

Abstract
Objective
Cerium oxide (CeO2) nanoparticles have been shown to protect cells in culture from lethal stress. Cardiac-specific expression of monocyte chemoattractant protein (MCP)-1 in mice causes ischemic cardiomyopathy associated with activation of endoplasmic reticulum (ER) stress. The aim of this study was to assess the effects of CeO2 nanoparticles on cardiac function and remodeling as well as ER stress response in this murine model of cardiomyopathy.
Methods
MCP-1 transgenic mice (MCP mice) and wild-type controls were administered intravenously 15 nmoles of CeO2 nanoparticles or vehicle only twice a week for two weeks. Cardiac function, myocardial histology, nitrotyrosine formation, expression of cytokines, and ER stress-associated genes were evaluated.
Results
Treatment with CeO2 nanoparticles markedly inhibited progressive left ventricular dysfunction and dilatation in MCP mice and caused a significant decrease in serum levels of MCP-1, C-reactive protein, and total nitrated proteins. The infiltration of monocytes/macrophages, accumulation of 3-nitrotyrosine, apoptotic cell death, and expression of proinflammatory cytokines, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 in the myocardium were markedly inhibited by CeO2 nanoparticles. Expression of the key ER stress-associated genes, including glucose-regulated protein 78 (Grp78), protein disulfide isomerase (PDI), and heat shock proteins (HSP25, HSP40, HSP70), were also suppressed by CeO2 nanoparticles.
Conclusions
CeO2 nanoparticles protect against the progression of cardiac dysfunction and remodeling by attenuation of myocardial oxidative stress, ER stress, and inflammatory processes probably through their autoregenerative antioxidant properties.
Keywords: MCP-1, Cardiomyopathy, ER stress, CeO2 nanoparticles, Transgenic animal model
1. Introduction
Over the recent years, inflammation and oxidative stress have been widely implicated in the pathogenesis of ischemic heart disease [1–3]. An increase in levels of many pro-inflammatory cytokines, such as tumor necrosis factor-α, interleukin (IL)-1, IL-6, monocyte chemoattractant protein-1 (MCP-1), and oxidative stress that results from an increased generation of reactive oxygen species or from a reduction of antioxidants are thought to contribute to the initiation and progression of cardiac dysfunction and remodeling of the extracellular matrix in the heart [2–5]. Recently, endoplasmic reticulum (ER) dysfunction/stress which in turn evokes the unfolded protein responses, such as induction of molecular chaperones in the ER, translational attenuation, and ER-associated protein degradation [6, 7], has been also reported to be associated with a range of diseases, including neurodegeneration [8, 9], diabetes mellitus [9, 10], hepatic disease [11, 12], and heart disease [13–18]. It can be activated by various stresses such as oxidative stress, hypoxia, and ischemia [19, 20], and lead to initiate of systemic inflammatory response and pathological cell death and dysfunction [9, 21]. We recently reported that transgenic mice with cardiac-specific expression of MCP-1 display a well-defined syndrome of ischemic cardiomyopathy as evidenced by myocardial inflammation (monocyte/macrophage infiltration and production of proinflammatory cytokines), thrombolic vasculopathy, cardiac remodeling (left ventricular chamber dilatation, interstitial fibrosis, and cardiac myocyte degeneration), ventricular dysfunction and mortality with heart failure by about 6 months of age [22–25]. Microarray analysis of gene expression profile changes during the development of heart failure in this mouse model revealed transcriptional activation of a set of ER stress-associated genes in the myocardium [13]. Quantitative real-time polymerase chain reaction (PCR) validated the upregulation of several of ER stress-associated genes. The expressions of the key ER stress proteins glucose-regulated protein 78 (Grp78 also known as Bip) and protein disulfide isomerase (PDI) as well as 25-kDa-, and 40-kDa heat shock proteins (HSP25, HSP40) were also found to be induced in the hearts, suggesting activation of ER stress in the development of heart disease in this model [13].
Cerium oxide, CeO2 nanoparticles are widely used in various applications such as oxygen sensors [26, 27] and automotive catalytic converters [28]. The best-known mechanism underlying the action of these nanoparticles is generally thought to be their dual oxidation state [29–30]. The loss of oxygen and the reduction of Ce4+ to Ce3+ are accompanied by creation of an oxygen vacancy. Recently, this group of nanoparticles has been used to modulate oxidative stress in biological system as a free radical scavenger [30]. For example, studies have demonstrated that CeO2 nanoparticles are able to rescue HT22 cells from oxidative stress-induced cell death [31], and protect normal human breast cells from radiation-induced apoptotic cell death [32]. However, no research has addressed the effects of this group of nanoparticles on any stress induced diseases in vivo. Here, we report that intravenously administration of CeO2 nanoparticles attenuates myocardial oxidative and/or ER stress and inflammatory processes and thereby inhibits the progression of left ventricular dysfunction and remodeling in this murine model of heart failure.
2. Materials and methods
2.1. Animals
Transgenic mice with cardiac-specific expression of MCP-1 were produced from the FVB/N strain and were bred, and maintained at the transgenic animal facility of the University of Central Florida. Detailed descriptions for the development and characterization of these transgenic mice line were reported previously [22]. The age-matched FVB/N strain mice purchased from Harlan Laboratories (Indianapolis, Indiana) were used as wild-type controls. Male mice were used for all experiments. The experimental procedures in mice and protocols used in this study were approved by Animal Care and Use Committee of the University of Central Florida, conforming with the Guide for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
2.2. Cerium oxide nanoparticles
A desired amount of NanoActive™ cerium oxide nanoparticles (NanoScale materials, Inc, USA) average particles size 7 nm in diameters were added in phosphate-buffered saline (PBS) solution. PBS was degassed in argon gas prior to use. CeO2 was mixed, vortexed and sonicated using Branson Sonifier 450 (Danbury, CT) for 2 minutes. The CeO2-PBS solution was sterile filtered through a 0.2 μm syringe filter.
2.3. Study protocol
The MCP-1 transgenic mice (referred to MCP mice) and wild-type FVB/N mice (referred to WT mice), 5-week-old, body weight of 20–25g, were divided into four experimental groups as follows: (a) MCP + vehicle group (n=6): MCP mice received 100 μl of PBS intravenously via tail vein twice a week for 2 weeks starting at 5 weeks of age. (b) MCP + CeO2 group (n=6): MCP mice received 100 μl of 0.15mM CeO2 nanoparticles intravenously via tail vein twice a week for two weeks starting at 5 weeks of age. (c) WT + vehicle group (n=6): FVB/N mice received 100 μl of PBS intravenously via tail vein twice a week for 2 weeks starting at 5 weeks of age. (d) WT + CeO2 group (n=6): FVB/N mice received 100 μl of 0.15mM CeO2 nanoparticles intravenously via tail vein twice a week for two weeks starting at 5 weeks of age. The timing of the CeO2 nanoparticles treatment was based on our previous studies that showing ER stress-associated genes were induced significantly in the early stage of disease in this model [13], and the dosage of the CeO2 nanoparticles was based on our preliminary experiments that showing there was no dose-dependent effects observed in CeO2 nanoparticles treatment, and some animals died at early stage after treatment with the highest concentrations of CeO2 nanoparticles.
