body motions within the Sec14p molecule accompany closing and opening of an A10/T4/A11 helical gate, and that “state-of-closure” of this helical gate determines access to the Sec14p phospholipid binding cavity. The data also project that conformational dynamics of the helical gate are controlled by a hinge unit (residues F212, Y213, K239, I240, and I242) that links to the N- and C-terminal ends of the helical gate, and by a novel gating module (composed of the B1LB2 and A12LT5 substructures) through which conformational information is transduced to the hinge. The 114TDKDGR119 motif of B1LB2 plays an important role in that transduction process. These simulations offer new mechanistic possibilities for an important half-reaction of the Sec14p phospholipid exchange cycle that occurs on membrane surfaces after Sec14p has ejected bound ligand, and is reloading with another phospholipid molecule. These conformational transitions further suggest structural rationales for known disease missense mutations that functionally compromise mammalian members of the Sec14-protein superfamily.
INTRODUCTION
Phosphatidylinositol transfer proteins (PITPs) play important biological roles in defining the identities of individual lipid signaling pools that regulate specific biological processes. It is in this capacity that PITPs are suggested to represent components of lipid metabolic nanoreactors that promote specific signaling reactions (Ile et al., 2006). How PITPs couple exchange of phosphatidylinositol (PtdIns) or phosphatidylcholine (PtdCho) monomers to physiological function remains unresolved (Phillips et al., 2006). PITPs fall into two families (i.e., the Sec14 family and the metazoan PITP family) on the basis of primary sequence homology and structural fold. Members of one family share no primary homology or structural similarity with members of the other family (Bankaitis et al., 1989; Dickeson et al., 1989; Sha et al., 1998; Yoder et al., 2001).
The SEC14 gene product (Sec14p) is the major PITP of budding yeast, and it is required for efficient transport of secretory cargo from the yeast trans-Golgi network (TGN; Bankaitis et al., 1989, 1990; Cleves et al., 1991a). Genetic and biochemical evidence demonstrates Sec14p coordinates lipid metabolism with activities of proteins required for biogenesis of transport vesicles on yeast TGN membranes (Cleves et al., 1991b; McGee et al., 1994; Xie et al., 1998; Li et al., 2002; Yanagisawa et al., 2002; Phillips et al., 2006). Sec14p is of additional interest in that it is the prototype for a eukaryotic protein superfamily of Sec14-like proteins consisting of >500 known members distributed across the Eukaryota (Phillips et al., 2006). Sec14-like proteins regulate membrane trafficking, membrane biogenetic pathways, or both, that include polarized membrane trafficking in yeast (Carmen-Lopez et al., 1994; Nakase et al., 2001; Rudge et al., 2004) and in higher plants (Vincent et al., 2005). Other Sec14-like proteins are implicated in control of specific stress responses (Kearns et al., 1998; Monks et al., 2001). Finally, loss-of-function mutations in members of the Sec14 superfamily are associated with inherited human disorders (Aravind and Koonin, 1999; Bomar et al., 2003; Meier et al., 2003; Panagabko et al., 2003; Stocker and Baumann, 2003).
Although not formally proven, phospholipid binding and exchange are presumed to be of relevance to Sec14p function in vivo (Ile et al., 2006; Phillips et al., 2006). This reaction is of biochemical interest in that Sec14p ejects bound phospholipid upon encountering a membrane surface and reloads with another phospholipid molecule before disengaging from that surface. Studies in purified systems using defined liposomes and recombinant Sec14p demonstrate the exchange reaction involves efficient abstraction of phospholipid from a stable membrane bilayer and that Sec14p executes this interfacial reaction in the absence of cofactors or ATP (Phillips et al., 1999). Thus, Sec14p couples conformational change to a cycle of phospholipid exchange. The crystal structure of detergent-bound Sec14p suggests how this may occur. The Sec14p hydrophobic cavity is bounded by a hybrid α-/310-helix proposed to function as a gate (Figure 1). The conformational status of this gate is proposed to determine whether Sec14p is in an “open” or a “closed” conformation (Sha et al., 1998; Phillips et al., 1999).
Figure 1.

Schematic representation of the Sec14p crystal structure. (A) Ribbon and α-carbon backbone renditions of the Sec14p monomer. Structural elements include α-helices (A1–A12), 310-helices (T1–T8), and β-strands (B1–B6). Relevant substructures include helix tripod motif (copper), the A7T3 hybrid helix (gray), α-helix A9 (purple), the A10T4 A11 hybrid helix, A12, T5–T8 (green), the β1–β6 β-strands (gold), and loops (black). The opening of the Sec14p hydrophobic pocket is oriented toward the reader. (B) Sec14p rendition as described in A except viewed from the posterior. Residue G266 is represented by a blue sphere.
Herein, we report the results obtained from unrestrained molecular dynamics (MD) simulations and biochemical analyses of Sec14p conformational dynamics. These analyses simulate oscillations between open and closed Sec14p conformers, and they implicate three Sec14p structural elements in control of this conformational dynamics pathway. Finally, the data suggest the Sec14p conformational circuitry described here is broadly conserved across the Sec14 protein superfamily and that defects in this circuitry may represent the molecular basis for some disease-causing mutations in mammalian Sec14-like proteins.
MATERIALS AND METHODS
Preparation of Starting Structures
The 2.5-Å resolution coordinates of the open Sec14p conformation (PDB 1AUA; Sha et al., 1998) was obtained from the Protein Data Bank (Berman et al., 2000). The two bound β-octylglucoside molecules were removed, and crystallographic waters were retained. Hydrogen atoms to both amino acid residues and water molecules were added using the LEaP module of AMBER 8.0 (Case et al., 2004). LEaP was also used to place eight sodium ions at positions of minimum electrostatic potential to neutralize the protein system. The system was solvated in 10,414 TIP3P water molecules (Jorgensen et al., 1983) with a buffer distance of 12.5 Å around the solute. In this manner, an octahedral box with initial dimensions of 81.5 × 81.5 × 81.5 Å, and containing a total of 36,156 atoms, was generated as input to the MD simulations. For purpose of simplicity, we loosely refer to this starting Sec14p structure as apo-Sec14p to reflect it is not a phospholipid-bound conformer.
Glycine 266 was replaced with an aspartate while selecting the lowest energy rotamer by using the Biopolymer module of Insight II (www.accelrys.com) to generate a starting structure for simulations of Sec14pG266D. This protein system was neutralized with nine sodium ions and solvated in 10,420 TIP3P molecules in a manner similar to the wild-type structure. The final Sec14pG266D system contained a total of 36,180 atoms in an octahedral box with initial dimensions similar to the Sec14p system.
Molecular Dynamics Simulation
MD simulations were carried out using the SANDER module of AMBER version 8.0. (Case et al., 2004) with the ff03 forcefield of Duan et al. (2003) as defined in parm99.dat and frcmod.ff03 parameter files (Cornell et al., 1995). Long-range electrostatic interactions were estimated using the particle mesh Ewald method (Toukmaji et al., 2000), whereas bonds involving protons were constrained using the SHAKE algorithm (tolerance = 00001 Å). A 2-fs time step was used throughout the simulation. A 10-Å cutoff was applied to Lennard–Jones interactions, and the nonbonded list was updated every 25 steps. Periodic boundary conditions were applied. All MD production runs were carried out at constant temperature and pressure.
Protein systems were minimized for 5000 steps. The first 2000 steps were of steepest descent followed by 3000 steps of conjugate gradient minimization. The minimization was followed by 20-ps constant volume dynamics with heating from 50 K to 300 K. Pressure and temperature were then held constant for 100 ps for the system to reach a density of 1 g/cm3. This was followed by a 40-ps regime at constant volume with reheating from 200 to 300 K, yielding a total equilibration time of 160 ps. The production run was continued for more than 32 ns, and snapshots of the coordinates were written out every 1 ps.
Molecular Dynamics Data Analyses
The PTRAJ module of AMBER 8.0 was used for trajectory processing and analyses of root mean square deviation (rmsd) values, interatomic distances, hydrogen bonding, and atomic positional fluctuations. The φ and Ψ dihedral angles measuring residue backbone conformations were also reported using PTRAJ. Unless otherwise stated, data analyses are reported for the 15- to 32-ns time frames of production simulations so that analyses could be focused on a time window where large conformational changes were evident in the Sec14p molecular dynamics simulation.
Correlation coefficients (Cohen, 1988; Blaikie, 2003) were calculated between local backbone angular conformations and Sec14p “state-of-closure” according to the following equation:
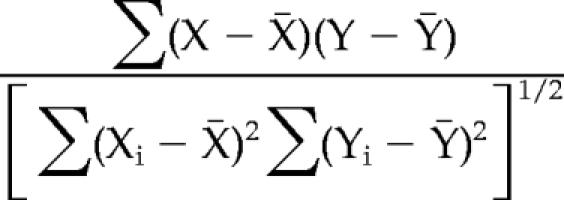 |
where xi represents the local backbone angular conformation (φi or Ψi) of a given residue at time i. The variable yi represents state-of-closure at time i as measured by distance between the Cα atoms of residues K195 and F231. The summations are over all snapshots included in the analysis. Snapshots between 15 and 32 ns were used for the Sec14p analysis, because conformational changes of a gate-like movement (including both opening and closing) occurred during this entire period. For Sec14pG266D, only snapshots between 2.5 and 4 ns were included in the analysis, because no “gating-like” motion occurred after this period. In addition, the 2.5- to 4.0-ns period included a closing motion without a subsequent opening motion. Correlation coefficients were calculated, and they are reported for every backbone dihedral angle for all residues.
Yeast Strains and Methods
Standard media, genetic techniques, and plasmid shuffle assays were described previously (Ito et al., 1983; Rothstein, 1983; Sherman et al., 1983; Cleves et al., 1991b; Phillips et al., 1999; Li et al., 2002). Strains included CTY182 [MATa ura3-52, Δhis3-200, lys2-801, SEC14], CTY1079 [MATa ura3-52, Δhis3-200, lys2-801, sec14-1ts spo14Δ::HIS3], CTY558 [MATα ade2 ade3 leu2 Δhis3 ura3-52 sec14Δ1::HIS3 YCp(SEC14, URA3)], and CTY303 [MATa ura3-52, Δhis3-200, cki1, sec14ΔP::hisG]. Experiments were performed at 30 and 37°C. All transformants were selected based on acquisition of uracil prototrophy.
