

Abstract
Nitroalkane oxidase (NAO) from Fusarium oxysporum catalyzes the oxidation of neutral nitroalkanes to the corresponding aldehydes or ketones with the production of H2O2 and nitrite. The flavoenzyme is a new member of the acyl-CoA dehydrogenase (ACAD) family, but it does not react with acyl-CoA substrates. We present the 2.2 Å resolution crystal structure of NAO trapped during the turnover of nitroethane as a covalent N5–FAD adduct (ES*). The homotetrameric structure of ES* was solved by MAD phasing with 52 Se-Met sites in an orthorhombic space group. The electron density for the N5-(2-nitrobutyl)-1,5-dihydro-FAD covalent intermediate is clearly resolved. The structure of ES* was used to solve the crystal structure of oxidized NAO at 2.07 Å resolution. The c axis for the trigonal space group of oxidized NAO is 485 Å, and there are six subunits (1½ holoenzymes) in the asymmetric unit. Four of the active sites contain spermine (EI), a weak competitive inhibitor, and two do not contain spermine (Eox). The active-site structures of Eox, EI, and ES* reveal a hydrophobic channel that extends from the exterior of the protein and terminates at Asp402 and the N5 position on the re face of the FAD. Thus, Asp402 is in the correct position to serve as the active-site base, where it is proposed to abstract the α proton from neutral nitroalkane substrates. The structures for NAO and various members of the ACAD family overlay with root-mean-square deviations between 1.7 and 3.1 Å. The homologous region typically spans more than 325 residues and includes Glu376, which is the active-site base in the prototypical member of the ACAD family. However, NAO and the ACADs exhibit differences in hydrogen-bonding patterns between the respective active-site base, substrate molecules, and FAD. These likely differentiate NAO from the homologues and, consequently, are proposed to result in the unique reaction mechanism of NAO.
Nitrochemicals are widely distributed throughout the environment from both natural and human sources. They are present in rocket fuels, wastes from the chemical industry, the exhaust from internal combustion engines, and tobacco smoke. They are also used as explosives, herbicides, pesticides, biocides, and drugs (1). Moreover, nitrated proteins, amino acids, and metabolites often possess antibiotic activity and can mitigate bacterial and/or fungal infection (2, 3). Some organisms, such as Penicillium atrovenetum (4) and Hippocrepis comosa, exploit these characteristics and produce nitroalkane compounds as a putative chemical defense mechanism (5, 6). Typically, the organisms that produce nitroalkane toxins also produce enzymes that transform the nitroaliphatic internally and thus protect the host from the toxic effects of the agent (4, 5). Likewise, some of the organisms targeted by nitroalkanes have also evolved strategies to detoxify these compounds. In fact, some microbes are able to exploit nitrochemicals so effectively that they can utilize these toxins as a sole source of nitrogen, carbon, and/or energy (7, 8). The soil fungus, Fusarium oxysporum, is an organism that effectively metabolizes a variety of nitroaliphatic compounds (9-12).
There are several examples of flavoenzymes that transform nitroalkanes via oxidative reactions. These include 2-nitropropane dioxygenase (2-NPD) from Hansenula mrakii (13) or Neurospora crassa (14), nitroalkane dioxygenase from Streptomyces ansochromogenes (15), 3-nitropropionate oxidase from either Penicillium atrovenetum or Hippocrepis comosa (4, 5), nitroalkane oxidase (NAO)1 from Fusarium oxysporum (9), glucose oxidase from Aspergillus niger (16, 17), and d-amino acid oxidase from mammals (18). However, NAO reacts preferentially with neutral nitroalkanes (9, 19), whereas all of the other nitrite-eliminating enzymes react with the anionic form of the nitro substrate. The reactions of glucose oxidase and d-amino acid oxidase with nitro-containing substrates are not physiological (16, 18). In contrast, the reactions catalyzed by each of the other oxidases do provide obvious advantages, such as the ability to obtain all nitrogen required for growth from a variety of nitrochemicals.