The animals were sacrificed at 6 months of age. The histological and biochemical parameters and mRNA levels of ER stress-associated genes and proinflammatory cytokines in the hearts were evaluated.
2.4. Assessment of cardiac function and structure
To evaluate the effect of CeO2 nanoparticles treatment on the progression of cardiac remodeling and ventricular dysfunction in MCP mice, LV dimensions and left ventricular fractional shortening were measured by noninvasive transthoracic echocardiography with 15-MHz high frequency transducer (Agilent Technologies SONOS 4500, Philips Medical System, Germany) at the beginning of the treatment and at 4, 5, and 6 months of age. Mice were lightly anesthetized via a nose cone and maintained with 0.5~2.0% isoflurane (AErrane, Baxter, USA) mixed with oxygen. Normothermia was maintained with a heating pad. A two-dimensional short-axis view of the left ventricle was obtained at the level of the papillary muscles, and two-dimensionally targeted M-mode tracings were recorded at a sweep speed of 100 mm/s. Parameters were determined using the American Society for echocardiography leading-edge technique. The percentage of left ventricular fractional shortening (FS) was calculated as: FS (%) = [(LVEDD-LVESD)/LVEDD]× 100, where LVEDD and LVESD indicate left ventricular end-diastolic and end-systolic dimension, respectively. Data from three to five consecutive selected -cardiac cycles were analyzed and averaged.
2.5. Histology, immunohistochemistry and TUNEL assay
At 6 months of age, following functional analyses, all animals were sacrificed and the hearts were removed, weighed, and fixed by immersion in 10% phosphate-buffered formaldehyde or snap-frozen in liquid nitrogen for RNA/protein analysis. The hearts fixed in 10% phosphate-buffered formaldehyde were routinely processed and paraffin embedded. Sections (5 μm) were stained with hematoxylin and eosin (H.E), and Masson’s trichrome using standard protocols for histopathological analysis.
Immunohistochemistry was performed in sections to determine cellular infiltration, expression of 3-nitrotyrosine (a biomarker of peroxynitrite formation and indicator for oxidative stress), and proinflammatory cytokines in site. Briefly, after deparaffinization and rehydration, sections (5 μm) were pretreated with 3% H2O2/methanol solution to quench endogenous peroxidase activity, and incubated with blocking buffer (PerkinElmer, Boston, MA) to block non-specific binding. Sections were then incubated with anti-mouse monocyte/macrophage (MAB1852, Chemicon), anti-3-nitrotyrosine (3-NT, Upstate Biotechnology), anti-interleukin (IL)-1β, anti-IL-6, and anti-tumor necrosis factor (TNF)-α antibodies (Santa Cruz Biotechnology, CA) overnight at 4°C, followed by reaction with TSA™ fluorescence systems (PerkinElmer, Boston, MA) or diaminobenzing visualizing system. Primary antibody omission incubation with blocking solution was performed as controls. On control sections, no specific immunoreactivity was detected. Photomicrographs were obtained at 400 × magnification. Monocyte/macrophage infiltration was quantified in five randomly selected fields of view for each animal and five animals were studied per group. Expression of 3-nitrotyrosine was quantified from 5 sections per animal, and the positive area of staining were measured and expressed as the percentage of total LV area by using the Metamorph Series 6.2 image analysis program (University Imaging, West Chester, PA).
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling (TUNEL) staining was performed in sections with an in site cell death detection kit, TMR red (Roche, Indianapolis, USA) according to the manufacture’s instructions. In this procedure, nuclei undergoing apoptosis were labeled with red fluorescence. The number of TUNEL-positive cells was manually counted in five randomly selected fields of view under 400× magnification for each animal and five animals were studied per group.
2.6. Reverse transcriptase-polymerase chain reaction (RT-PCR) for proinflammatory cytokines
Total RNA was isolated from frozen cardiac tissues obtained from MCP mice and age-matched WT controls using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA (1 μg) was reverse transcribed to cDNA using the SuperScript First-Strand Synthesis System (BioRad-IScript, USA) in 40 μl reverse transcription reaction mix (42 °C/30 min). The reaction was stopped by heat inactivation (85 °C/5 min) and the mixture was chilled on ice. Subsequently 1 μl of the resulting cDNA was amplified with the following primers for detecting TNF-α, IL-1β, and IL-6 genes. Primer pairs used were as follows: TNF-α: forward 5′-ACTCAACAAACTGCCCTTCTGAG-3′, reverse 5′-TTACAGCTGGTTTCGATCCATTT-3′; IL-1β: forward 5′-TGTGGCTGTGGAGAAGCTGT-3′, reverse 5′-CAGCTCATATGGGTCCGAGA-3′; IL-6: forward 5′-CACGGCCTTCCCTACTTCAC-3′, reverse 5′-TGCAAGTGCATCATCGTTGT-3′. Similarly, β-actin primers: forward 5′-AAATCGTGCGTGACATCAAAG-3′, reverse 5′-TGTAGTTTCATGGATGCCACAG-3′ were used in PCR reactions under the same conditions which included an initial denaturation (94 °C/5 min), followed by a cycle of denaturation (94 °C/30 sec), annealing (60 °C/30 sec), and extension (72 °C/30 sec). Each sample was subjected to 35 cycles followed by a final extension (72 °C/10 min). PCR products were separated and visualized on 1.5% agarose ethidium bromide stained gel. Band intensity was assessed using imaging software (Alphaimager 2200), normalized to β-actin expression in each sample. The relative gene expressions expressed as the ratio of β-actin expression.
2.7. Real-time PCR assay for ER stress-associated genes
Total RNA samples used in RT-PCR analysis were also used for quantitative real-time PCR. cDNA was synthesized from one nonogram of RNA using the SuperScript First-Strand Synthesis System (BioRad-IScript, USA). A total of three samples were run for each experimental group and all PCR sample were in duplicate. Relative levels of mRNA transcripts for ER stress-associated genes, Grp78, PDI, HSP25, HSP40 were quantified by real-time PCR as we described previously [13]. Primer pairs used were as follows: Grp78 (accession number: NM022310; size: 232bp), forward 5′-ACCTGGGTGGGGAAGACTTT-3′, reverse 5′-TCTTCAAATTTGGCCCGAGT-3′; PDI (accession number: AW045202; size: 156bp), forward 5′-AACGGGAGAAGCCATTGTA-3′, reverse 5′-AGGTGTCATCCGTCAGCTCT-3′; HSP25 (accession number: NM013560; size: 276bp), forward 5′-CCTCTTCGATCAAGCTTTCG-3′, reverse 5′-GCCTTCCTTGGTCCTCACTG-3′; HSP40 (accession number: NM018808; size: 148bp), forward 5′-CGGTTCCGAATCAAAGTTGT-3′, reverse 5′-GGGAAATATGGACCTTGTG-3′; HSP70 (accession number: AJ002387; size: 157bp), forward 5′-TGCAGCAGGACATCAAGTTC-3′, reverse 5′-TACGCCTCAGCAGTCTCCTT-3′; β–actin, forward 5′-GAAACAGCTTCAGCCCAAA-3′, reverse 5′-ACGTCAGGTGACTGCTGAA-3′.
