

Tyrosine hydroxylase (TyrH) catalyzes the hydroxylation of tyrosine to dihydroxyphenylalanine (DOPA) using O2 and a tetrahydropterin (PH4) as cosubstrates.1 TyrH belongs to a family of aromatic amino acid hydroxylases which also includes phenylalanine hydroxylase (PheH) and tryptophan hydroxylase (TrpH); all three contain virtually identical mononuclear iron sites. In the mechanism proposed for TyrH1b (Scheme 1), the attack on the aromatic ring by an Fe(IV)O intermediate causes a change in hybridization at the site of oxygen addition; however, no secondary isotope effect is found for wild-type TyrH when 3,5-2H2-tyrosine is used as a substrate.2 In contrast, a DVmax value of 0.93 is seen when 5-2H-tryptophan is used as a substrate for TrpH, consistent with a change in hybridization in that case.3 This suggests that this chemical step in the TyrH reaction is masked by slower steps.4,5
In wild-type TyrH, there is a 1:1 stoichiometry between PH4 oxidation and amino acid hydroxylation. Recently, several active site mutations of TyrH have been characterized which alter this stoichiometry, such that a large excess of PH4 is consumed relative to the amount of amino acid hydroxylated.6 A kinetic model for such uncoupling is shown in Scheme 2.7 Here, TyrH binds all three substrates before the hydroxylating intermediate (X) is formed. The reaction may then follow one of two paths, amino acid hydroxylation with k1 as the net rate constant or unproductive breakdown of the hydroxylating intermediate with k2 as the net rate constant. The stoichiometry of amino acid hydroxylation to PH4 oxidation, or coupling C, is equal to k1/(k1 + k2), independently of whether slow steps precede these chemical steps. An isotope effect on k1 will be reflected in the coupling if k2 ≥ k1; the magnitude of the observed isotope effect should increase to the value of the intrinsic isotope effect on k1 as k2 becomes much greater than k1.8 Consequently, isotope effects which are masked in the wild-type enzyme could be unmasked by measuring product formation for enzymes which exhibit substantial uncoupling of PH4 oxidation and tyrosine hydroxylation. In the present study, this approach has been applied to detect kinetic isotope effects on the hydroxylation of tyrosine by TyrH.
The TyrH mutant enzymes F309A, E326A, and H336E oxidize a large excess of PH4 relative to the amount of DOPA produced; for the first two enymes, the Km values for tyrosine and 6-MePH4 are changed less than 2-fold by the mutations, while both Km values increase about 5-fold in the H336E enzyme.6a,c-d As shown in Table 1, the ratio of DOPA formed to PH4 consumed shows a significant inverse isotope effect in the case of the F309A and E326A enzymes, while the high Km value for 6-MePH4 of the H336E enzyme results in very low precision for the isotope effect with that enzyme. The unproductive breakdown of the hydroxylating intermediate can also result in isotope effects on the rate of DOPA formation if the rate constant for uncoupling exceeds all other first-order rate constants in the reaction. Consistent with this prediction, the Vmax values for DOPA formation by the E326A and H336E enzymes show significant inverse deuterium isotope effects, while that for F309A TyrH shows a small but statistically insignificant isotope effect. Thus, use of mutant enzymes with altered product partitioning has indeed allowed unmasking of chemical steps. The magnitudes of the isotope effects are very similar to the value of 0.93 found for TrpH,3 supporting the proposal that all three aromatic amino acid hydroxylases share a common mechanism.
Table 1.
Isotope Effects for Uncoupled TyrH Mutant Enzymes
| TyrH | Ca | DC | DVmaxb | D2OCc | D2OVmax |
|---|---|---|---|---|---|
| F309A | 0.20 ± 0.01 | 0.93 ± 0.03 | 0.97 ± 0.04 | 0.77 ± 0.05 | 0.87 ± 0.10 |
| E326A | 0.13 ± 0.01 | 0.93 ± 0.04 | 0.88 ± 0.03 | 0.49 ± 0.02 | 0.53 ± 0.03 |
| H336E | 0.04 ± 0.01 | 0.98 ± 0.20 | 0.90 ± 0.03 | nd | 0.62 ± 0.05 |
Nanomoles of DOPA produced per nanomole of PH4 oxidized. An HPLC-based assay similar to that previously described7 was used to measure directly the stoichiometry of amino acid hydroxylation and 6-MePH4 oxidation.
Isotope effects on the rate of DOPA formation were determined by direct comparison of the initial rates using 3,5-2H2-tyrosine or tyrosine as the substrate; the data were analyzed assuming identical DV and DV/K values.