NAO from F. oxysporum has been very well-characterized in recent years (reviewed in ref 20). The enzyme is produced in large quantities when cultures are grown on nitroethane (9), and reasonable physiological roles for the enzyme include (a) protecting the fungus from nitroalkane toxins produced by plants (6), (b) detoxifying antibiotics produced by the competing soil microbes such as Pseudomonas fluorescens (21), and/or (c) scavenging nitrogen from a variety of nitrochemicals. The molecular weight of the 439-residue monomeric subunit is 47 955, but analytical ultra-centrifugation suggests that the enzyme exists in solution as a homotetramer/homodimer mixture (22). Amino acid sequence comparisons indicate that NAO is homologous to the acyl-CoA dehydrogenase (ACAD) family of flavoproteins, with identities that range from 23 to 27% and similarities as high as 46% (23). In contrast to most mitochondrial ACAD family members, NAO has amino acid sequence homology in the C-terminal region that likely targets it for the peroxisome (24-27). Moreover, NAO does not oxidize acyl-CoA substrates but rather has a marked preference for unbranched, primary nitroalkanes containing four or more carbon atoms (10, 28).
Mechanistic studies of NAO, in particular with nitroethane as the substrate, have established many of the details of the catalytic cycle (20). The reductive half-reaction is initiated by the abstraction of the α proton of the substrate by Asp402 (step 1 in Scheme 1), and the formation of the nitroethane anion is rate-limiting (28, 29). The substrate carbanion has been definitively established as an intermediate in the catalytic cycle (30), a unique feature of the NAO reaction among flavoenzymes. The nitroethyl-anion adds to the flavin N5 to form a covalent intermediate (step 2). The loss of nitrite forms an electrophilic imine cation, a reaction of which with hydroxide leads to aldehyde formation and reduced enzyme (steps 3–5). To complete the reaction cycle, the reduced flavin is rapidly oxidized by O2 to yield H2O2 (step 6). The electrophilic imine cation formed in step 3 can be trapped by alternative nucleophiles, and trapping by cyanide has been used to demonstrate directly the involvement of this species in catalysis (31). The addition of a nitroethane anion to the electrophilic imine intermediate formed during turnover with nitroethane (step 8) yields inactive enzyme (ES*). Mass spectrometric analysis of the flavin species released from ES* at low pH is consistent with a N5-(2-nitrobutyl)-1,5-dihydro-FAD adduct (32). The ES* complex is thus a trapped reaction cycle intermediate. The remarkable stability of ES* at 4 °C and neutral pH enabled us to crystallize the ES* form of the enzyme. We solved the crystal structure with selenomethionine (Se-Met)-enriched protein and MAD-phasing techniques (33, 34) using the 52 Se-Met sites of the homotetramer. To our knowledge, this is the first crystal structure of a flavoprotein trapped during the turnover of authentic substrates as a covalent adduct at N5–FAD. We also report the structure of oxidized NAO, in which we observe a spermine molecule bound in some of the active sites. Together, these structures provide insights into (a) the substrate preference and access to the FAD, (b) the reaction mechanism, and (c) the features that differentiate NAO from structural but not functional homologues in the ACAD family.
Scheme 1.

Proposed NAO Reaction Mechanism with Nitroethane as a Substrate (29)a
a The imine reaction intermediate after step 3 can react with hydroxide to release the aldehyde followed by oxidation by O2 (steps 4–6). Alternatively, the intermediate can react with a nitroalkane anion (step 8) to generate the ES* complex.