Protein Stability
Appropriate derivatives of yeast strain CTY303 carrying SEC14 expression plasmids of interest were grown to mid-logarithmic phase at 25°C and shifted to 37°C for 1, 3, and 5 h. Cell densities were normalized to an OD600 = 1.5, protein extracts were prepared by cell disruption with glass beads, and extracts were subsequently analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting.
Site-directed Mutagenesis
The various mutagenic primers and their corresponding reverse complementary sequences are available from us by request. Mutagenesis of SEC14 sequences was performed on a 2.6-kb EcoR1-Sph1 SEC14 fragment resident in either plasmid pRE246 (Alb et al., 1995) or the pQE31-based HIS6SEC14 plasmid pRE526 (Skinner et al., 1995). Mutagenized SEC14 sequences were confirmed by DNA sequence analysis and correctly mutagenized gene cassettes were individually subcloned into the centromeric and episomal yeast–Escherichia coli shuttle vectors YCplac33 and YEplac195, respectively (Geitz and Sugino, 1988).
Sec14p Purification
Recombinant Sec14p and selected mutant derivatives were purified from E. coli as described previously (Phillips et al., 1999) with modification. Briefly, cultures of E. coli strain KK2186 were grown aerobically at room temperature to an OD600 of 0.5 in superbroth medium supplemented with 60 μg/ml ampicillin. Sec14p expression was induced by addition of isopropyl β-thiogalactoside to 1 mM final concentration, and cultures were incubated for an additional 5 h with aeration. Cells were harvested by centrifugation at 7000 × g (10 min), and resuspended in lysis buffer (50 mM sodium phosphate, pH 7.1, 300 mM sodium chloride, 1 mM NaN3, and 0.2 mM phenylmethanesulfonyl fluoride). Lysozyme was added, and the suspension was incubated at room temperature for 10 min with occasional agitation. Cells were disrupted using 0.1- to 0.3-mm glass beads and a bead beater (Biospec Products, Bartlesville, OK), and cell-free lysate was clarified by serial centrifugation at 7000 and 130,000 × g, respectively. The supernatant was applied to Talon Sepharose resin, and bound protein was eluted with a linear imidazole gradient (0–200 mM in lysis buffer). Peak fractions were identified by SDS-PAGE, pooled, and buffer was exchanged with lysis buffer by using Centripore 10 spin columns (Millipore, Billerica, MA).
PtdCho Transfer Assays
[14C]PtdCho transfer from liposomes to bovine heart mitochondria by using either recombinant protein or yeast cytosol was assayed as described previously (Phillips et al., 1999; Li et al., 2000). When yeast cytosol was used, Sec14p species were expressed in strain CTY303 from the episomal pDR195 vector where Sec14p expression was driven by the PMA1 promoter (Rentsch et al., 1995). This system drives significant overexpression of Sec14p species and increases signal to noise in transfer assays and compensates for trivial effects associated with mutant protein instability.
Limited Proteolysis
Lyophilized trypsin (650275; Calbiochem, San Diego, CA) was reconstituted in 50 mM Tris, pH 7.5, and 10 mM CaCl2 and stored as 0.1 μg/μl aliquots. Reactions were run in 50 mM Tris, pH 7.5, and 10 mM CaCl2 with a 100:1 M ratio of recombinant Sec14p (15 μg):trypsin in 50 μl and incubated for 5 min at 25°C. The products were evaluated by SDS-PAGE. Individual proteolytic products were identified by staining with Coomassie blue, excised from destained SDS-polyacrylamide gels, and analyzed by matrix-assisted laser desorption ionization/time of flight (MALDI-TOF) mass spectrometry at the University of North Carolina Proteomics Core Facility (Chapel Hill, NC).
RESULTS
Helix A10/T4 Fluctuates between Open and Closed States in Sec14p
The rmsd values comparing a series of structural snapshots, taken at 1-ps intervals of Sec14p during the course of a 32-ns production run, to the first apo-Sec14p snapshot are reported in Figure 2A. A structural transition is apparent for Sec14p at ∼9 ns, and additional rmsd fluctuations in excess of 1 Å continued throughout the simulation. The fluctuating rmsd values report an ensemble of Sec14p conformers where flexible domains are undergoing large motions. To identify such mobile Sec14p structural elements, the atomic positional fluctuation (apf) for all Sec14p Cα atoms were monitored during the 15- to 32-ns simulation time window. In all references to amino acid residues herein, we define the initiator Met as residue 1. Plots of the root mean square fluctuation (rmsf) data indicated the largest atomic positional motions of Sec14p occurred within helix A10/T4 around F231 (Figure 2B). Residue F231 is located at the C terminus of helix A10/T4 and exhibited the greatest rmsf value (5.4 Å). The rmsf values that averaged in excess of 2.0 Å included residues 223–238 that span helices A10/T4 and A11 (referred to as the A10/T4/A11 helical gate; see below). Similarly, the 271DESK274 cluster positioned immediately C terminal to helix T5 in the string motif also showed average rmsf values in excess of 2.0 Å. The significance of these string motif dynamics is discussed below.
Figure 2.

Mobile regions of the Sec14p molecule. (A) All atom RMSD plot for Sec14p (black) and Sec14pG266D (gray) simulations as a function of simulation time (nanoseconds). (B) Average Cα atomic position fluctuation plot for Sec14p (black) and Sec14pG266D (gray) simulations as a function of residue (plotted from N to C terminus). Two major areas of distinction between the Sec14p and Sec14pG266D plots are centered around residues K116 and F231. The position of G266 is identified with the arrow for reference.
Evidence to indicate helix A10/T4/A11 was closing during the simulation was obtained from monitoring of interatomic distances between positional references on helix A10/T4 and on helix A9, i.e., the structural element that lies opposite from helix A10/T4 across the opening to the Sec14p hydrophobic cavity. Distance-monitoring plots demonstrated that the Cα atoms of residues R195 (helix A9) and F231 (helix A10/T4) approached to within 10.9 Å of each other and then separated to distances in excess of 25 Å during the simulation (Figure 3, A and B). These motions proceeded in an oscillating manner characterized by periods of proximity followed by periods of distance. Periods of proximity were characterized by a stepwise progression with interatomic distances of 18.7, 14.0, and 10.9 Å measured at 3.6, 8.9, and 18.1 ns of the simulation, respectively (snapshots 3616, 8937, and 18130). The lifetime of each period of proximity increased progressively (0.3, 0.6, and 0.85 ns). The smallest R195-F231 interatomic Cα distance (10.9 Å) was recorded first at 18.1 ns of the simulation. It was recorded again during the 4-ns period of proximity attained after helix A10/T4 closed at 24 ns of the simulation, and this 10.9-Å distance was maintained throughout that entire 4-ns period of proximity. The R195-F231 interatomic Cα distances increased again at 28 ns.
Figure 3.

Conformational transitions in the helical gate and Sec14p status-of-closure. (A) Distance monitoring plot between the Cα atoms for residues R195 and F231 for Sec14p (black) and Sec14pG266D (gray) simulations as a function of time (nanoseconds). (B) Cα backbone rendition of apo-Sec14p highlighting the relative starting positions of residues R195 (gray sphere) and F231 (black sphere). (C) PtdCho transfer activity of Sec14p cysteine pair mutants. Recombinant His6-Sec14p, His6- Sec14pR195C,F231C, and His6-Sec14pY188C,F231C were purified from E. coli and assayed for PtdCho transfer activity under nonreducing (open bars) and reducing (solid bars) conditions. E. coli cytosol served as background. Activity is expressed as the percentage of total radiolabeled PtdCho transferred in 30 min at 37°C after subtraction of background. Data represent the averages of at least three independent experiments and a working range of 0.25–2.0 μg of recombinant protein. Total input of [14C]PtdCho averaged 37,320 cpm per assay. The average background was 1325 cpm per reaction. (D) Purified recombinant Sec14p was challenged with trypsin and 0.1% SDS as indicated below. Lane 1 represents mass standards and the various forms of Sec14p; the N-terminal Sec14p tryptic fragment (Sec14p'), and the C-terminal Sec14p tryptic fragment (‘Sec14p) are identified at right.
The apf plots suggest the motion was concentrated in the gate with helix A10/T4/A11 moving toward helix A9. As indicated above, the largest rmsf value (5.4 Å) for any Sec14p residue in the simulation was recorded for the gate residue F231. In contrast, the rmsf values for helix A9 residue R195 and for residues on helix A12 were only 0.8 and ∼1.0 Å, respectively. These data suggest helices A9 and A12 undergo only small positional fluctuations during the simulation, whereas helix A10/T4/A11 engaged in much larger motions relative to these two structural elements. Distance monitoring plots recorded for the Cα atoms of residues K229 and E250 as well as R195 and E250 are also consistent with this conclusion (Supplemental Figure S1, A and B). K229 marks the C terminus of the A10 element of helix A10/T4 and is an invariant residue among Sec14-domain proteins, whereas E250 resides in helix A12. The interatomic distances between Cα atoms of K229 and E250 fluctuated inversely as a function of time relative to fluctuations recorded between R195 and F231. In addition, distances on average of 29 Å are maintained between helix A9 residue R195 and helix A12 residue E250 throughout the simulation.
Together, unrestrained MD simulations of apo-Sec14p in explicit solvent reveal an oscillatory pattern for Sec14p between open and closed conformations. The data further suggest that conformational oscillations are associated with “gate-like” motions of the A10/T4/A11 helices. For purposes of simplicity, we refer to the Sec14p conformation characterized by R195-F231 interatomic Cα distance of 10.9 Å as closed, although it is likely this represents a “partially closed” conformation (see Discussion).
Biochemical Evidence for Gate Closure
We sought direct validation that 1) the trajectory of gate closure for Sec14p described by the MD simulation models a relevant conformational pathway and 2) that gate-mediated closure of the Sec14p hydrophobic pocket occurs by constriction of distances between the A10/T4/A11 helical gate and helix A9. To that end, opposing pairs of Cys residues were positioned on helices A9 and A10/T4, and each pair was evaluated for competence to form a disulfide bond. The double Cys mutant proteins were subsequently purified and assayed in parallel for PtdCho transfer activity under nonreducing and reducing conditions.