2.8. Western blot analysis
Tissue samples from the LV were homogenized in a buffer containing 20 mM Tris-HCl, pH 6.8, 1 mM EDTA, 1% SDS, 1 mM PMSF, and 1× protease inhibitor cocktail (Roche, Mannheim, Germany). Equal amounts of protein from each sample were separated with a 10% or 12.5% SDS-PAGE as we described previously [13]. The separated proteins were transferred onto polyvinylidene difluoride membranes (Immobilon-P, Millipore, MA), and the membranes were blocked for 1hr with 5% milk, then incubated overnight at 4 °C with one of the following antibodies: Grp78, diluted 1:5000; PDI, diluted 1:1000; HSP25, diluted 1:4000; HSP40, diluted 1:1000 (Stressgene Biotechnologies, Canada) and MCP-1, diluted 1:1000 (Santa Cruz Biotechnology, CA). After being washed, the membrane was incubated with the corresponding secondary antibody conjugated to horseradish peroxidase. Immunoreactive bands were visualized with the SuperSignal West Pico enhanced chemiluminescence kit (Pierce, Rockford, IL) according to the manufacture’s instructions. Protein loading was evaluated by the glyceraldehydes-3-phosphate dehydrogenase (GAPDH) band visualized with a monoclonal antibody (Novus Biological, Littleton, CO). Band intensities were quantified using a densitometer equipped with a multi-analyst software program (Alpha innotech, San Leandro, CA).
2.9. Circulating levels of CRP, MCP-1, and NOX
Whole blood was collected at sacrifice. The circulating levels of C-reactive protein (CRP) and MCP-1 were determined with the commercially available ELISA kit (Kamiya Biomedical Company, seattle, WA) according the manufacture’s recommendations. Circulating levels of NOx, evaluated as total nitrated proteins (nitrite and nitrate), were measured by Griess reagent kit (Invitrogen)
2.10. Statistical analysis
All values are presented as mean ± standard error. The results were analyzed by one-way analysis of variance (ANOVA) followed by a Bonferroni post-test for multiple comparisons. A p-value of less than 0.05 was considered significant.
3. Results
3.1. Effects of CeO2 nanoparticles on cardiac function
To evaluate the effects of CeO2 nanoparticles treatment on the progression of cardiac dysfunction, LVEDD and FS were measured by echocardiography. All animals showed the same echocardiographic parameters at the beginning of the experiment when they were 5 weeks of age. LVEDD, and FS were similar in WT, vehicle-treated MCP, and CeO2 –treated MCP mice (Fig. 1, A, B). Serial echocardiographic examination revealed that as animal aged the progressive LV dilatation and contractile impairment occurred in the vehicle-treated MCP mice compared to wild-type controls, whereas these changes were attenuated by the treatment with CeO2 nanoparticles (Fig. 1, A and B). LV end-diastolic diameter increased and LV fractional shortening decreased in a time-dependent manner in the vehicle-treated MCP mice. CeO2 nanoparticles treatment significantly inhibited the progressive increase in LV end-diastolic diameter and the decrease in LV fractional shortening. This observation was also reflected by the decreased heart/body weight ratio in CeO2-treated MCP mice compared to vehicle-treated MCP mice, although the magnitude of reduction in HW/BW ratio did not reach that observed in WT controls (Fig. 1C).
Fig. 1.
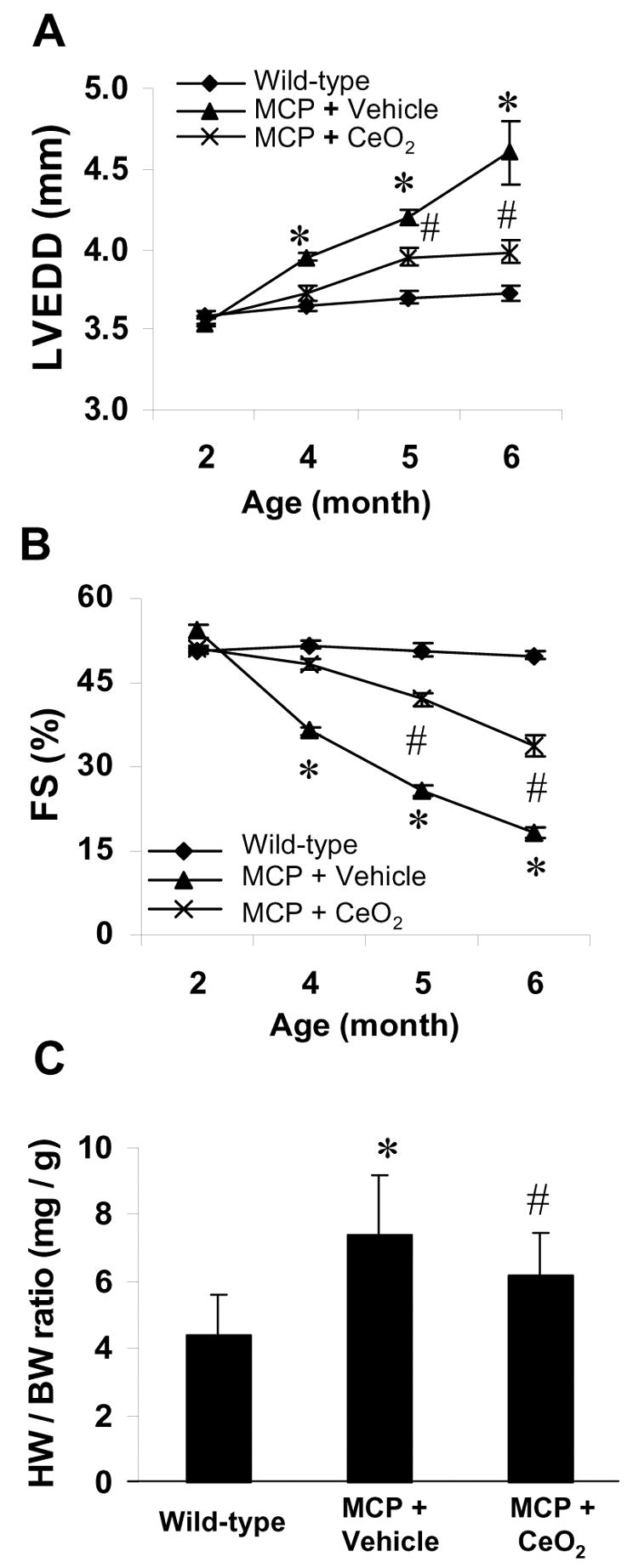
Effects of CeO2 nanoparticles on left ventricular function and remodeling. LV end-diastolic dimensions (LVEDD) (A) and percent fractional shortening (%FS) (B) were depicted from 2 to 6 months of age. The progressive LV dilatation and LV dysfunction in the vehicle-treated MCP mice were attenuated by treatment with CeO2 nanoparticles. *P<0.001 versus wild-type controls; #P < 0.05 versus vehicle-treated MCP mice; n=6 per group and per time point. Heart weight to body weight ratio (HW/BW) was depicted at 6 months of age (C). *P<0.001 versus wild-type controls; #P < 0.05 versus vehicle-treated MCP mice; n=5 per group.