MATERIALS AND METHODS
Crystallization
Recombinant nitroalkane oxidase was expressed and purified from Escherichia coli strain BL21 (DE3) transformed with pETNAO4 as previously described (23, 35). The purified enzyme was stored in 20 mM HEPES at pH 8.0, 1 mM EDTA, and 5% glycerol, and aliquots were flash-frozen with liquid N2. Se-Met-enriched NAO was obtained by expression in the methionine auxotroph E. coli B834 (DE3) (Novagen) as previously described (36). Each subunit of NAO contains 13 methionine residues, excluding the N-terminal residue (23). To generate the covalent adduct, 25 μM NAO was incubated with 28 mM neutral nitroethane and 9 mM anionic nitroethane in 500 mM HEPES at pH 8.0 for 1 h at 25 °C. The enzyme was then exchanged into 20 mM HEPES at pH 8.0, 1 mM EDTA, and 5% glycerol at 4 °C using an Ultrafree-15 centrifugal filter device (Millipore Co., Bedford, MA), and aliquots were flash-frozen with liquid N2. Crystals of oxidized NAO in the trigonal space group were obtained by mixing 2 μL of 10 mg/mL protein solution (10 mM sodium cacodylate at pH 7.5) with 2 μLof reservoir solution containing 25% (w/v) PEG 4000 and 35% (v/v) glycerol, sodium cacodylate trihydrate (200 mM, pH 7.5), and 1 mM spermine hydrochloride at 4 °C, as previously reported (36). Microcrystalline showers of NAO–ES* were initially obtained from a flexible sparse matrix screen (37). These conditions were optimized, and diffraction quality crystals were grown by mixing 2 μL of 10 mg/mL protein solution (10 mM PIPES at pH 7.5) with 2 μL of reservoir solution containing 20% (w/v) PEG 1500 and 300 mM ammonium sulfate and 150 mM PIPES at 4 °C. The crystals typically grew within 20 days with the hanging drop, vapor diffusion method. They were essentially colorless, in contrast to oxidized NAO, which yields yellow crystals. Single crystals of the Se-Met-enriched form of ES* were grown from similar conditions, except for the addition of 10 mM tris(2-carboxyethyl) phosphine hydrochloride to the crystallization drop. Single crystals were mounted in nylon loops (no cryoprotectant was necessary) and flash-frozen by quick submersion into liquid N2. All X-ray diffraction data were collected from crystals held at approximately 100 K.
Data Collection, Phasing, and Refinement
Data collection and analysis for the natural form of oxidized NAO in space group P3221 is described elsewhere (36). Additional data collection, phasing, and refinement statistics are presented in Table 1. The unit cell of the crystals of oxidized NAO contains a c axis edge of 485 Å, has a low mosaic spread (0.3°), and diffracts to at least 1.6 Å resolution. However, the complete data have been collected to 2.07 Å because of the complexity of data collection with crystals that contain a very long unit cell edge. The Se-Met enriched ES* complex was used to collect a complete and highly redundant three wavelength MAD dataset at the IMCA-CAT beamline 17-ID using an ADSC Q210 detector. The data were integrated, merged, and scaled using MOSFLM/SCALA (38, 39). The three-wavelength MAD dataset of Se-Met ES* was further scaled and analyzed with FHSCALE (40) and SCALEIT (41) from the CCP4 suite of programs (42). MAD analysis and phase determination were carried out in SOLVE (43). Initially, 30 Se-Met sites were located in space group P212121 and produced a figure of merit of 0.47 and a z score of 251. An additional 22 Se-Met sites were located with SHARP (44) by analyzing maps phased with the original 30 Se-Met sites. All 52 Se-Met sites were then refined to convergence to 2.2 Å resolution in SHARP. Density modification was with SOLMON (45)in CCP4, and initial model building was with RESOLVE giant version 2.06 (46). The remaining portions of the model were built manually in O (47). The FAD covalent adduct was modeled using PRODRG server (48). The anomalous Se-Met library was derived from experimental f and f ″ values. The final structure of ES* was refined with the 0.9794 Å X-ray diffraction data set using maximum likelihood refinement in CNS version 1.1 (49). The refined structure of ES* has a R factor of 20.2% and a Rfree of 23.2% to 2.2 Å resolution. The structure of EI was solved by molecular replacement in space group P3221 using MOLREP (50). An ES* subunit was used as the search model, and six subunits or 1½ holoenzymes were located in the asymmetric unit. The structure of EI was refined with REFMAC 5.1 (42) to a R factor of 18.8% and a Rfree of 22.5% at 2.07 Å resolution. Our crystal structure of EI is among the highest resolution structure with a c axis greater than 485 Å deposited in the PDB to date. Except for Thr90 in all of the chains, all main-chain angles fall into most favored and additionally allowed regions of the Ramachandran plot. Eight residues at the C-terminal end of each subunit are not visible in the electron-density maps, suggesting that they are disordered. Figures were prepared with ALSCRIPT (51), PyMol (52), SwissPDBviewer (53), and Pov-Ray version 3.6.
Table 1.