Sec14p contains four endogenous Cys residues, all of which are buried within the interior of the protein and are not solvent accessible (Sha et al., 1998). This is reflected by the robust PtdCho transfer activity of Sec14p in both nonreducing and reducing conditions (Figure 3C) and by our inability to modify Sec14p with N-ethylmaleimide without first denaturing the protein (our unpublished data). In the open apo-Sec14p conformation, residues R195 and F231 are separated by 23.82 Å (Sha et al., 1998). When recombinant Sec14pR195C,F231C was assayed under nonreducing conditions, a 72% decrease in specific PtdCho transfer activity was recorded for the mutant protein relative to Sec14p (Figure 3C). This diminution of activity was not the result of some trivial and undefined protein denaturation. Sec14pRF195,231CC PtdCho transfer activity was substantially resuscitated upon introduction of β-mercaptoethanol into the transfer assay. These results demonstrate Sec14p tolerates the R195C,F231C substitutions so long as Sec14pR195C,F231C is in a reducing environment.
We interpret these data to indicate that the R195C and F231C side chains efficiently form disulfide bonds in nonreducing environments and that the resultant “forced closure” of the Sec14p hydrophobic pocket is inconsistent with phospholipid transfer activity. That inhibition of Sec14pR195C,F231C activity under nonreducing conditions was quantitatively constant over a 40-fold range of protein concentration in the assay, strongly argues against formation of intermolecular disulfide bonds (our unpublished data). Such intermolecular reactions are expected to be exquisitely sensitive to protein concentration, whereas formation of intramolecular disulfide bonds should not be so sensitive. As significant distance constraints govern disulfide bond formation (i.e., participating sulfhydryl groups must be within ∼3 Å of each other; Careaga and Falke, 1992), these data indicate the side chains of helix A9 residue R195 and A10/T4 residue F231 must lie in proximity to each other in the closed Sec14p conformation. In support, a mutant Sec14pY188C,F231C (where the interatomic distance for the Cα atoms of these helix A9-helix A10/T4 Cys pairs is 16 Å in the closed Sec14p conformation defined by MD simulation; compared with 10.9 Å in the case of Sec14pR195C,F231C) exhibited wild-type PtdCho transfer activities in both oxidative and reducing environments (Figure 3C).
Limited proteolysis was used as an independent probe for changes in the proximity of helices A9 and A10/T4/A11 as Sec14p transitions from closed to open conformations. Opening of the A10/T4/A11 helical gate evokes large conformational changes, and we sought to detect proteolytic events unique to open Sec14p conformations. These experiments were aided by the fact that nondenatured Sec14p adopts a compact fold and is highly resistant to proteolytic challenge with trypsin (Figure 3D) and a variety of other proteases (our unpublished data). In the presence of SDS micelles, however, trypsin rapidly and quantitatively cleaves Sec14p to generate two significant degradation products (Sec14p' and 'Sec14p; Figure 3D). In-gel trypsinolysis of each species coupled with MALDI-TOF mass spectrometry unambiguously identified the more rapidly migrating 'Sec14p species as being derived from the C-terminal 109 residues of Sec14p. The N terminus of 'Sec14; is defined by the 196EASYISQNYYP ER208 tryptic peptide, and all subsequent major tryptic fragments C-terminal to this peptide were identified. Only the short 209MGK211, 250ELLK253 and 264FGGK267 peptides were not detected in these analyses (our unpublished data).
The more slowly migrating Sec14p' represents the N-terminal Sec14p fragment and the most C-terminal tryptic peptide identified from this fragment was 146NLVWEYESVVQYR LPACSR164. In the intervening 31 residues between Sec14p' and 'Sec14p, the only possible site(s) of trypsinolysis are limited to the amide linkages between helix A9 residues K180/G181 and/or R195/E196, and A8 residues R164/A165. Although we have not yet been able to distinguish several viable possibilities for pattern(s) of cleavage (i.e., whether only one or several of these positions define the precise site(s) of cleavage; we know similar trypsin cleavage patterns are obtained for Sec14p and Sec14pR195C), we note all three candidate positions surround the opening of the hydrophobic pocket in the open Sec14p conformer. We interpret these data to suggest SDS micelles induce transition of Sec14p from the closed to the open conformation with the result that the K180, R195, and/or R164 amide bonds are rendered exquisitely susceptible to trypsin cleavage. Together, the Cys-scanning and trypsinolysis data independently indicate that transition from the open to the closed Sec14p conformation involves closure of the A10/T4/A11 helical gate with respect to helix A9.
A Hinge for Rigid Body Motions of the A10/T4/A11 Helical Gate
Distance monitoring between the Cα atoms of the highly conserved A10/T4 residue F221 and the B4 residue I214 suggest restricted motion of the N-terminal region of the helix A10/T4 with respect to the β-strands. Similarly, interatomic Cα distance monitoring between F221 and helix A9 residue A187 suggest that relatively constant distances averaging ca. 14.9 Å are maintained between these A9 and A10/T4 residues in Sec14p throughout the simulation (Supplemental Figure S2, A and B). A superimposition of average structures from different time frames of the MD simulation is depicted in Supplemental Figure S3. This superimposition illustrates the displacement of helix A10/T4 in the transition of Sec14p from an open conformation to a closed conformation during the simulation and that the N-terminal aspect of A10/T4 remains relatively fixed during this transition.
To identify candidate structural elements that might be involved in supporting the transitions between open and closed Sec14p conformations, a set of correlation coefficients between Sec14p state-of-closure parameters and backbone (ϕ, Ψ) angle variations were calculated for each residue along the polypeptide chain (Supplemental Figure S4). Distances between the R195 and F231 Cα atoms in each snapshot reported the state-of-closure parameter, whereas (ϕ, Ψ) angles in each snapshot reported the backbone conformation for each residue. Changes in the (ϕ, Ψ) angles of F212 and Y213 (both reside within the B4 strand) as well as K239 (immediately precedes B5), I240 (N-terminal residue of B5), and I242 (immediately follows B5) correlated most strongly with the state-of-closure for the A10/T4/A11 helical gate (less than −0.34 or >0.34). Interestingly, the B4 and B5 structural elements flank the A10/T4 and A11 helices, i.e., these residues occupy both the appropriate positions and exhibit the appropriate conformational motions to hinge the A10/T4/A11 gate (Figure 4A). For purposes of definition, we identify the overall gating region as residues G210-I242, and we refer to residues F212, Y213 as the FY component of the hinge unit and residues K239, I240, I242 as the KII component, respectively.
Figure 4.

Functional analysis of Sec14p hinge residue mutants. (A) Hinge residues (gray spheres) are identified in the context of a Sec14p α-carbon backbone trace at left. The hinge is depicted in greater detail at right where the specific residues of the hinge are identified and rendered as black spheres. The gating helix is the rightmost structural element in these depictions. (B) PtdCho transfer activity of purified recombinant Sec14p (open bars) and Sec14pF221A,S222A (solid bars) as a function of protein concentration (see Materials and Methods). Activity is expressed as the percentage of total [14C]PtdCho transferred after subtraction of background (determined by a mock assay using an equivalent concentration of E. coli cytosol). Data represent the averages of at least three independent experiments. (C) PtdCho transfer assays for yeast cytosol derived from YEp(SEC14), YEp(sec14Y213A) and YEp(sec14Y213A,F221A,S222A) derivatives of the sec14Δ strain CTY303. The first condition serves as positive control. Background values measured for the negative control [YEp(URA3) cytosol] were subtracted from values measured for YEp(SEC14), YEp(sec14 Y213A) and YEp(sec14Y213A,F221A,S222A) cytosols. With regard to the PtdCho transfer assays in B and C, total input of [14C]PtdCho averaged 15,755 cpm per assay. The background averaged 642 cpm per reaction. In these assays, under the conditions of protein expression used (see Materials and Methods), saturation is achieved at ∼0.5 μg of purified wild-type Sec14p and 50 μg/ml Sec14p-containing yeast cytosol. The data are presented to emphasize the strong decrement in transfer activity of the mutant proteins.
The hinge residue F212 orients its side chain toward the interior of the hydrophobic pocket, whereas the side chain of Y213 is disposed toward the posterior of the Sec14p molecule. The orientation of Y213 allows the hinge to be supported on the exterior of the phospholipid binding pocket by permitting the side chain of Y213 to participate in π-stacking interactions with B5 residue F241 (Supplemental Figure S5A). In addition, the Y213 side chain packs neatly into a pocket formed by the side chains of helix A12 residues L251 and Q254. Structural support for the hinge unit (and contribution to rigidity of the N-terminal aspect of helix A10/T4 within the Sec14p) is construed from interactions on the pocket surface where I242 sits in a cleft between F221 and S222 of helix A10/T4 (Supplemental Figure S5B).
Together, the MD simulations suggest the structural elements that undergo large-scale motions during gating of the Sec14p hydrophobic pocket move as a rigid body. The data further suggest that such rigid-body conformational motion is facilitated by “hinge-like” local backbone angular changes in the KII component of the hinge that interdigitates with the N terminus of the A10/T4/A11 helical gate and the FY component of the hinge that resides within the phospholipid binding pocket itself.
Hinge Function and Efficient Sec14p Activity
To assess whether the hinge unit is a functionally important element, the ability of Sec14p derivatives compromised for this element to catalyze PtdCho transfer was assessed in vitro. With regard to the KII hinge component, Sec14pF221A,S222A is defective in interdigitation of hinge residue I242 with the N terminus of the A10/T4/A11 helical gate and exhibits an ∼80% reduction in PtdCho transfer activity relative to Sec14p when the corresponding purified recombinant proteins are assayed (Figure 4B). With regard to the FY component of the hinge, the single mutant Sec14pY213A protein similarly exhibits a 40% reduction in PtdCho activity in vitro (Figure 4C), but combination of individual defects in the KII and FY hinge components levies more dramatic defects in general hinge function than those observed for either individual defect alone. Sec14pY213A,F221A,S222A exhibits no measurable PtdCho transfer activity in vitro (Figure 4C) and is nonfunctional as a Sec14p in vivo (Table 1), even though it is a stable protein both in vivo and in cytosol fractions used in transfer assays.
Table 1.