3.2. Effects of CeO2 nanoparticles on MCP-1 expression-induced inflammation
To evaluate the effect of CeO2 nanoparticles on MCP-1 expression-induced myocardial inflammation, we investigated the cardiac histology and the production of proinflammatory cytokines. Shown in Fig. 2A are representative photomicrographs of H&E and Masson’s staining. Staining patterns demonstrated that the cellular infiltration, interstitial fibrosis, and cardiac myocytes degeneration occurred in the heart of MCP mice were significantly suppressed by CeO2 treatment. As shown in Fig. 2B, MAB1852-positive monocyte/macrophage diffusely infiltrated the interstitial space of the myocardium in vehicle-treated MCP mice. The number of immunopositive cells per section was significantly greater in the myocardium of vehicle-treated MCP mice than in WT controls, whereas the number of MAB1852-positive cells were markedly reduced in the myocardium of CeO2-treated MCP mice (Fig.2C). Quantitative analysis apoptotic cell death in TUNEL-stained sections showed that there was an almost 6-fold increase in the number of TUNEL-positive nuclei in the myocardium of vehicle-treated MCP mice (20.2 ± 2.8 cells/per field, n=5) compared with age-matched wild-type controls (3.8 ± 1.2 cells/per field, n=3), whereas the number of TUNEL-positive cells were significantly decreased in the myocardium of CeO2-treated MCP mice (9.3 ± 2.6 cells/per field, n=5) (Fig. 2D).
Fig. 2.
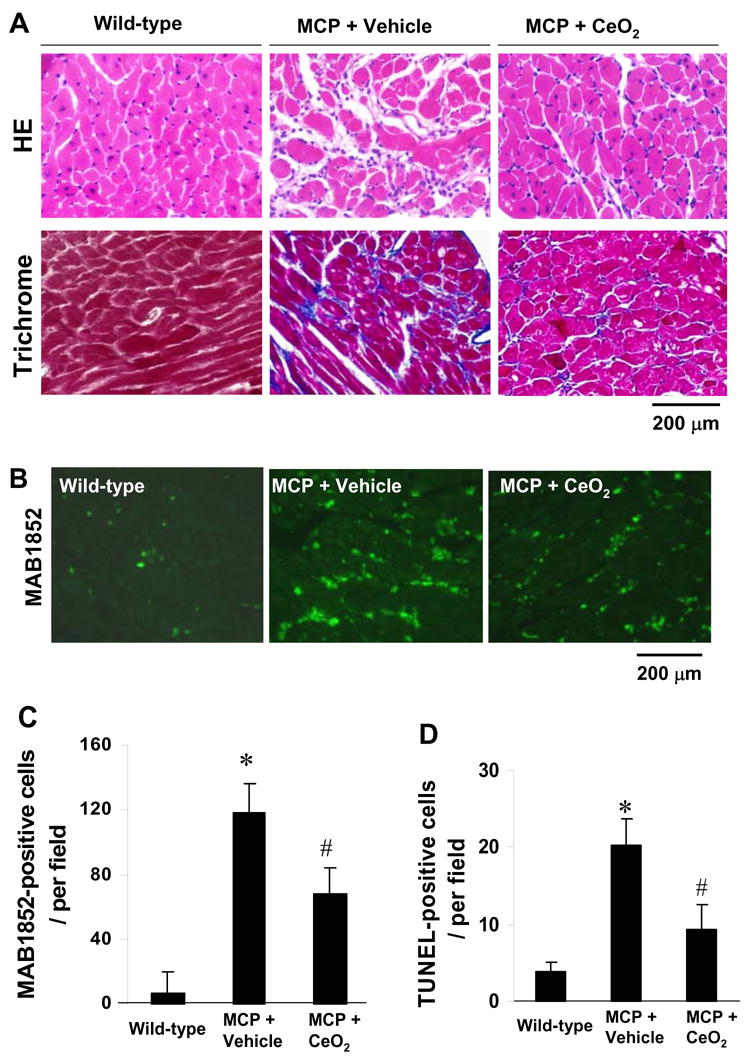
Effects of CeO2 nanoparticles on myocardial inflammation. (A) Histopathological photomicrographs of LV obtained from wild-type control, vehicle- and CeO2-treated MCP mice. Upper panel, H.E staining; Lower panel, Masson’s trichrome staining, blue represents collagen deposition. (B) Representative immunohistochemical photomicrographs of monocyte/macrophage infiltration in the myocardium. Green denotes infiltrated monocyters/macrophages. (C, D) Histograms showing the number of MAB1852-positive cells and TUNEL-positive cells in the myocardium of different group of mice, respectively. *P < 0.001 versus wild-type controls; #P<0.05 versus vehicle-treated MCP mice; n=5 per group.
Production of proinflammatory cytokines TNF-α, IL-1β, and IL-6 in mice myocardium were evaluated by measuring the levels of their mRNA transcripts using RT-PCR. The expression of these proinflammatory cytokines in the myocardial tissues markedly increased in the vehicle-treated MCP mice compared to the wild-type controls, whereas the elevation of mRNA levels of these cytokines was significantly reduced in the myocardium of CeO2 treated MCP mice (Fig. 3, A to D). The expression of these proinflammatory cytokines in the myocardium was confirmed by immunohistochemistry. The infiltrated inflammatory cells in the interstitial of myocardium were defined as the main cellular source of TNF-α and IL-1β production; however, cardiac myocytes were a main cellular source of IL-6 production (Fig. 3, E). Furthermore, CeO2 treatment markedly suppressed the expression of MCP-1 in the myocardium of MCP mice (Fig. 4, A and B) and inhibited the production of circulating levels of MCP-1 and CRP, although the magnitude of the reduction in circulating levels of MCP-1 and CRP did not reach exactly the levels observed in WT controls (Fig. 4, C and D).
Fig. 3.