Data Collection, Phasing, and Model Refinement Statistics for NAO–ES* and Oxidized NAO
| data collection | NAO–ES* covalent adduct (Se-Met) | oxidized NAO | ||
|---|---|---|---|---|
| wavelength (Å) | 0.9794 | 0.9611 | 0.9795 | 0.9 |
| space group | P212121 | P212121 | P212121 | P3221 |
| a (Å); b (Å); c (Å) | 90.4; 163.9; 173.4 | 90.4; 163.2; 173.5 | 90.5; 163.9; 173.8 | 103.4;103.4; 485.1 |
| resolution range (Å) | 40–2.2 | 40–2.64 | 40–2.85 | 50–2.07 |
| (high resolution shell) | (2.32–2.2) | (2.78–2.64) | (3.00–2.85) | (2.15–2.07) |
| total reflections | 1 496 015 | 1 023 723 | 850 177 | 570 374 |
| unique reflections | 130 876 | 76 209 | 61 184 | 371 128 |
| completeness (%) | 99.9 (99.9) | 99.9 (99.9) | 99.9 (99.9) | 93.5 (80.0) |
| redundancy | 11.4 | 13.4 | 13.9 | 1.5 |
| Rsym (%) | 0.105 (0.53) | 0.106 (0.422) | 0.097 (0.491) | 0.139 (0.30) |
| I/σ | 6.0 (1.5) | 5.4 (1.7) | 6.8 (1.5) | 12.6 (3.0) |
| phasing statistics of NAO–ES* after refining 52 Se-Met sites in SHARP | ||||
| Rcullis centric (iso) | 0 | 0.99 | 0.99 | |
| Rcullis accentric (iso/ano) | 0/0.758 | 0.978/0.840 | 1.0/0.839 | |
| phasing power centric (iso) | 0 | 0.351 | 0.138 | |
| phasing power (iso/ano) | 0/1.405 | 0.361/0.974 | 0.136/0.80 | |
| FOM | 0.13 (centric reflections) 7950; 0.32 (accentric reflections) 119 500 0.94 after solvent flattening in SHARP |
|||
| model refinement statistics | NAO–ES* | oxidized NAO | ||
| number protein residues | 1720 | 2580 | ||
| number water molecules | 694 | 1136 | ||
| resolution range (Å) | 40–2.2 | 50–2.07 | ||
| number of reflections | 130 649 | 171 484 | ||
| Rcryst (%) | 20.14 | 18.95 | ||
| Rfree (%) | 23.22 | 22.51 | ||
| Ramachandran statistics | ||||
| most favored (%) | 91.8 | 93.3 | ||
| additional allowed (%) | 7.7 | 6.2 | ||
| generously allowed (%) | 0.2 | 0.3 | ||
| disallowed (%) | 0.3 | 0.3 | ||
| average B values (Å2) | ||||
| protein | 33.95 | 26.73 | ||
| water | 37.62 | 31.71 | ||
| FAD | 34.16 | 19.86 | ||
| 2-nitrobutyl | 46.31 (spermine) | |||
| ligand | 100% occupancy: 52.7 (C atoms); 75.7 (nitro group) |
50.23 (glycerol) | ||
| 80% occupancy: 46.4 (C atoms); 66.2 (nitro group) |
51.45 (PEG 4000) | |||
| rmsd from ideal geometry | ||||
| bond lengths (Å) | 0.006 | 0.013 | ||
| bond angles (deg) | 1.1 | 1.4 | ||
RESULTS AND DISCUSSION
Overview of the NAO Structures
The trapped intermediate of NAO, ES*, crystallizes at 4 °C in space group P212121 (Table 1) with one homotetramer per asymmetric unit [VM, 3.4 Å3/Da; 63% solvent (54, 55)]. The ES* structure has 222 local symmetry relating the four subunits to each other (Figure 1), and consequently, the holoenzyme approximates a tetrahedron shape. The α4 aggregate is approximately 100 Å wide (measured between Cα of Gly158a and Gly158b; the naming convention is residue type, number, subunit; for example, Asp402a is from subunit A) and approximately 80 Å tall (measured between Cα of Thr34b and Thr34c). The oxidized form of NAO crystallizes in space group P3221 (Table 1) from slightly different conditions that include spermine tetrahydrachloride (36). The volume of the trigonal unit cell is greater than 4 570 000 Å3 and contains six monomers per asymmetric unit (VM, 2.6 Å3/Da; 53% solvent). These are arranged as one complete homotetramer and one-half of another holoenzyme. The additional dimer is oriented with a local 2-fold symmetry axis coincident with a crystallographic 2-fold axis, which results in a homotetramer spanning the unit cell boundary.
Figure 1.