Functional evaluation of mutant sec14 proteins
| Sec14p species | Structural element | Rescue of sec14Δ |
|---|---|---|
| WT | +++ | |
| Y213A | Hinge | +++ |
| F221A | Hinge | ++ |
| F221A,S222A | Hinge | + |
| Y213A,F221A,S222A | Hinge | —** |
| K116P | B1LB2 | ++ |
| K116G | B1LB2 | +++ |
| D115A | B1LB2 | +++ |
| D115G | B1LB2 | +++ |
| D115A,K116P | B1LB2 | —** |
| D115A,K116G | B1LB2 | +++ |
A plasmid shuffle assay that reports whether physiological levels of expression of the indicated mutant sec14 protein rescues the lethality associated with sec14 nullizygosity (sec14▵). This plasmid shuffle assay employs CTY558 as parental yeast strain (see Materials and Methods) and has been described (Phillips et al., 1999; Li et al., 2000). Expression of wild-type Sec14p (WT) served as positive control, and ability to rescue identifies proteins that retain at least some function. The structural element disturbed for each mutant sec14 protein is identified. Robustness of plasmid shuffle was scored after 72 h of incubation at 26°C on YPD medium, and scores are assigned as qualitative comparisons relative to the WT control. Scores were assigned in a range from +++ (best) to + (poor) to — (failure to rescue).
** indicates relevant sec14 proteins are stable under the conditions of the experiment.
Altered Dynamics of the A10/T4/A11 Helical Gate in Sec14pG266D
The rmsf values plotted for apo-Sec14p in the 15- to 32-ns window of the MD simulation suggested a relationship between A10/T4/A11 motion and the dynamics of the C-terminal string motif as rmsf values in excess of 2 Å were recorded for residues 271DESK274 of the Sec14p string motif (Figure 2B). In that regard, the sec14-1ts gene product (Sec14pG266D) harbors a G266D missense substitution that resides in the string motif immediately N-terminal to the 271DESK274 cluster (Cleves et al., 1989; Sha et al., 1998). To explore this issue further, an unrestrained 32-ns MD simulation was run for Sec14pG266D after a starting structure for the mutant protein was calculated (see Materials and Methods). Conformational equilibration of Sec14pG266D occurred rapidly in the simulation as evidenced by the structural transition that occurs between 2.9 to 3.8 ns in the rmsd plot (Figure 2A). Similar to Sec14p, the rmsd values between the average Sec14pG266D structure recorded in the last 20 ns of the simulation, and the Sec14p reference structure, were large (4.0 Å). Monitoring of atomic positional fluctuations and interatomic distances between helix A9 residue R195 and helix A10/T4 residue F231 were consistent with the large rmsd values reporting closure of the Sec14pG266D A10/T4/A11 helical gate.
In contrast to the oscillations between open and closed conformations evident in the Sec14p simulation, however, rmsf calculations indicated the A10/T4/A11 helical gate failed to undergo the large-scale conformational motions throughout the 15- to 32-ns simulation time window that were observed for Sec14p. This is reflected in the reduced rmsf value (1.5 Å) for residue F231 in Sec14pG266D relative to the value of 5.4 Å calculated for F231 in Sec14p MD simulations (Figure 2B). Distance-monitoring analyses indicate that, after 3.4 ns, the interatomic distances between Cα atoms of R195 and F231 converged to an average value of 10.5 Å. This interatomic distance was then maintained throughout the remainder of the 32-ns simulation (Figure 3A). These measurements suggest apo-Sec14pG266D collapses to a closed structural conformation and cannot efficiently reopen. Closure of the helical gate in the Sec14pG266D simulation recapitulated the trajectory observed in the unrestrained Sec14p MD simulation. That is, helix A10/T4 residue K229 receded from residue E250 on helix A12 at the same time that helix A10/T4 residue F231 approached residue R195 on helix A9 (Supplemental Figure S1C).
The rmsf values in the immediate vicinity of the G266D missense substitution were altered in the Sec14pG266D simulation relative to those observed for that region in Sec14p. An additional region of Cα fluctuation also became apparent in the Sec14pG266D simulation. This new region of conformational mobility involved residues D115-R119 that reside within the loop element between β-strands B1 and B2. This unique conformational mobility was maximal for residue K116 which was displaced 1.7 Å in Sec14pG266D compared with 0.8 Å for Sec14p. The significance of these observations is discussed below.
Hinge Function and Gate Closure in Sec14pG266D
The unrestrained MD simulations of apo-Sec14pG266D suggested this mutant protein is defective in gate opening. We therefore inspected the conformational dynamics of the FY and KII hinge components to assess their functionality in the context of Sec14pG266D. As for Sec14p, distance monitoring plots of the F221 and I214 Cα atoms suggest that rigidity of the N-terminal region of helix A10/T4 with respect to the β-strands is maintained (Supplemental Figure S2). However, in contrast to Sec14p, distance monitoring of F221 and A187 Cα atoms reveal the N-terminal region of helix A10/T4 approaches helix A9 during the first 7.0 ns of simulation (Supplemental Figure S2). An average interatomic distance of 8.5 Å is then maintained throughout the remainder of the Sec14pG266D MD production run. This is in contrast to the average interatomic distance of 15 Å for the F221 and A187 Cα atoms maintained in the Sec14p simulation. The contrast between the conformational dynamics of Sec14p and Sec14pG266D is highlighted by superimposition of the two average structures (Supplemental Figure S3).
Insight into the mechanism of the gate opening defect was gleaned from correlations of backbone (ϕ, Ψ) angle variations with R195-F231 distance monitoring plots that report state-of-closure of the A10/T4/A11 helical gate. These analyses identified candidate bonds, rotations around which permit conformational changes of the helical gate. The strong correlations between (ϕ, Ψ) angle changes and gate closure we observed in the Sec14p MD simulations were preserved in the Sec14pG266D context for hinge residues K239 and I242 (−0.48 and −0.59, respectively; Supplemental Figure S4). Unlike the case of Sec14p, however, changes in (ϕ, Ψ) angles for hinge residues F212 and Y213 were no longer correlated with closure of the A10/T4/A11 helical gate. These collective data suggest the FY component of the hinge functions primarily to open the Sec14p A10/T4/A11 helical gate, whereas the KII hinge component functions in gate closure.
A Novel Gating Module That Regulates Opening and Closing of the A10/T4/A11 Helical Gate
How does the G266D missense substitution, which resides in the string motif, compromise C-terminal hinge function given these two structural elements are not in proximity to each other? As described below, the 310-helix T5 (i.e., the structural element directly impacted by G266D) is one component of a novel gating module that we posit regulates opening and closing of the A10/T4/A11 helical gate (overall distance monitoring schematic is illustrated in Figure 5). This gating module, designated the Sec14G-module, is made up of two independent substructures that are themselves interdigitated via a hydrogen bond (H-bond) network. The first substructure (A12LT5) includes helix A12, extends slightly beyond 310-helix T5 and encompasses residues Q248–S268. The second substructure is made up of the B1 and B2 β-strands along with the intervening loop region. It includes residues P108-E125 and is designated B1LB2. Sec14p residues 114TDKDGR119 of the B1LB2 motif cluster with Q254 of the A12LT5 substructure to form a polar, solvent-exposed patch on the Sec14p surface. The function of the Sec14G-module in the context of Sec14p and Sec14pG266D simulations is compared below. For purposes of comparison, we begin with a dynamics analysis of the hydrogen bond network that involves helix T5. We then consider the dynamics of the B1LB2 component and its interaction with the loop separating helix A12 from 310-helix T5.
Figure 5.

General distance-monitoring schematic of the Sec14p G-module. The Sec14G-module consists of two independent substructures. The first substructure, A12LT5, includes helix A12 and extends slightly beyond helix T5. The second substructure, B1LB2, consists of the β1- and β2 β-strands and the intervening loop region. The Sec14G-module conformation depicted here is that which exists in the open Sec14p conformer. Sec14p residue G266 is highlighted as a blue sphere, and relevant interatomic distances to be monitored are identified by the dashed lines. Reference residues that report status-of-closure include helix A10/T4 residue F231 (magenta sphere) and A9 residue R195 (green sphere).
Backbone H-Bond Interactions and the A12LT5 Component of the Sec14G-Module
The A12LT5 composite is stabilized by a series of H-bond interactions within the substructure itself. Helix T5 is made up of three core and two flanking residues (261PVKFG265). P261 and G265 reside at the two terminal turns of 310-helix T5 and cap it at its N and C terminus (Figure 6A). These capping residues engage in the 4 → 1 H-bond interactions typical of 310-helices (Richardson and Richardson, 1988; Penel et al., 1999a,b; Pal et al., 2002; Pal et al., 2003, 2005). The P261 carbonyl also engages in a strong H-bond interaction with the F264 amide. F264 exhibits (φ, Ψ) angles of −104 and 14°, respectively, and these angles are again typical for C-terminal residues of 310-helical helices. The backbone angular conformation of F264 orients both the G265 amide and the V262 carbonyl in a geometrically favorable configuration for a stable H-bond interaction (Figure 6A).
Figure 6.

Backbone hydrogen bond network of the helix T5 component of the Sec14G-module. All backbone interactions are read from the amide nitrogen to the backbone carbonyl oxygen. (A) Schematic diagram of the hydrogen bond network helix T5 and vicinal residues of Sec14p (top). The two consecutive 4 → 1 hydrogen bonds are characteristic of 310-helices. Helix capping residues are designated as Nc and Cc. Backbone hydrogen bond interactions are indicated by the dashed lines. An atomic stick rendition of that network within Sec14p is presented at bottom. (B) Schematic diagram of the hydrogen bond network helix T5 and vicinal residues of Sec14pG266D is presented at top. Backbone hydrogen bond interactions are indicated by the dashed lines. An atomic stick model of that network within Sec14pG266D is presented at bottom.