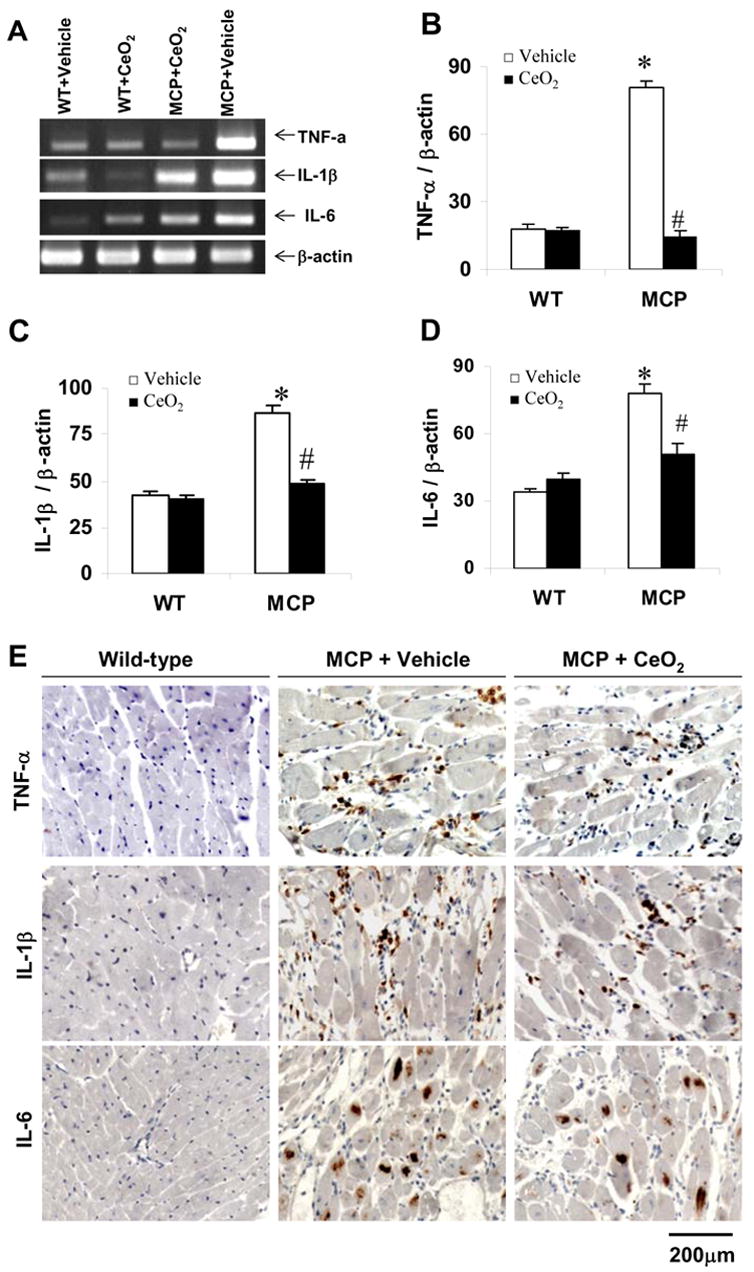
Effects of CeO2 nanoparticles on TNF-α, IL-1β, and IL-6 gene expression in the myocardium of MCP mice. (A) Expression of TNF-α, IL-1β, and IL-6 mRNA in the myocardium of wild-type control, vehicle- and CeO2-treated MCP mice were assayed by RT-PCR. (B–D) Bands were quantified by densitometric analysis and normalized by β-actin. *P < 0.001 versus wild-type controls; #P<0.05 versus vehicle-treated MCP mice; n=5 per group. (E) Representative photomicrographs of in site immunohistochemical staining demonstrating production of proinflammation cytokines TNF-α, IL-1β, and IL-6 in the myocardium. Positive-stained cells were visualized with diaminobenzidine (brown).
Fig. 4.
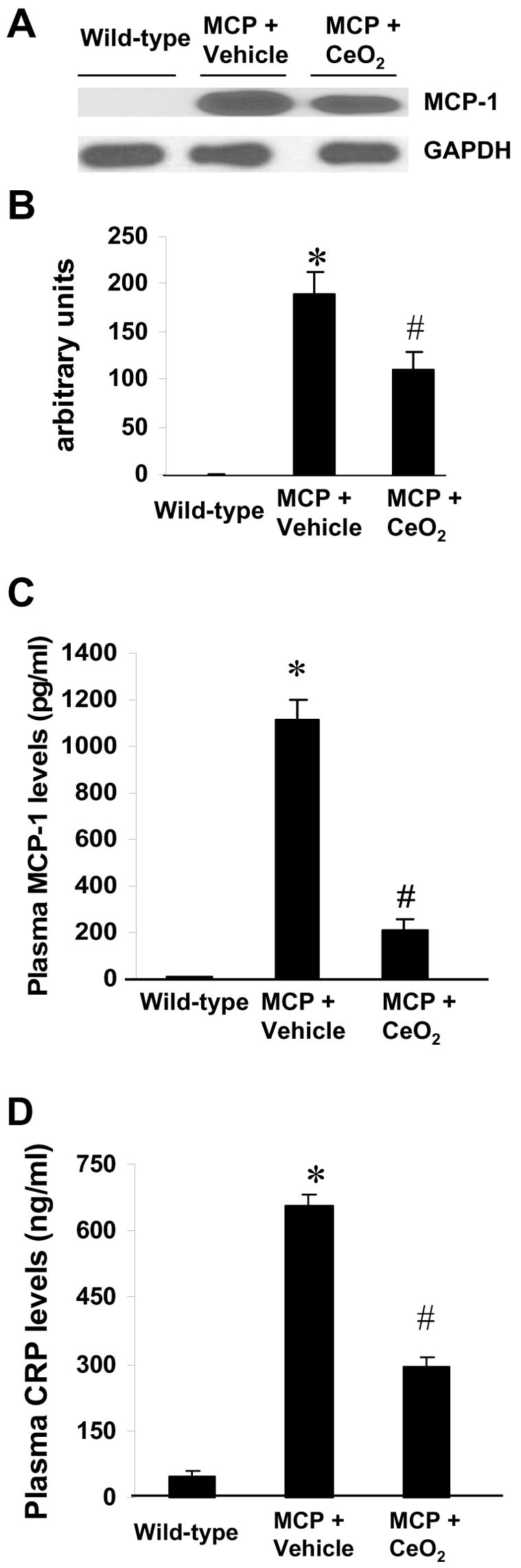
Effects of CeO2 nanoparticles on MCP-1 expression and release. (A) Immunoblot showing levels of MCP-1 in the myocardium of wild-type control, vehicle-, and CeO2-treated MCP mice. (B) Summary of densitometric analysis of data in A. *P < 0.001 versus wild-type control; #P<0.05 versus vehicle-treated MCP mice and wild-type controls; n=5 per group. (C–D) Circulating levels of MCP-1 and CRP assayed by ELISA. *P < 0.001 versus wild-type control; #P<0.05 versus vehicle-treated MCP mice and wild-type controls; n=6 per group.