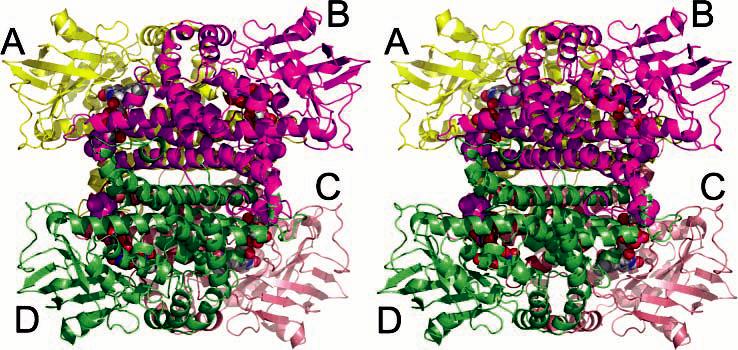
Divergent stereoview of the NAO homotetramer in the ES* structure. Each subunit is depicted as a differently colored Cα ribbon trace, and the FAD covalent adducts are shown in CPK atoms within each subunit. The A/B dimer consists of one purple and one yellow subunit.
All subunits in the asymmetric units of the oxidized NAO and the ES* structures have very similar structures with an average root-mean-squared difference for 430 Cα atoms between 0.22 and 0.33 Å, despite the fact that noncrystallographic symmetry was not restrained during the refinements. Previous biochemical studies (22, 32) and our crystal structures suggest that NAO is a homotetramer with homology similar to that of the medium-chain acyl-CoA dehydrogenase (MCAD) (56-58). The labeling of the secondary structure of the NAO subunits is also analogous to the MCAD (Figure 2A and Supplementary Figure S1 in the Supporting Information). Each subunit of NAO is comprised of two α-helical domains, one at the N-terminal region (residues 1–125) and one at the C-terminal region (residues 260–438). A β-sheet domain (residues 125–260) is located in the central region. The C-terminal H, I, J, and K α helices stabilize the α4 quaternary structure. In contrast, the β-sheet domains are located on the exterior, approximately at the points of the tetrahedron.
Figure 2.
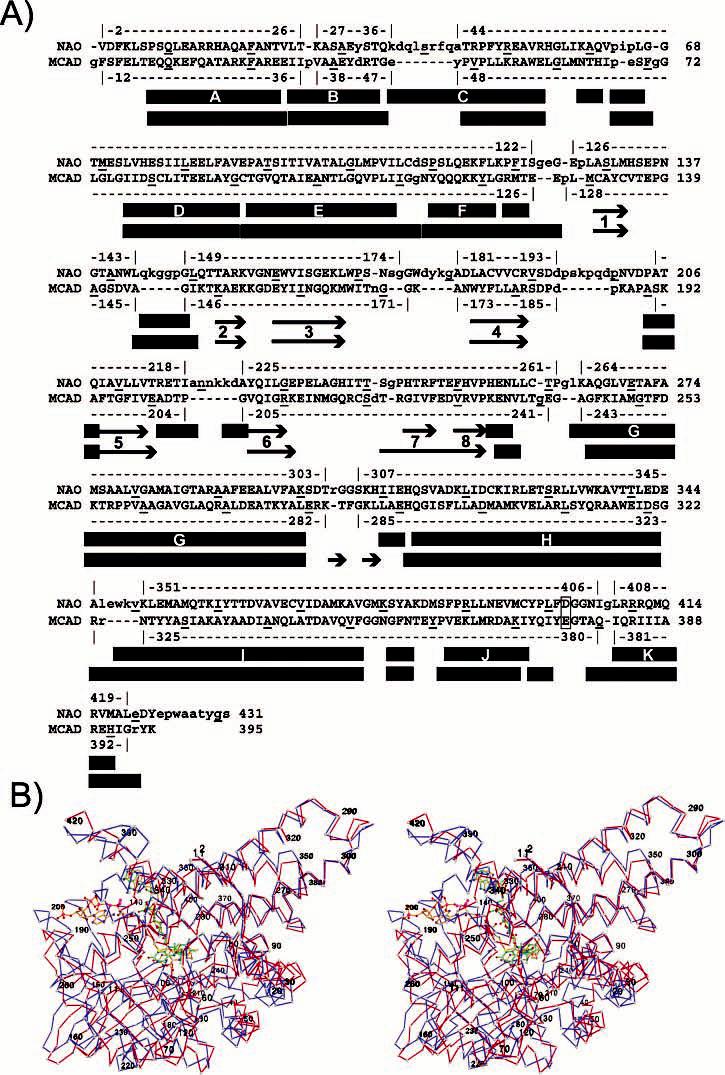
Structure-based amino acid sequence alignment between NAO–ES* and MCAD–C8S (PDB code 1udy) (76). (A) α4 holoenzyme for each was superimposed with the secondary-structure matching web server (75). The structural homology used for the rmsd comparison is indicated above and below each sequence, where rectangles and arrows indicate α helices and β strands, respectively, for NAO (top) and MCAD–C8S (bottom). Every 10th residue is underlined, and the active-site base is highlighted in yellow. A complementary CLUSTALW sequence alignment of NAO and other family members is shown in Supplementary Figure S1 in the Supporting Information. (B) Divergent stereoview of the Cα chain trace for subunit A of NAO–ES* (blue) superimposed upon subunit A of the MCAD–C8S complex (red). The NAO–ES* trace is labeled at residues 2, 20, 40, etc., and the N5–FAD adduct is shown in a ball-and-stick model with green bonds. The MCAD–C8S complex is labeled at residues 11, 31, 50, etc., and the FAD and substrate analogue are shown as a ball-and-stick model with orange bonds.