Solvent isotope effects on the Vmax value for DOPA formation were determined by direct comparison of the rates of DOPA formation in H2O and D2O at several pL's spanning the pH optimum (6.5–8.5); the maximal values were used to calculate the solvent isotope effect.
Scheme 1.

Scheme 2.

The proposed heterolytic cleavage of the peroxypterin–Fe bridge in the mechanism of Scheme 1 will require the donation of a proton. This could result in a normal solvent isotope effect on DOPA formation. The solvent isotope effects on the Vmax values and on the coupling of DOPA formation and PH4 oxidation for the mutant enzymes are given in Table 1. While wild-type TyrH shows a solvent isotope effect of unity,4 the mutant enzymes show inverse solvent isotope effects. Figure 1 shows the proton inventory for the solvent isotope effect on the ratio of amino acid hydroxylation to PH4 oxidation. The effect on the proton inventory of a solvent isotope effect in the transition state on k1 in Scheme 2 is given by eq 1. A fit of the data to eq 1 yields a line which is concave up (Figure 1). Alternatively, the effect of a solvent isotope effect on k2 is described by eq 2. A fit of the data to eq 2 yields a significantly better fit, as shown by the straight line of Figure 1, with a solvent isotope effect in 100% D2O of 2.38 ± 0.04 on k2.
| (1) |
| (2) |
Figure 1.
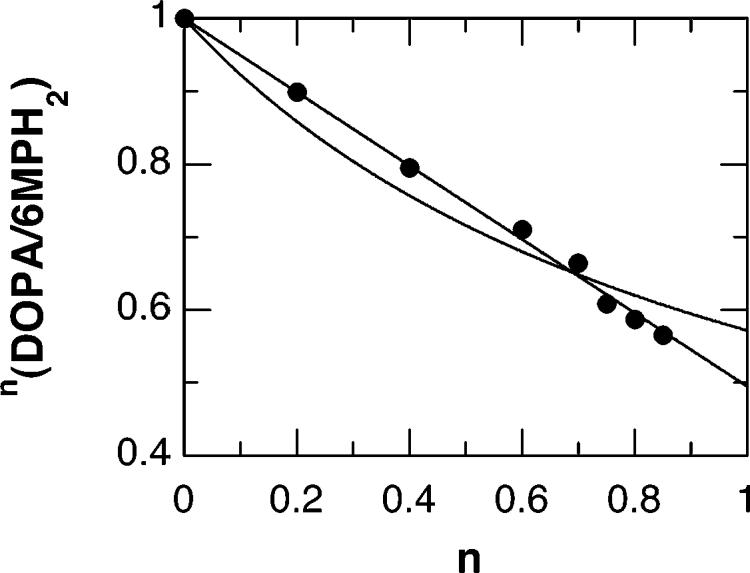
Proton inventory for E326A TyrH. The curved line is from a fit of the data to eq 1; the straight line is a fit to eq 2. The relative stoichiometries of PH4 oxidation and DOPA formation were determined by HPLC as described for Table 1. Buffers were made up at pH 7.0 and pD 7.4, the respective pH optima, and mixed to obtain the indicated mole ratios of D2O.
There are two steps in Scheme 1 at which unproductive breakdown of an intermediate would result in PH4 oxidation without amino acid hydroxylation. The first possibility is breakdown of the peroxypterin intermediate due to protonation of the distal oxygen to form a peroxypterin. Loss of peroxide would produce a quinonoid pterin and H2O2. This reaction would be similar to the uncoupling of the flavin monooxygenases9 and the cytochrome P450 enzymes10 and to the autoxidation of tetrahydropterins.11 An alternate possibility is the unproductive decay of the putative Fe(IV)O intermediate due to attack of a water molecule. Both possibilities are consistent with the solvent inventory results. One characteristic that distinguishes breakdown of the ferryl-oxo intermediate is that the branch point occurs at the same step as amino acid hydroxylation. As shown in Figure 2, there is a good correlation between the solvent isotope effect and the secondary deuterium isotope effect for the enzymes described here, consistent with both isotope effects arising from a single step. This suggests that the uncoupled reaction of E326A TyrH is due to inappropriate access of water to the iron. The structures of TyrH and PheH are consistent with a role of Glu326 in preventing access of solvent to the active site. When the amino acid and PH4 are both bound to PheH, a flexible loop moves to cover the active site.12 In the closed form, Ser137 forms a water-mediated hydrogen bond with Glu280 (Glu326 of TyrH). This serine is replaced by lysine in TyrH. Modeling of lysine in place of Ser137 in PheH shows that it could form an ionic interaction with Glu326 when the loop is closed over the active site but not when the loop is open (results not shown). This suggests that this glutamate plays a key role in keeping the active site closed, preventing access of solvent to the active site.