3.3. CeO2 nanoparticles attenuates myocardial oxidative stress
We performed immunohistochemistry to examine peroxynitrite formation (a marker of oxidative stress) by detecting nitrosylated tyrosine residues on proteins with anti-3-nitrotyrosine (3-NT) antibody [33]. As shown in Fig. 5A, nitrated proteins were positively stained mainly in the cardiac myocytes as well as the infiltrated polymorphonuclear leukocytes. Staining with 3-NT was barely detectable in the myocardium of WT controls, whereas the intensity and the extent of immunoreactivity was prominent in the myocardium of vehicle-treated MCP mice. However, treatment with CeO2 nanoparticles resulted in markedly decreased intensity of 3-NT staining. Quantitative analysis revealed that CeO2 treatment significantly suppressed peroxynitrite formation in the myocardium of MCP mice compared to the vehicle-treated MCP mice (Fig. 5B). Furthermore, the administration of CeO2 nanoparticles also significantly attenuated the levels of NOx, evaluated as serum levels of total nitrated proteins (Fig. 5C), suggesting that treatment with CeO2 nanoparticles attenuated myocardial oxidative stress.
Fig. 5.
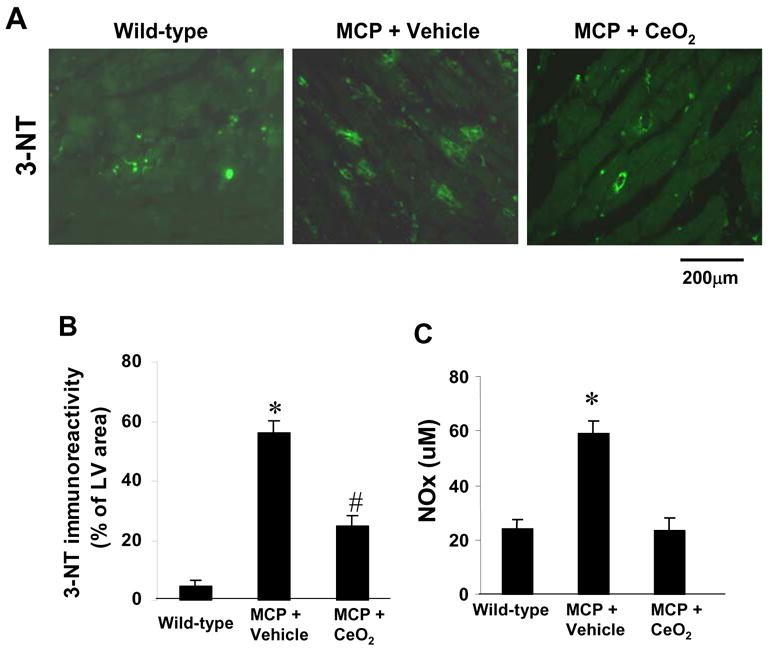
Effects of CeO2 nanoparticles on myocardial oxidative stress. (A) Representativeimmunohistochemical photomicrographs of 3-NT in the myocardium of wild-type control, vehicle-, and CeO2-treated MCP mice. (B) Histograms showing the accumulation of nitrotyrosine formation in the myocardium of different group of mice. *P < 0.001 versus wild-type controls; #P<0.05 versus vehicle-treated MCP mice and wild-type controls; n=5 per group. (C) Serum levels of NOx (total nitrate and nitrite in proteins) assayed by Griess reagent. *P < 0.001 versus wild-type controls and CeO2-treated MCP mice; n=6 per group.
3.4. CeO2 nanoparticles attenuates myocardial ER stress
To determine whether CeO2 nanoparticles attenuated myocardial ER stress we measured the expression levels of the key ER stress-associated genes. As shown in Fig. 6A, real-time PCR analysis demonstrated that expression of ER stress-associated genes, Grp78, PDI, HSP25, HSP40, and HSP70 mRNA was significantly upregulated in the myocardium of the vehicle-treated MCP mice compared to the WT controls (P<0.001). The increased expression of these ER stress-associated genes mRNAs in the myocardium of MCP mice was significantly suppressed by treatment with CeO2 nanoparticles (P<0.05 versus vehicle-treated group).
Fig. 6.
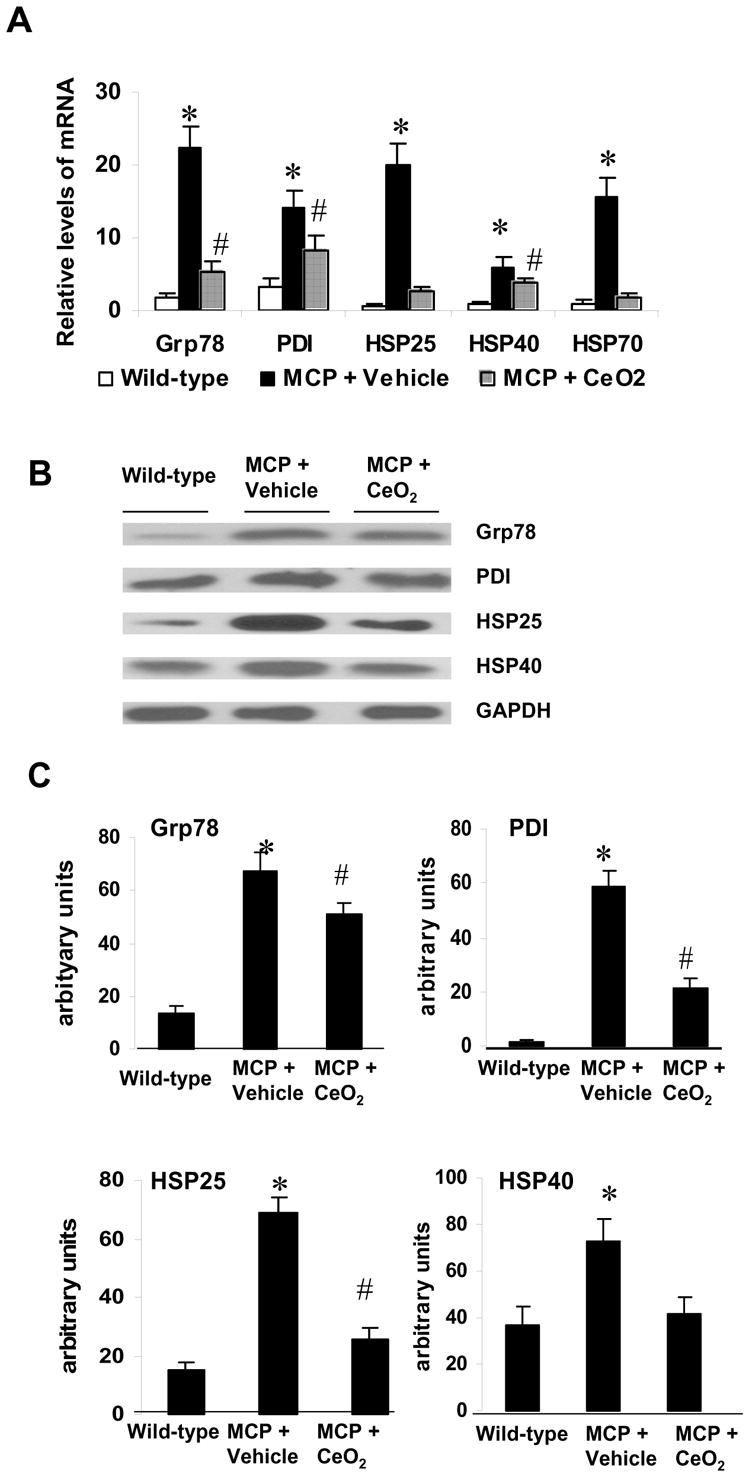
Effects of CeO2 nanoparticles on myocardial ER stress. (A) Relative mRNA levels of ER stress-associated genes Grp78, PDI, HSP25, HSP40, and HSP70 were measured by real-time PCR. *P < 0.001 versus wild-type controls; #P<0.05 versus vehicle-treated MCP mice and wild-type controls; n=5 per group. (B) Immunoblot showing levels of ER stress-associated proteins Grp78, PDI, HSP25, and HSP40 in the myocardium of wild-type controls, vehicle-, and CeO2-treated MCP mice. (C) Summary of densitometric analysis of data in B. *P < 0.001 versus wild-type controls; #P<0.05 versus vehicle-treated MCP mice and wild-type controls; n=5 per group.