Helix T5 residues engage in contacts with both the intervening loop region between helix A12 and T5 as well as with the region downstream of T5, in the A12LT5 substructure (Figure 6A). The F264–P261 and G265–V262 interactions that involve the capping residue for helix T5 are preserved throughout the simulation (Supplemental Figure S6, A and B). In addition to H-bonding interactions with components of helix T5, V262 also bridges the helical and post-T5 regions of the A12LT5 substructure by forming a 6 → 1 H-bond with S268. Although the integrity of the V262–S268 interaction fluctuates during the MD simulation, the S268 amide engages in a stable H-bond interaction with the L260 carbonyl (Supplemental Figure S6, C and D). An additional H-bond interaction exists between the K267 amide and the A257 carbonyl (Supplemental Figure S6E), and these residues flank the T5 310-helix. The last H-bond interaction within this unit engages the T5-proximal N259 amide with the carbonyl of the helix A12 proximal residue P256 (Supplemental Figure S6F). MD simulation data project five of these six H-bonds are stable in the Sec14p context, because these persist in >90% of the snapshots collected during the 32-ns simulation time. The remaining H-bond (V262–S268) is present in >50% of the snapshots collected.
Interface between A12LT5 and B1LB2 in the Sec14G-Module
Several categories of H-bond interactions are evident between the A12LT5 and B1LB2 substructures in Sec14p MD simulations (Figure 5). B1LB2 residues T114, D115, and K116 interact with A12LT5 residues Q254 and N259 to stabilize the Sec14G-module. R119 is a candidate for several intra- and interstructural side chain solvent interactions as well. The panels displayed in Supplemental Figure S7 overlay distance-monitoring plots for the indicated residue pairs within the Sec14G-module. Those results suggest helix A12 residue Q254 engages in two H-bond interactions; the backbone interaction with K116, and a side chain interaction with D115. A side chain to main chain H-bond interaction between residues N259 and T114 further stabilizes this configuration. As detailed below, rearrangements in this H-bond network correlate with dynamics of the A10/T4/A11 helical gate.
Sec14G-Module Conductance Correlates with Motions of the A10/T4/A11 Helical Gate
Gate motions (as reported by distance monitoring between the Cα atoms of R195 and F231) correlate with the atom-to-atom H-bond switch involving the Q254-D115 side chains. Although the side chain interactions are maintained throughout the Sec14p MD simulation, the interactions alternate between the two H-bond acceptor atoms of the D115 side chain (OD1 and OD2) (Figure 7A). In the open conformation, H-bonds from Q254 NE2 form with either D115 OD1 or OD2. When Sec14p transitions to closed conformations, only the Q254 NE2::D115 OD1 H-bond is evident. A Q254 NE2 H-bond switch from D115 OD1 to D115 OD2 is also registered at the onset of every gate-opening event that occurs once the R195 and F231 Cα atoms approach to within 14 Å of each other.
Figure 7.

Sec14G-module transitions and gate dynamics. (A) Interatomic distances between the Cα atom of residues R195 and F231 report status of A10/T4/A11 gating helix closure (gray). Distances between the Q254 side chain amine and D115 OD1 (red) and D115OD2 (black) are also plotted. A hydrogen bond interaction between the side chains of Q254 and D115 persists throughout the simulation, but this interaction switches between OD1 and OD2 of residue D115 when Sec14p is in the open conformation, and it is restricted to OD1 when Sec14p is in the closed conformation. (B) Correlation of Sec14G-module to the fluctuating interaction between residues V262 and S268. Segments of time exist (3.5–8.9, 8.9–17.2, and 17.2–22.9 ns) when V262 and S268 (green) are bound or unbound, defining clusters that show transitions occurring at times when the side chains of Q254 and D115 (black) switch between the OD1 and OD2 atoms of D115 as well as transitions in the opening and closing of the A10/T4/A11 helical gate (R195-F231; gray). (C) First 5 ns of unrestrained Sec14pG266D MD simulation of the status of three hydrogen bond interactions between the B1LB2 and A12LT5 substructures during the transition from the open-to-closed Sec14p conformers at ∼3 ns. The T114–N258 interaction breaks at 0.882 ns but reforms again. Main chain hydrogen bonding interaction (blue) between the K116 amide and the Q254 carbonyl is lost during the transition and fails to reestablish during the simulation.
Although the S268 carbonyl interaction with the V262 amide fluctuates throughout the Sec14p simulation, the S268 amide maintains an H-bond interaction with the L260 carbonyl throughout (Supplemental Figure S6, C and D). These fluctuations in V262–S268 interaction coincide with discreet clusters in opening and closing of the A10/T4/A11 gate. The V262–S268 fluctuations also coincide with the H-bond switch between Q254 NE2 and the D115 OD1 and OD2 acceptors (Figure 7B). Finally, S268 lies immediately N terminal to a region of significant conformational mobility (residues 271–274) as indicated by the Sec14p atomic fluctuation plot (Figure 2B). These data suggest that the conformational dynamics in this region are linked to conformational events within the Sec14G-module.
Rearrangements in the T5 Helical Component of Sec14pG266D
The closed Sec14pG266D conformation is suggested to exhibit significant rearrangements within the helix T5 backbone conformation. These rearrangements minimize the steric problems otherwise imposed by the G226D missense substitution, while maximizing favorable interactions of the G266D side chain with solvent. The projected consequence of the G266D substitution for the string motif is loss of four H-bonds normally found within the Sec14p T5 helical component, and formation of a new H-bond not present in Sec14p (Figure 6B). The G265–V262 interaction is weakened, and the V262–S268 and S268–L260 H-bonding interactions are subsequently lost in Sec14pG266D (Supplemental Figure S6, B–D). In Sec14p, the K267 carbonyl engages in an H-bond with the A257 amide. By contrast, that specific H-bond interaction breaks early in the Sec14pG266D simulation and is replaced by an alternate interaction of the A257 amide with the backbone carbonyl of the mutant D266 residue (Supplemental Figure S6G). This alternate A257–D266 interaction is quite long-lived; it persists for almost 13 ns. The F264–P261 and N259–P256 interactions observed for Sec14p are preserved in the Sec14pG266D MD simulations (Supplemental Figure S6, A and F).
Disengagement of Sec14G-Module Structural Elements in Sec14pG266D and Closure of the A10/T4 Helical Gate
The loops of the two Sec14G-module substructures move apart during the simulated transition of Sec14pG266D to the closed conformation, and all H-bonds that link A12LT5 and B1LB2 of the Sec14G-module are broken (Supplemental Figure S7, A–D). The transition from open to closed conformations occurs early (between 3 and 4 ns) in the Sec14pG266D simulations. During this transition period, side chain distances between Q254 and D115 increase in a manner inconsistent with maintenance of H-bond interactions. The distance monitoring plot between these two specific amino acids then remains erratic throughout the remaining 28 ns of the MD simulation (Supplemental Figure S7, B and C). The N259 side chain to T114 main chain interaction breaks first (at 0.882 ns of the simulation; snapshot 882), but the interaction forms again (Figure 7C). The next dissociation is the backbone H-bond interaction between K116 and Q254, and this bond is not recovered again in the simulation. Loss of this H-bond interaction suggests a rationale for the unique site of conformational flexibility that involves residues of the 114TDKDGR119 motif (particularly K116) that is apparent in the Sec14pG266D apf plot (Figure 2B). The K116–Q254 dissociation is accompanied by strong correlation between changes in (φ, Ψ) angles of B1 residues Y111 and H112 and closure of the A10/T4/A11 helical gate (Supplemental Figure S4). These specific correlations uniquely occur in Sec14pG266D simulations and not in Sec14p MD production runs.
H-Bond Interactions between Loops of the Sec14G-Module Are of Functional Consequence
The MD simulation data implicate residue 114TDKDGR119 of the B1LB2 substructure as playing an important role in activity of the Sec14G-module. Specifically, the data suggest 114TDKDGR119 helps maintain the association of the B1LB2 and A12LT5 substructures and that this component of the B1LB2 motif serves as an inflection point, or swivel, that transduces B1LB2 conformational motions to helix A10/T4/A11 gating function (see Figure 8A for relative positions of K116 and G266). The MD simulations suggest the backbone H-bond between K116 amide (B1LB2) and the Q254 carbonyl (A12LT5), and that side chain interactions between D115 (B1LB2) and Q254, play important roles in execution of these functions. One prediction of this hypothesis is that removal of the backbone H-bond formed by K116 to the Q254 carbonyl and side chain H-bond interactions between residues D115 and Q254 will be of functional consequence. This prediction was tested in silico with a Sec14K116P mutant. The K116P missense substitution obviates formation of the relevant backbone H-bond with the Q254 carbonyl. A 32-ns Sec14pK116P MD simulation reproduced key features of the Sec14pG266D simulations described above. Distance monitoring plots for interatomic R195-F231 Cα distances show Sec14pK116P begins to transition toward the closed state at 10 ns of simulation and that a closed state is achieved at 14 ns (average interatomic R195-F231 Cα distance of 10.5 Å; Figure 8B). Sec14pK116P remains closed for the remainder of the simulation.
Figure 8.

B1LB2–A12LT5 interactions and efficient Sec14p function. (A) α-Carbon backbone rendition of Sec14p highlighting residues K116 (gray sphere) and G266 (black sphere). (B) Distance monitoring plot between the Cα atoms for helix A9 residue R195 and helix A10/T4 residue F231 that report helical gate status-of-closure for Sec14pK116P (black trace) as a function of simulation time (nanoseconds). The same plot for Sec14p is presented as the gray trace. (C) PtdCho transfer activity of cytosol from yeast strains expressing Sec14pK116G, Sec14pK116P, Sec14pD115A, Sec14pD115G, Sec14pD115A,K116P, and Sec14pD115A,K116G as sole Sec14p species. Transfer assays were performed as described in the legend to Figure 4. Total input of [14C]PtdCho averaged 18,803 cpm per assay, whereas assay background averaged 950 cpm per reaction. (D) Stabilities of mutant Sec14p proteins at 30 and 37°C. Cytosol was prepared from the sec14Δ cki1 yeast strain CTY303 expressing individual Sec14p species from the YEplac195-derived overexpression vector as for phospholipid transfer assays (see Materials and Methods). Aliquots of each cytosol were split and incubated in parallel at 30 or 37°C for 30 min. Reactions were terminated by boiling in SDS (1% final concentration), resolved by SDS-PAGE, transferred to nitrocellulose, and decorated with monoclonal anti-Sec14p antibody. Sec14p species were visualized by the enhanced chemiluminescence system developed by GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom). The Sec14p species are identified at top, and 10 μg of total protein was loaded for each sample.