The homotetramer can also be described as a dimer of dimers, in which an A/B dimer interacts with a C/D dimer. However, subunit A forms extensive interfaces with both subunits B and C. In contrast, only the side chains from Gln314a, Ser315a, and Asp318a form hydrogen bonds across a 2-fold axis with residues from subunit D. The buried surface area at the A/C dimer interface is slightly greater (∼5270 Å2; 16.5% of the total solvent-accessible surface area for the subunit) than that in the A/B dimer (∼4560 Å2; 14% of the total). The interface between the A/C and B/D dimers is stabilized by several types of interactions. For example, residues from subunit A along helix K and at the C-terminal region form hydrogen bonds with residues from helix H and G of subunit C. In addition, four salt bridges stabilize the A/C interface: Lys324a–Glu353c, Arg410a–Asp318c, Arg410a–Asp322c, and Arg411a–Asp318c.
Architecture of the Active Site
The active site within each subunit is clearly established by the isoalloxazine ring of the FAD. The 2mFo – DFc electron density for the covalent adduct in the ES* structure is resolved in each active site (parts A and B of Figure 3). The electron-density map of oxidized NAO indicates that four of the active sites contain spermine (EI), a weak competitive inhibitor (Ki ∼ 200 mM at pH 8) and two do not contain spermine (Eox) (parts C and D of Figure 3). Together, these structures establish the essential features of each active site, which is comprised of residues donated principally from one subunit and a 2-fold related subunit.
Figure 3.
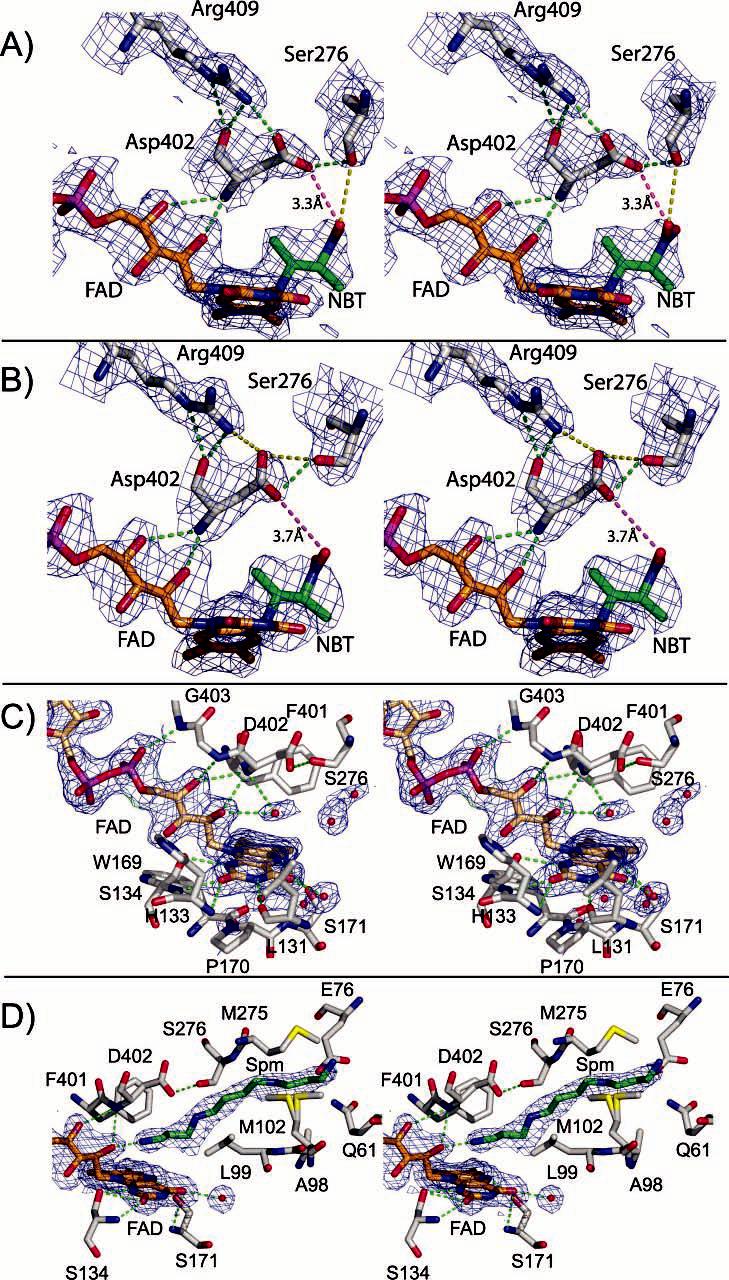