Western blot analysis was also performed to see whether the changes in the transcript levels were reflected in the protein levels. Consistent with the transcript levels, expression of ER stress-associated proteins Grp78, PDI, HSP25, and HSP40 in the myocardium of MCP mice was significant downregulated by treatment with CeO2nanoparticles (Fig. 6, B and C).
4. Discussion
Recent studies have provided evidence that cerium and yttrium oxide nanoparticles act as direct antioxidants to protect cells from various forms of lethal stress in vitro [30–32]. Here we demonstrate that treatment with CeO2 nanoparticles inhibited monocyte/macrophage infiltration and suppressed the production of proinflammatory cytokines (TNF-α, IL-1β, IL-6, and MCP-1) in the myocardium. Furthermore, we show that CeO2 nanoparticles inhibited peroxynitrite formation and levels of NOx, and suppressed the induction of ER stress-associated genes in the hearts. Thus, it is likely that CeO2 nanoparticles suppresses myocardial inflammatory process and oxidative and ER stress and thereby inhibits the progression of left ventricular dysfunction and remodeling in this transgenic mouse model. Yttrium oxide nanoparticles, that have also been reported to protect neurons from death because of their antioxidant activity, were not tested in the current murine model.
A pathogenic role for oxidative stress in acute myocardial injury as well as progressive cardiac and vascular disease is becoming increasingly apparent [2–4]. Oxidative stress could lead to activation of redox-sensitive transcription factors and induction of transcription of proinflammatory cytokines TNF-α, IL-6 and chemokine MCP-1 and IL-8 [5, 34, 35]. We have previously demonstrated that monocyte/macrophage infiltration and increased transcription of proinflammatory cytokines play a critical role in the development of heart failure in this mouse model [24, 25]. Macrophages are sites of production of reactive oxygen and reactive nitrogen species [36]. Nitration of protein tyrosine residues, producing 3-nitrotyrosine (3-NT), is an unique feature of reactive nitrogen species that has been recognized as a key oxidant species in the initiation and/or progression of a wide array of cardiac pathologies [33, 37, 38]. In the present study, we observed a significant increase in the number of monocyte/macrophage and the presence of protein-3NT in the myocardium of MCP mice with heart failure compared with age-matched WT controls. Treatment with CeO2 nanoparticles significantly decreased the circulating levels of NOx, the infiltration of monocyte/macrophage, and the accumulation of 3-NT in the myocardium resulting in preservation of cardiac function, suggesting that cardiac oxidative stress may contribute to the progression of heart failure in this model. Our data are consistent with the previous observations regarding the effect of CeO2 nanoparticles on oxidative stress [30–32]. It has been suggested that the potent antioxidant property of CeO2 nanoparticles may be because of its presence of the mixed valence state of Ce3+ and Ce4+ that could result in an autoregenerative reaction cycle (Ce3+ → Ce4+ → Ce3+) [30–32].
Oxidative stress activates redox-sensitive transcription factors (NF-kB and activating protein-1), which in turn lead to the transcription of many inflammation-related genes. Proinflammatory cytikines as TNF-α, IL-1β and IL-6 have been identified as contributors to the underlying cardiomyopathyic processes of progressive cardiac dysfunction [1, 5, 39].These cytokines are produced by all nucleated cell types residing in the myocardium possibly through mechanisms such as reactive oxygen species (ROS)-induced intracellular signal transduction (e.g., NF-kB activation) [34, 35]. Both TNF-α and IL-6 are also the central mediator of the acute phase response and the main inducer of C-reactive protein production in liver, which, along with TNF-α and IL-6, has been associated with coronary heart disease and heart failure (39). Our data showed that the infiltrated inflammatory cells in the interstitial of myocardium were the main cellular source of TNF-α and IL-1β production, whereas cardiac myocytes were a main cellular source of IL-6 production. The production of cytokines TNF-α, IL-1β, and IL-6 were significantly increased at mRNA transcript in the myocardium of MCP mice, and the increased production of these cytokines in the myocardium of MCP mice were significantly suppressed by treatment with CeO2 nanoparticles. We also showed that the increased levels of circulating CRP and MCP-1 in the MCP mice were also markedly reduced by CeO2 nanoparticles treatment. The circulating levels of MCP-1 may be produced by targeted expression of MCP-1 transgene in the myocardium and as the result of the inflammatory process caused by the expression of the MCP-1 gene. The observed reduction in circulating MCP-1 levels as well as the reduction in CRP levels probably represent the anti-inflammatory effects of CeO2 nanoparticles due to its potential autoregenerative antioxidant properties.
Recently, ER stress pathways have been reported to also be activated by various stresses such as oxidative stress, hypoxia, and ischemia [19, 20]. Studies have demonstrated that pressure overload induces prolonged ER stress which contributes to cardiac myocyte apoptosis during the progression of cardiac hypertrophy to failure [16]. We previously showed that ER stress is involved in the pathophysiology of the development of heart failure in this mouse model [13]. Although the causative role has been demonstrated in most of these disease conditions, possible mechanisms by which ER stress could initiate or promote a disease state is not completely understood. ER stress has been shown to promote apoptosis through the activation of the transcription factor GADD153/CHOP and caspase-12 [9, 40, 41]. In the present study, ER stress-associated genes Grp78, PDI, HSP25, HSP40 as well as HSP70 mRNA and protein levels were significantly increased in the myocardium of the vehicle-treated MCP mice that is consistent with the histological finding showing extensive apoptotic cell death in the myocardium. Treatment with CeO2 nanoparticles markedly suppressed the expression of above ER stress-associated genes at mRNA and protein levels and attenuated apoptotic cell death in the myocardium, resulting in significant preservation of LV dimension with less ventricular dilatation and slightly decreased fractional shortening. Since CeO2 nanoparticles acts as a free radical scavenger, the important findings that emerged in the present study is that treatment with CeO2 nanoparticles might prevent pathological inflammatory disorders by blocking oxidative stress and the resulting ER stress. This hypothesis was supported by the recent findings that ER stress activates cleavage of CREBH to induce a systemic inflammatory response [21].