Biochemical and in vivo assays reinforce the collective importance of the K116-Q254 main chain and D115-Q254 side chain H-bond interactions in Sec14p function. PtdCho transfer activities for Sec14pK116P and Sec14pK116G were assessed in vitro as a biochemical test of the prediction that Sec14pK116P will exhibit functional defects, whereas Sec14pK116G will not. The latter alteration mimics the secondary structural disruptions associated with the proline substitution, while maintaining both the relevant backbone H-bond with the Q254 carbonyl and the side chain H-bond interactions between D115 and Q254. The biochemical data indicate Sec14pK116P exhibits a ∼50% reduction in PtdCho transfer activity in vitro, whereas Sec14pK116G is as active as Sec14p in those assays (Figure 8C). Similarly, both Sec14pD115A and Sec14pD115G (ablated for the side chain H-bond interactions between D115 and Q254 but retain the backbone H-bond between the K116 amide and the Q254 carbonyl) exhibited significant PtdCho transfer activity in vitro when assays were performed at 30°C to limit destabilization of the mutant proteins (Figure 8C; see Materials and Methods). Thus, K116-Q254 backbone and D115-Q254 side chain H-bond interactions are individually sufficient to maintain a reasonably functional interface between the B1LB2 and A12LT5 substructures. We also note that the PtdCho transfer activity of Sec14pK116P was more strongly reduced (but nonetheless detectable) under the conditions we employ when assays were performed at 37°C. Those exaggerated defects for Sec14pK116P at 37°C correlate its degradation in vitro; even when cytosol from yeast overproducing Sec14pK116P is used (Figure 8D). In that regard, Sec14pK116P, Sec14pD115A, and Sec14pD115G all experience significant postlysis lability in cytosol prepared from yeast not engineered for overproduction of these proteins (our unpublished data).
To fully ablate the interaction between the loops of the B1LB2 and A12LT5 substructures, the K116P and D115A missense mutations were combined. The resultant Sec14pD115A,K116P exhibits no measurable PtdCho transfer activity in vitro, whereas Sec14pD115A,K116G retains wild-type levels of PtdCho transfer activity (Figure 8C). Although we present only the PtdCho transfer data, essentially the same results were obtained in PtdIns transfer assays (our unpublished data). These biochemical data are recapitulated by functional complementation analyses in vivo. Sec14pK116G, Sec14pD115A, Sec14pD115G, and Sec14pD115A,K116G all score as functional in complementation tests, whereas Sec14pD115A,K116P is nonfunctional in those in vivo assays (Table 1). Finally, we note the 115DKDGRPV121 motif is conserved in even distant members of the Sec14 family, e.g., the plant SSH PIP binding proteins (Kearns et al., 1998). It is of functional importance in those contexts as well. The relatively conservative D115N substitution inactivates the Sec14-like activities of the soybean PITP Ssh2p (our unpublished data).
DISCUSSION
Herein, are described a series of conformational transitions that we propose accompany phospholipid exchange by the major yeast PITP Sec14p. These findings were obtained from a combination of unrestrained MD simulations of Sec14p and Sec14pG266D conformational dynamics and functional analyses that test the predictive power of the simulations. Because simulations derive from a starting structure that approximates an open conformer of an apo-Sec14p, the conformational trajectories described most likely simulate those that occur on membrane surfaces once Sec14p has ejected bound phospholipid and is poised to reload with another phospholipid molecule. The major intramolecular motion simulated for Sec14p involves closing and opening of the A10/T4/A11 helical gate, i.e., the structural element proposed to regulate access to the hydrophobic phospholipid binding cavity of Sec14p. The conformational dynamics of the helical gate are projected to involve large rigid body motions within the Sec14p molecule, and these motions are controlled by two additional structural elements: 1) a hinge unit that interfaces with the N- and C-terminal ends of the helical gate and controls status-of-closure of the A10/T4/A11 helical gate; and 2) a gating module, or G-module through which conformational information is transduced to the hinge. A normal mode simulation of Sec14p conformational dynamics that coarsely depicts the motions described in these studies is provided in the Supplemental Figure S8.
The possibility that helix A10/T4 gates the phospholipid binding cavity of Sec14p was first raised when the crystal structure of an open Sec14p conformer bound to two detergent (β-octylglucoside; βOG) molecules was solved (Sha et al., 1998; Phillips et al., 1999). Subsequent crystallographic studies of closed conformers of distant members of the Sec14 protein superfamily provided additional support for this idea. In those structures, the cognate A10/T4 and A9 helices lie adjacent to each other, thereby sealing the opening to the hydrophobic cavity of Sec14-like proteins (Stocker et al., 2002; Min et al., 2003). The Sec14p MD simulations report oscillating motions of the A10/T4/A11 helical gate that are consistent with this structural element regulating access of phospholipid to the Sec14p interior. Moreover, the transitions between the closed and open states in the MD simulations involve large movements that occur on fast time scales. The rapidity of these substantial conformational transitions suggests Sec14p is designed to undergo an intrinsic high-frequency “breathing” regime when it is in the apo-conformer on membrane surfaces. These breathing motions may prime the apo-Sec14p for efficient reloading with phospholipid. The rapid motion of the A10/T4/A11 helical gate is consistent with the rapid rate with which Sec14p catalyzes PtdIns and PtdCho exchange reactions in vitro.
That the MD simulation is approximating conformational motions relevant to Sec14p phospholipid exchange activity is strongly supported by three lines of experimental evidence. First, trypsinolysis and Cys pair cross-linking experiments demonstrate that holo-Sec14p exists in a closed conformation in solution and that open and closed Sec14p conformers are distinguished by the distances between helix A9 and the A10/T4/A11 helical gate. The Cys pair experiments further demonstrate that covalent forced closure of the A10/T4/A11 helical gate is incompatible with phospholipid exchange. These forced closure data provide a powerful first demonstration that mobility of the A10/T4/A11 helical gate is required for phospholipid exchange by Sec14p. Third, both in vitro and in vivo data demonstrate that a functional hinge unit is essential for Sec14p to catalyze phospholipid transfer in vitro, and for Sec14p functionality in cells.
The MD simulations suggest state-of-closure of the A10/T4/A11 helical gate is controlled by N- and C-terminal hinges that flank the gate itself. The N-terminal aspect of the helical gate is projected to remain reasonably fixed during transitions between open and closed conformations, whereas the C-terminal region is simulated to swing open and shut, much like a windshield wiper blade. We find it most interesting that the hinge residue F212 contacts the acyl chain of βOG in the detergent-bound Sec14p structure, which we loosely refer to as the apo-Sec14p conformer, whereas a combination of genetic, biochemical, and crystallographic data identify an involvement of the hinge residue K239 in binding of the inositol headgroup of PtdIns (Phillips et al., 1999; our unpublished data). These physical interactions raise the attractive possibility that activities of the hinge subcomponents are instructed not only by intramolecular transitions within the Sec14p molecule but also by the phospholipid ligand itself. Given that the MD simulation data suggest a primary role for the KII component of the hinge in gate closure, and residue K239 resides within this component, the ligand-dependent regulation of conformational transitions may restrain the helical gate from reopening. Such a restraint could thereby direct apo-Sec14p from a “shallow breathing” conformational dynamics regime down a trajectory to a stable closed conformer. In that regard, the unrestrained MD simulation for Sec14p almost certainly approximates a “partially closed” structure. The closest approach of the helix A10/T4 residue F231 Cα atom to the residue R195 Cα atom on helix A9 in the Sec14p MD simulation is 10.21 Å (snapshot 27653). The corresponding interatomic distance in the closed phospholipid-bound conformer of a yeast Sec14-like protein must be smaller given our ability to induce disulfide bond formation in Sec14pR195C,F231C.
An unanticipated outcome from comparisons of MD simulations of Sec14p and Sec14pG266D was identification of a novel gating module, the Sec14G-module, that we propose transduces conformational information to the hinges, and, ultimately, to the A10/T4/A11 helical gate. This Sec14G-module includes the A12LT5 and B1LB2 composites, and these are linked by an intricate H-bond network. The data suggest a primary function of the Sec14G-module is conductance of conformational change to the hinges to open the A10/T4/A11 helical gate. A movie depicting key elements of this conductance is presented in the Supplemental Figure S9A. This finding begs reconsideration of the defects associated with the sec14-1ts mutation, and of the role of the Sec14p string motif itself. Originally, it was suggested the string motif functions merely to stabilize the Sec14-fold and that the sec14-1ts mutation results in structural destabilization of the protein (Sha et al., 1998). Although the stability of the sec14-1ts gene product (Sec14pG266D) is clearly affected, the mutant protein is functionally defective even under conditions where stability is not yet compromised. The Sec14pG266D MD simulations suggest this intrinsic flaw reflects an inability of Sec14pG266D to efficiently open the A10/T4/A11 helical gate because of a defect in the B1LB2 substructure of the Sec14G-module. In this regard, the MD simulations reported herein make a strong prediction with respect to the types of conformational differences we expect to discern in the G-modules of open and closed conformers of Sec14p. Specifically, the twist of the β2 strand, and, subsequently, the β1 strand around the D115/K116/Q254 swivel depicted in the Supplemental Figure S9B, is expected in the closed phospholipid-bound form of Sec14p. This transition from open to closed conformations will also alter the relative orientations of the loops of the B1LB2 and A12LT5 substructures to each other in a specific way. Determination of whether these key predictions are fulfilled is forthcoming given recent progress in the crystallization of phospholipid-bound forms of the yeast Sec14p homologue Sfh1p (Schaaf et al., 2006).