Figure 2.
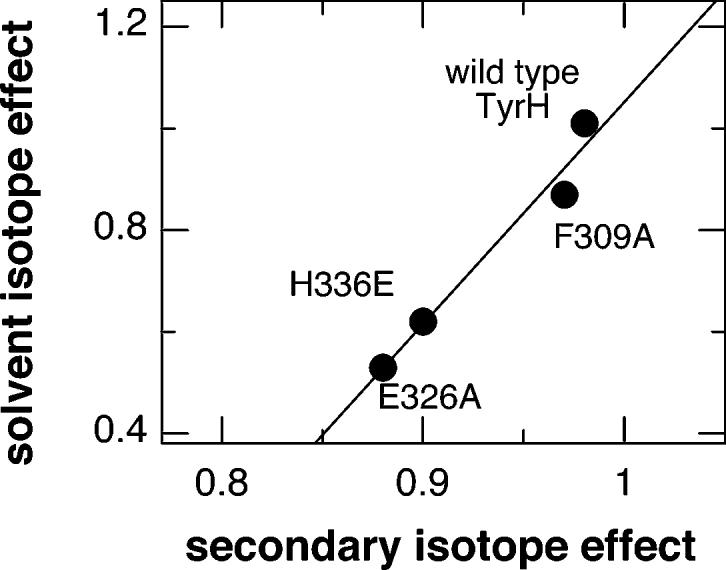
Correlation between secondary and solvent isotope effects for wild-type TyrH2,4 and mutant enzymes.
Supplementary Material
Acknowledgment
This work was funded in part by NIH grants R01-GM47291 (to P.F.F.) and T32-GM08523 (P.A.F.).
Footnotes
Supporting Information Available: Derivations of eqs 1 and 2 as well as more complete equations for the isotope effects (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1. (a) Fitzpatrick PF. Annu. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]; (b) Fitzpatrick PF. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fitzpatrick PF. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 3.Moran GR, Derecskei-Kovacs A, Hillas PJ, Fitzpatrick PF. J. Am. Chem. Soc. 2000;122:4535–4541. [Google Scholar]
- 4.Fitzpatrick PF. Biochemistry. 1991;30:6386–6391. doi: 10.1021/bi00240a006. [DOI] [PubMed] [Google Scholar]
- 5.Francisco WA, Tian G, Fitzpatrick PF, Klinman JP. J. Am. Chem. Soc. 1998;120:4057–4062. [Google Scholar]
- 6. (a) Ellis HR, Daubner SC, McCulloch RI, Fitzpatrick PF. Biochemistry. 1999;38:10909–10914. doi: 10.1021/bi991160u. [DOI] [PubMed] [Google Scholar]; (b) Ellis HR, Daubner SC, Fitzpatrick PF. Biochemistry. 2000;39:4174–4181. doi: 10.1021/bi9928546. [DOI] [PubMed] [Google Scholar]; (c) Daubner SC, Fitzpatrick PF. Biochemistry. 1999;38:4448–4454. doi: 10.1021/bi983012u. [DOI] [PubMed] [Google Scholar]; (d) Fitzpatrick PF, Ralph EC, Ellis HR, Willmon OJ, Daubner SC. Biochemistry. 2003;42:2081–8. doi: 10.1021/bi0271493. [DOI] [PubMed] [Google Scholar]
- 7.Hillas PJ, Fitzpatrick PF. Biochemistry. 1996;35:6969–6975. doi: 10.1021/bi9606861. [DOI] [PubMed] [Google Scholar]
- 8. (a) Miwa GT, Garland WA, Hodshon BJ, Lu AYH, Northrop DB. J. Biol. Chem. 1980;255:6049–6054. [PubMed] [Google Scholar]; (b) Jones JP, Korzekwa K, Rettie AE, Trager W. J. Am. Chem. Soc. 1986;108:7074–7078. [Google Scholar]; (c) Korzekwac KR, Trager WF, Gillette JR. Biochemistry. 1989;28:9012–9018. doi: 10.1021/bi00449a009. [DOI] [PubMed] [Google Scholar]
- 9.Massey V. J. Biol. Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 10.Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 11.Eberlein GA, Bruice TC, Lazarus RA, Henrie R, Benkovic SJ. J. Am. Chem. Soc. 1984;106:7916–7924. [Google Scholar]
- 12. (a) Andersen OA, Flatmark T, Hough E. J. Mol. Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]; (b) Andersen OA, Flatmark T, Hough E. J. Mol. Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.