In summary, this represents the first demonstration that treatment with CeO2 nanoparticles attenuated the progressive cardiac dysfunction and remodeling in a murine model of ischemic cardiomyopathy. The beneficial effect of CeO2 nanoparticles is a result of its inhibition of myocardial oxidative stress, ER stress and inflammatory processes through its potential autoregenerative antioxidant properties.
Acknowledgments
This work was supported by grant HL-69458 from the National Institutes of Health. We thank Isagani Santos and Charlene Ross for the care of the transgenic animals used in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aukrust P, Gullestand L, Ueland T, Damas JK, Yndestad A. Inflammatory and anti-inflammatory cytokines in chronic heart failure: potential therapeutic implications. Ann Med. 2005;37:74–85. doi: 10.1080/07853890510007232. [DOI] [PubMed] [Google Scholar]
- 2.Grieve DJ, Byme JA, Cave AC, Shah AM. Role of oxidative stress in cardiac remodeling after myocardial infarction. Heart Lung Circ. 2004;13:132–8. doi: 10.1016/j.hlc.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Molavi B, Mehta JL. Oxidative stress in cardiovascular disease: molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr Opin Cardiol. 2004;19:488–93. doi: 10.1097/01.hco.0000133657.77024.bd. [DOI] [PubMed] [Google Scholar]
- 4.Kameda K, Matsunaga T, Abe N, Hanada H, Ishizaka H, Ono H, et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease: possible role for left ventricular remodeling. Eur Heart J. 2003;24:2180–5. doi: 10.1016/j.ehj.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol. 2003;65:81–101. doi: 10.1146/annurev.physiol.65.092101.142249. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman RJ. Stress signaling from the lumen of endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–33. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 7.Morri K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–4. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 8.Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–83. doi: 10.1016/s0143-4160(03)00139-8. [DOI] [PubMed] [Google Scholar]
- 9.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type II diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 11.Emadali A, Nguyen DT, Rochon C, Tzimas GN, Metrakos PP, Chevet E. Distinct endoplasmic reticulum stress responses are triggered during human liver transplantation. J Pathol. 2005;207:111–8. doi: 10.1002/path.1798. [DOI] [PubMed] [Google Scholar]
- 12.Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol. 2004;10:1699–708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291:H1411–20. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–17. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martindole JJ, Fernansez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. ER stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 16.Okada KI, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–12. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 17.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infracted mouse heart and hypoxic cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 18.Weeks J, Morrison A, Mullen R, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–16. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 19.Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, et al. Activating transcription factor 4 is translationally regulated by hypoxia stress. Mol Cell Biol. 2004;24:7469–82. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilmore WJ, Kirby GM. Endoplasmic reticulum stress due to altered cellular redox status positively regulates murine hepatic CYPZA5 expression. J Pharmacol Exp Ther. 2004;308:600–8. doi: 10.1124/jpet.103.060111. [DOI] [PubMed] [Google Scholar]
- 21.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–99. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 22.Kolattukudy PE, Quach T, Bergese S, Breckenridge S, Hensley J, Altschuld R, et al. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–11. [PMC free article] [PubMed] [Google Scholar]
- 23.Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, Kolattukudy PE. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res. 2000;87:378–84. doi: 10.1161/01.res.87.5.378. [DOI] [PubMed] [Google Scholar]
- 24.Niu J, Azfer A, Deucher MF, Goldschmidt-Clermont PJ, Kolattukudy PE. Targeted cardiac expression of soluble Fas prevents the development of heart failure in mice with cardiac-specific expression of MCP-1. J Mol Cell Cardiol. 2006;40:810–20. doi: 10.1016/j.yjmcc.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu J, Azfer A, Kolattukudy PE. Monocyte-specific Bcl-2 expression attenuates inflammation and heart failure in monocyte chemoattractant protein-1 (MCP-1)-induced cardiomyopathy. Cardiovasc Res. 2006;71:139–48. doi: 10.1016/j.cardiores.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izu N, Shin W, Matsubara I, Murayama N. Development of resistive oxygen sensors based on cerium oxide thick film. J Electroceramics. 2004;13:703–6. [Google Scholar]
- 27.Jasinski P, Suzuki T, Anderson HU. Nanocrystalline undoped ceria oxygen sensor. Sensors and Actuators B. 2003;95:73–7. [Google Scholar]
- 28.Masui T, Ozaki T, Machida K, Adachi G. Preparation of ceria-zirconia sub-catalysts for automotive exhaust cleaning. J Alloys Compd. 2000;303:49–55. [Google Scholar]
- 29.Robinson RD, Spanier JE, Zhang F, Chan SW, Herman IP. Visible thermal emission from sub-band gap laser excited cerium dioxide particles. J Appl Phys. 2002;92:1936–41. [Google Scholar]
- 30.Chung D. Nanoparticles have health benefits too. New Scientist. 2003;179:2410–6. [Google Scholar]
- 31.Schubert D, Dargusch R, Raitano J, Chan SW. Cerium and yttrium oxide nanoparticles are neuroprotective. Biochem Biophy Res Commun. 2006;342:86–91. doi: 10.1016/j.bbrc.2006.01.129. [DOI] [PubMed] [Google Scholar]
- 32.Tarnuzzer RW, Colon J, Patil S, Seal S. Vacancy engineered ceria nanostructures for protection from radiation-induced cellular damage. Nano Lett. 2005;5:2573–7. doi: 10.1021/nl052024f. [DOI] [PubMed] [Google Scholar]
- 33.Viappiani S, Schulz R. Detection of specific nitrotyrosine-modified proteins as a marker of oxidative stress in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2006;96:H2167–8. doi: 10.1152/ajpheart.00128.2006. [DOI] [PubMed] [Google Scholar]
- 34.Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–24. [PubMed] [Google Scholar]
- 35.Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–74. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- 36.Sandhu JK, Robertson S, Birnboim HC, Goldstein R. Distribution of protein nitrotyrosine in synovial tissues of patients with rheumatoid arthritis and osteoarthritis. J Rheumatol. 2003;30:1173–81. [PubMed] [Google Scholar]
- 37.Cai L, Wang L, Zhou G, Chen T, Song Y, Li X, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–97. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 38.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87:241–7. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 39.Pai JK, Pischon T, Ma J, Manson JE, Hankinson SE, Joshipura K, et al. Inflammation markers and the risk of coronary heart disease in men and women. N Engl J Med. 2004;351:2599–610. doi: 10.1056/NEJMoa040967. [DOI] [PubMed] [Google Scholar]
- 40.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2004;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 41.Oyadomari S, Mori M. Role of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]