With regard to the B1LB2 substructure of the Sec14G-module, we are compelled to comment on the recent work of Griac and coworkers. They reported the D115G missense substitution ablates Sec14p PtdCho transfer activity and simultaneously effects a strong reduction in PtdIns transfer activity (Tahotna et al., 2007). Although we concur that Sec14pD115G reduces Sec14p PtdCho and PtdIns transfer activity, and now provide the first mechanistic explanation for why this missense substitution exerts these effects, we cannot agree with the claim of Griac and coworkers that Sec14pD115G is devoid of PtdCho transfer activity (Tahotna et al., 2007). In our hands, Sec14pD115G exhibits readily detectable PtdCho transfer activity, particularly when precautions are taken to ensure that levels of this polypeptide are not limiting in the phospholipid transfer assay. Among other issues, we note Griac and coworkers used crude yeast cytosol clamped at a rather low concentration in the assays upon which they base their claim, and they prepared those cytosols from yeast where they apparently assumed that Sec14pD115G and Sec14p levels were similar. We find Sec14pD115G (as well as Sec14pD115A and Sec14pK116P) produced under such conditions is subject to significant postlysis lability at 37°C. Because Tahotna et al. (2007) do not report what effect increased Sec14pD115G input has on measured transfer activities, or actual levels of mutant protein under conditions of assay, we suspect these issues form the basis of the dissonance between our conclusions and theirs. Obviously, if Sec14pD115G is not absolutely devoid of PtdCho transfer activity (and in our hands, it retains measurable activity), the claim of Tahotna et al. (2007) that PtdCho transfer activity is dispensible for Sec14p function in vivo is premature. This issue of threshold activity assumes additional import when one considers that physiological levels of Sec14p are approximately an order of magnitude in excess of those required for in vivo function (Salama et al., 1990). It is for this reason that mutant Sec14p derivatives with strong reductions in in vitro PtdIns and PtdCho transfer activity score as functional in genetic complementation assays (case in point, the sec14-1ts gene product at permissive temperatures; Bankaitis et al., 1990).
The importance of the structural elements identified in this study, and particularly of the B1LB2 substructure, in conformational dynamics of Sec14-like proteins is amply emphasized by inspection of distant members of the Sec14 protein superfamily. Human Sec14-like proteins include the vitamin E binding proteins α-tocopherol transfer protein (αTTP) and SNPF, the neurofibromin 1 (NF1) ras GTPase activating protein, and cellular retinaldehyde binding protein (CRALBP). Either crystal structures, or structural models, are available for each (Stocker et al., 2002; Min et al., 2003; Liu et al., 2005; D'Angelo et al., 2006). We find the structural elements geraine to this study (i.e., the helical gate, the hinge, and the A12LT5 and B1LB2 substructures of the Sec14G-module) are represented in each of these proteins. In Sec14p, αTTP, NF1, and SNPF, the loop between cognate β1 and β2 elements (B1LB2) orients itself such that the respective β-strands bend away from the interior of the hydrophobic pocket and toward the posterior of the protein. A backbone H-bond (K116-Q254 in Sec14p) is observed between the cognate B1LB2 and A12LT5 substructures in three of the four structures. With regard to that H-bond interaction, αTTP is the single exception where the cognate K116 is substituted with a proline. Thus, αTTP represents a natural version of the K116P context that partially compromises Sec14p. Why is αTTP tolerant of this K → P substitution, but Sec14p is less tolerant? Inspection of the αTTP crystal structure suggests the C-terminal helix of this protein provides structural support to the atypical αTTP G-module, thereby obviating the necessity of a backbone hydrogen bond between the cognate αTTP B1LB2 and A12LT5 motifs (data not shown).
Are the cognate helical gate, hinge and A12LT5/B1LB2 substructures of the G-module of functional consequence for these distant members of the Sec14 superfamily? It seems so. αTTP, NF1 and SNPF, and CRALBP insufficiencies cause various human diseases, and the responsible disease mutations have been identified at the primary sequence level. Strikingly, a number of these mutations lie either within the hinge regions that flank the helical gate or within the B1LB2 substructure of the cognate G-module itself (Figure 9). Given the small size of the B1LB2 element (∼17 residues), it is impressive how well represented this motif is with regard to incidence of mutation. We suggest the disease mutations highlighted in Figure 9 resemble Sec14pG266D in functional crippling of the G-modules of their respective proteins. These defects in turn hinder opening of the corresponding helical gates that control access of the individual binding substrates to the hydrophobic ligand-binding cavities of each of these Sec14-like proteins. With regard to the atypical G-module of αTTP, a common truncation mutation found in families affected by severe ataxia with vitamin E deficiency is caused by a single base-pair insertion (Ouachi et al., 1995; Benomar et al., 2002). This insertion results in a frame shift that alters the last 30 residues of αTTP (Figure 9), thereby compromising the C-terminal αTTP helix we posit stabilizes the atypical G-module of this Sec14-like protein.
Figure 9.

Relationship between disease mutations in Sec14-like proteins and structural elements involved in conformational transitions. Primary sequences of Sec14p, human αTTP, and human NF1 were aligned on the basis of primary sequence homology and structural criteria. CRALBP was aligned according to an αTTP-based structural homology search (Liu et al., 2005). Relevant secondary structures for each conformational element are indicated at top. Key Sec14p residues in this study and disease-causing mutations in the indicated Sec14-like proteins are highlighted.
Although the experiments reported here project a pathway for conformational change in what is most likely a transition from an apo-Sec14p to a phospholipid-bound holo-Sec14p, it is virtually certain that the motions described represent an incomplete description of this transition. This incompleteness derives in part from the lack of provision in the MD simulations for how a phospholipid ligand may influence Sec14p conformational transitions. How these motions relate to those involved in ejection of bound phospholipid from the Sec14p hydrophobic cavity (i.e., are these perfectly symmetrical processes?) also remains unclear. These unresolved issues notwithstanding, we posit the Sec14p structural elements described herein are relevant to the execution of the major Sec14p conformational transitions that occur throughout a cycle of phospholipid exchange. Finally, these studies raise the question of how is the activity of the Sec14G-module regulated so that the dynamics of the helical gate are properly coordinated with the phospholipid exchange cycle?
Supplementary Material
ACKNOWLEDGMENTS
Oligonucleotide primer synthesis and DNA sequence analyses were performed at the University of North Carolina Lineberger Comprehensive Cancer Center Genome Analysis and Nucleic Acids Core Facility. The mass spectrometry was performed at the University of North Carolina–Duke Michael Hooker Proteomics Center, and we thank Carol Parker for assistance and guidance in these analyses. Finally, we are grateful to Jan Hermans (Department of Biochemistry and Biophysics, University of North Carolina) for helpful advice regarding the MD simulations. MD production runs were performed at the R. L. Juliano Structural Bioinformatics Core Facility. This work was supported by National Institutes of Health grant GM-44530 (to V.A.B.).
Abbreviations used:
- αTTP
α-tocopherol transfer protein
- βOG
β-octylglucoside
- apf
atomic positional fluctuation
- CRALBP
cellular retinaldehyde binding protein
- MALDI-TOF
matrix-assisted laser desorption ionization/time of flight
- MD
molecular dynamics
- NF1
neurofibromin 1
- PAGE
polyacrylamide gel electrophoresis
- PtdCho
phosphatidylcholine
- PtdIns
phosphatidylinositol
- rmsd
root mean square deviation
- rmsf
root mean square fluctuation.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E06-11-1024) on March 7, 2007.

 The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
REFERENCES
- Alb J. G., Jr, Gedvilaite A., Cartee R. T., Skinner H. B., Bankaitis V. A. Mutant rat phosphatidylinositol/phosphatidylcholine transfer proteins specifically defective in phosphatidylinositol transfer: implications for the regulation of phosphatidylinositol transfer activity. Proc. Natl. Acad. Sci. USA. 1995;92:8826–8830. doi: 10.1073/pnas.92.19.8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L., Koonin E. V. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- Bankaitis V. A., Aitken J. R., Cleves A. E., Dowhan W. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature. 1990;347:561–562. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- Bankaitis V. A., Malehorn D. E., Emr S. D., Greene R. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benomar M., et al. Clinical comparison between AVED patients with 744 del A mutation and Friedreich ataxia with GAA expansion in 15 Moroccan families. J. Neurol. Sci. 2002;198:25–29. doi: 10.1016/s0022-510x(02)00057-6. [DOI] [PubMed] [Google Scholar]
- Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaikie N. London, United Kingdom: SAGE Publications Ltd; 2003. Analyzing Quantitative Data: From Description to Explanation, [Google Scholar]
- Bomar J. M., et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat. Genet. 2003;35:264–269. doi: 10.1038/ng1255. [DOI] [PubMed] [Google Scholar]
- Careaga C. L., Falke J. J. Structure and dynamics of Escherichia coli chemosensory receptors. Engineered sulfhydryl studies. Biophys. J. 1992;62:209–216. doi: 10.1016/S0006-3495(92)81806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen-Lopez M., Nicaud J.-M., Skinner H. B., Vergnolle C., Kader J. C., Bankaitis V. A., Gaillardin C. A phosphatidylinositol/phosphatidylcholine transfer protein is required for differentiation of the dimorphic yeast Yarrowia lipolytica from the yeast to the mycelial form. J. Cell Biol. 1994;124:113–127. doi: 10.1083/jcb.125.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A, et al. AMBER 8. San Francisco, CA: University of California; 2004. [Google Scholar]
- Cleves A. E., McGee T. P., Bankaitis V. A. Phospholipid transfer proteins: a biological debut. Trends Cell Biol. 1991a;1:30–34. doi: 10.1016/0962-8924(91)90067-j. [DOI] [PubMed] [Google Scholar]