Divergent stereoviews of the typical 2mFo – DFc electron-density maps at the active sites. The 1σ maps are calculated to 2.07 and 2.2 Å resolution for oxidized NAO and NAO–ES*, respectively. (A) Subunit A of NAO–ES* with hydrogen bonds indicated by green dashed lines and ionic interactions indicated by a red dashed line and distance indicator. The 2-nitrobutyl moiety of the FAD adduct is labeled as NBT. (B) Subunit B of NAO–ES* illustrating an alternative orientation of Asp402 and Ser276. (C) Subunit A of oxidized NAO illustrating several solvent molecules in the active site. (D) Subunit B of oxidized NAO illustrating a spermine molecule within the active site.
The FAD binds noncovalently in an extended conformation to each NAO subunit (Figures 3 and 4). The isoalloxazine ring is located between the β-sheet domain on the si face and by α-helices E, G, F1, and the J–K loop on the re face. The FAD pyrophosphate moiety interacts with the guanidino group of Arg304b as well as the positive dipole at the N-terminal end of α-helix K from subunit A. The ribose, pyrophosphate, ribityl chain, and dimethylbenzene ring on one side of the FAD interact with a turn after α-helix I in subunit B, which contains residues Ala376b–Met379b. The adenine moiety extends to the dimer–dimer interface. At that location, it interacts with His313b via π-stacking and hydrogen bonds with the side chains of Gln314c and Asn141a. Residues from subunits A and B exclude approximately 63% (∼700 Å2) and 35% (∼400 Å2) of the FAD adduct from the solvent, respectively. Only about 2% (∼24 Å2) of the surface area of the FAD adduct in subunit A of NAO–ES* is exposed to the solvent. Approximately half of this is associated with the amine group on the adenine ring at the A–C subunit interface. The remaining 1% is associated with a solvent-excluded cavity at the si face of the dimethylbenzene ring of the FAD (see below).
Figure 4.