- Cleves A. E., McGee T. P., Whitters E. A., Champion K. M., Aitken J. R., Dowhan W., Goebl M., Bankaitis V. A. Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell. 1991b;64:789–800. doi: 10.1016/0092-8674(91)90508-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves A. E., Novick P. J., Bankaitis V. A. Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J. Cell Biol. 1989;109:2939–2950. doi: 10.1083/jcb.109.6.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. Hillsdale, NJ: Lawrence Erlbaum Associates; 1988. The significance of a product moment rs. [Google Scholar]
- Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Jr, Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- D'Angelo I., Welti S., Bonneau F., Scheffzek K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 2006;7:174–179. doi: 10.1038/sj.embor.7400602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickeson S. K., Lim C. N., Schuyler G. T., Dalton T. P., Helmkamp G. M., Jr, Yarbrough Y. R. Isolation and sequence of cDNA clones encoding rat phosphatidylinositol transfer protein. J. Biol. Chem. 1989;264:16557–16564. [PubMed] [Google Scholar]
- Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Geitz R. D., Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Ile K. E., Schaaf G., Bankaitis V. A. Phosphatidylinositol transfer proteins and cellular nanoreactors for lipid signaling. Nat. Chem. Biol. 2006;2:576–583. doi: 10.1038/nchembio835. [DOI] [PubMed] [Google Scholar]
- Ito H., Fukuda Y., Muratat K., Kimura A. Transformation of intact yeast cells with alkali cations. J. Bacterial. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- Kearns B. G., Monks D. E., Fang M., Rivas M. P., Sha B. D., Phillips S. E., Bankaitis V. A., Luo M. Crystallization and preliminary X-ray diffraction studies of the Saccharomyces cerevisiae phospholipid transfer protein Sec14p. Acta Crystallogr. 1998;D53:784–786. doi: 10.1107/S0907444997006872. [DOI] [PubMed] [Google Scholar]
- Li X., Rivas M. P., Fang M., Marchena J, Mehrotra B., Chaudhary A., Feng L., Prestwich G. D., Bankaitis V. A. Analysis of OSBP homolog Kes1p function in regulation of Sec14p-dependent protein transport from the yeast Golgi complex. J. Cell Biol. 2002;157:63–77. doi: 10.1083/jcb.200201037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Routt S., Xie Z., Cui X., Fang M., Kearns M. A., Bard M., Kirsch D., Bankaitis V. A. Identification of a novel family of nonclassical yeast PITPs whose function modulates activation of phospholipase D and Sec14p-independent cell growth. Mol. Biol. Cell. 2000;11:1989–2005. doi: 10.1091/mbc.11.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Jenwitheesuk E., Teller D. C., Samudrala R. Structural insights into the cellular retinaldehyde-binding protein (CRALBP) Proteins. 2005;61:412–422. doi: 10.1002/prot.20621. [DOI] [PubMed] [Google Scholar]
- McGee T. P., Skinner H. B., Whitters E. A., Henry S. A., Bankaitis V. A. A phosphatidylinositol transfer protein controls the phosphatidylcholine content of yeast Golgi membranes. J. Cell Biol. 1994;124:273–287. doi: 10.1083/jcb.124.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier R., Tomizaki T., Schulze-Briese C., Baumann U., Stocker A. The molecular basis of vitamin E retention: structure of human tocopherol transfer protein. J. Mol. Biol. 2003;331:725–734. doi: 10.1016/s0022-2836(03)00724-1. [DOI] [PubMed] [Google Scholar]
- Min K. C., Kovall R. A., Hendrickson W. A. Crystal structure of α-tocopherol transfer protein bound to its ligand: implications for ataxia with vitamin E deficiency. Proc. Natl. Acad. Sci. USA. 2003;100:14713–14718. doi: 10.1073/pnas.2136684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks D. E., Aghoram K., Courtney P. D., DeWald D. B., Dewey R. E. Hyperosmotic stress induces the rapid phosphorylation of a soybean phosphatidylinositol transfer protein homolog through activation of the protein kinases SPK1 and SPK2. Plant Cell. 2001;13:1205–1219. doi: 10.1105/tpc.13.5.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase Y., Nakamura T., Hirata A., Routt S. M., Skinner H. B., Bankaitis V. A., Shimoda C. The Schizosaccharomyces pombe spo20(+) gene encoding a homologue of Saccharomyces cerevisiae Sec14 plays an important role in forespore membrane formation. Mol. Biol. Cell. 2001;4:901–917. doi: 10.1091/mbc.12.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouachi K., Arita M., Kayden H., Faycal H., Hamida M. B., Sokol R., Arai H., Inoue K., Mandel J.-L., Koenig M. Ataxia with vitamin E deficiency is caused by mutations in the α-tocopherol transfer protein. Nat. Genet. 1995;9:141–145. doi: 10.1038/ng0295-141. [DOI] [PubMed] [Google Scholar]
- Pal L., Basu G., Chakrabarti P. Variants of 310-helices in proteins. Proteins. 2002;48:571–579. doi: 10.1002/prot.10184. [DOI] [PubMed] [Google Scholar]
- Pal L., Chakrabarti P., Basu G. Sequence and structure patterns in proteins from an analysis of the shortest helices: implications for helix nucleation. J. Mol. Biol. 2003;326:273–291. doi: 10.1016/s0022-2836(02)01338-4. [DOI] [PubMed] [Google Scholar]
- Pal L., Dasgupta B., Chakrabarti P. 3(10)-Helix adjoining alpha-helix and beta-strand: sequence and structural features and their conservation. Biopolymers. 2005;78:147–162. doi: 10.1002/bip.20266. [DOI] [PubMed] [Google Scholar]
- Panagabko C., Morley S., Hernandez M., Cassolato P., Gordon H., Parsons R., Manor D., Atkinson J. Ligand specificity in the CRAL-TRIO protein family. Biochemistry. 2003;42:6467–6474. doi: 10.1021/bi034086v. [DOI] [PubMed] [Google Scholar]
- Penel S., Hughes E., Doig A. J. Side-chain structures in the first turn of the α-helix. J. Mol. Biol. 1999a;287:127–143. doi: 10.1006/jmbi.1998.2549. [DOI] [PubMed] [Google Scholar]
- Penel S., Morrison R. G., Mortishire-Smith R. J., Doig A. J. Periodicity in α-helix lengths and C-capping preferences. J. Mol. Biol. 1999b;293:1211–1219. doi: 10.1006/jmbi.1999.3206. [DOI] [PubMed] [Google Scholar]
- Phillips S. E., et al. Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol. Cell. 1999;4:187–197. doi: 10.1016/s1097-2765(00)80366-4. [DOI] [PubMed] [Google Scholar]
- Phillips S. E., Vincent P., Rizzieri K., Schaaf G., Gaucher E. A., Bankaitis V. A. The diverse biological functions of phosphatidylinositol transfer proteins in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2006;41:1–28. doi: 10.1080/10409230500519573. [DOI] [PubMed] [Google Scholar]
- Rentsch D., Laloi M., Rouhara I., Scmelzer E., Delrot S., Frommer W. B. NTR1 encodes a high affinity oligopeptide transporter in Arabidopsis. FEBS Lett. 1995;370:264–268. doi: 10.1016/0014-5793(95)00853-2. [DOI] [PubMed] [Google Scholar]
- Richardson J. S., Richardson D. C. Amino acid preferences for specific locations at the ends of α-helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- Rothstein R. J. One step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Rudge S. A., Sciorra V. A., Iwamoto M., Zhou C., Strahl T., Morris A. J., Thorner J., Engebrecht J. Role of phosphoinositides and of Spo14p (phospholipase D)-generated phosphatidic acid during yeast sporulation. Mol. Biol. Cell. 2004;15:207–218. doi: 10.1091/mbc.E03-04-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama S. R., Cleves A. E., Malehorn D. E., Whitters E. A., Bankaitis. V. A. Cloning and characterization of the Kluyveromyces lactis SEC14: a gene whose product stimulates Golgi secretory function in S. cerevisiae. J. Bacteriol. 1990;172:4510–4521. doi: 10.1128/jb.172.8.4510-4521.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf G., Betts L., Garrett T. A., Raetz C.R.H., Bankaitis V. A. Crystallization and preliminary X-ray diffraction analysis of phospholipid-bound Sfh1p: a member of the Saccharomyces cerevisiae Sec14p-like phosphatidylinositol transfer protein family. Acta Crystallogr. F. 2006;62:1156–1160. doi: 10.1107/S1744309106041728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B., Phillips S. E., Bankaitis V. A., Luo M. Crystal structure of the Saccharomyces cerevisiae phosphatidylinositol transfer protein Sec14p. Nature. 1998;391:506–510. doi: 10.1038/35179. [DOI] [PubMed] [Google Scholar]
- Sherman F., Fink G. R., Hinks J. B. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 1–113. [Google Scholar]
- Skinner H. B., McGee T. P., McMaster C. R., Fry M. R., Bell R. M., Bankaitis V. A. The Saccharomyces cerevisiae phosphatidylinositol transfer protein effects a ligand-dependent inhibition of choline-phosphate cytidylyltransferase activity. Proc. Natl. Acad. Sci. USA. 1995;92:112–116. doi: 10.1073/pnas.92.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker A., Baumann U. Supernatant protein factor in complex with RRR-alpha-tocopherylquinone: a link between oxidized vitamin E and cholesterol biosynthesis. J. Mol. Biol. 2003;332:759–765. doi: 10.1016/s0022-2836(03)00924-0. [DOI] [PubMed] [Google Scholar]
- Stocker A., Tomizaki T., Schulze-Briese C., Baumann U. Crystal structure of the human supernatant protein factor. Structure. 2002;10:1533–1540. doi: 10.1016/s0969-2126(02)00884-5. [DOI] [PubMed] [Google Scholar]
- Tahotna D., Holic R., Poloncova K., Simockova M., Griac P. Phosphatidylcholine transfer activity of yeast Sec14p is not essential for its function in vivo. Biochim. Biophys. Acta. 2007;1771:83–92. doi: 10.1016/j.bbalip.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Toukmaji A., Sagui C., Board J., Darden T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000;113:10913–10927. [Google Scholar]
- Vincent P., Chua M., Nogue F., Fairbrother A., Mekheel H., Xu Y., Allen N., Bibikova T. N., Gilroy S., Bankaitis V. A. A Sec14p-nodulin domain phosphatidylinositol transfer protein polarizes membrane growth of Arabidopsis thaliana root hairs. J. Cell Biol. 2005;168:801–812. doi: 10.1083/jcb.200412074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Fang M., Rivas M. P., Faulkner A., Sternweis P. C., Engebrecht J., Bankaitis V. A. Phospholipase D activity is required for suppression of yeast phosphatidylinositol transfer protein defects. Proc. Natl. Acad. Sci. USA. 1998;95:12346–12351. doi: 10.1073/pnas.95.21.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa L., Marchena J., Xie Z., Li X., Poon P. P., Singer R., Johnston G., Randazzo P. A., Bankaitis V. A. Activity of specific lipid-regulated ARFGAPs is required for Sec14p-dependent Golgi secretory function in yeast. Mol. Biol. Cell. 2002;13:2193–2206. doi: 10.1091/mbc.01-11-0563.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder M. D., Thomas L. M., Tremblay J. M., Oliver R. L., Yarbrough L. R., Helmkamp G. M., Jr Structure of a multifunctional protein. Mammalian phosphatidylinositol transfer protein complexed with phosphatidylcholine. J. Biol. Chem. 2001;276:9246–9252. doi: 10.1074/jbc.M010131200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.