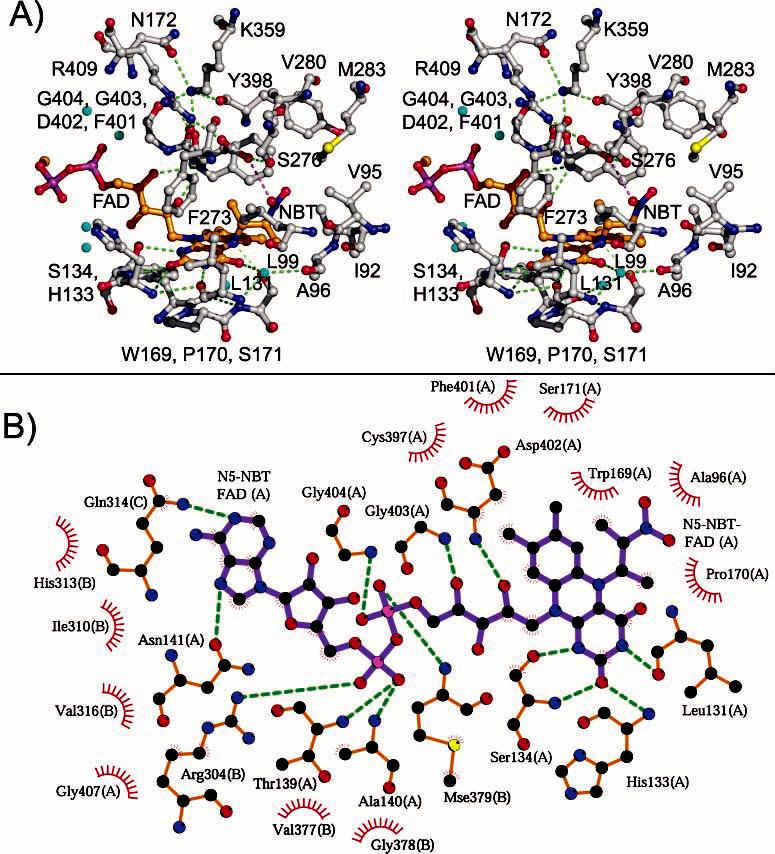
Environment around the N5-(2-nitrobutyl)-1,5-dihydro-FAD moiety. (A) Divergent stereoview with C, O, N, and S atoms colored gray, red, blue, and yellow, respectively. Bonds for the FAD are orange, and those of Asp402 and the nitrobutyl adduct are magenta. Hydrogen-bond interactions are indicated by green dashed lines. (B) Schematic view from LIGPLOT.
The crystal structures presented here correlate well with previous biochemical studies and confirm the presence of Cys397, Tyr398, and Asp402 in the active site (35, 59, 60). For example, Tyr398 was identified by tetranitromethane modification, which resulted in inactivation of the enzyme (60). Our structures show that the side chain of Tyr398 is located approximately 8 Å from the dimethylbenzene ring of the FAD and just over 10 Å from the nitro group of the nitrobutyl adduct in NAO–ES* (Figure 4). However, the residue is adjacent to Phe401 and Met283, both of which form van der Waals contacts with the nitrobutyl adduct. Therefore, modification of Tyr398 by tetranitromethane most likely inactivates the enzyme by either (a) forcing alternative side-chain orientations of these latter residues that subsequently prevent substrate association, (b) forcing the side chain of Asp402 into a nonproductive orientation, or (c) a combination of these effects. Similarly, Cys397, which was identified by chemical modification by N-ethylmaleimide (59), is less than 4 Å from both Phe401 and the FAD dimethylbenzene ring. Asp402 has been identified as the essential active-site base by several types of experiments (23, 30, 35). As illustrated by Figures 2A, 3, and 4, the backbone atoms of Asp402 are located within a loop connecting α-helices J and K (residues Pro399–Asn405). As a consequence of the distinct turn formed by this loop, the carbonyl oxygen atoms of Tyr398, Phe401, and Gly403 and side-chain oxygen of Asn405 are forced into relatively close proximity to each other. The resulting negative electrostatic potential is further enhanced by the C-terminal end of α-helix J. These influences are compensated for by the positively charged Nε atoms of Lys359 via hydrogen-bonding and Coulombic interactions (Figure 4). The turn positions the amide proton of Asp402 so that it can form a hydrogen bond with the O2 atom on the FAD ribityl chain. Together, these interactions orient the J–K loop such that Asp402 is directly above the re face and the N5 atom of the FAD.
Distortion of the FAD Isoalloxazine Ring
The electron-density maps for the isoalloxazine rings in Eox and EI indicate that they are distorted from planarity by approximately 15° (Figures 3 and 5 and Supplementary Table S1 in the Supporting Information). The piperizine ring in the ES* structure is also puckered at the N5 atom and, consequently, yields a larger distortion (∼21°). These features are consistent with a reduced FAD in the N5 adduct. In each case, the deviation from planarity is principally along the N5–N10 axis, which bisects the essentially planar dimethylbenzene and pyrimidine ring systems. The position of the FAD isoalloxazine ring is stabilized through several hydrogen bonds with the pyrimidine ring (Figures 3 and 4 and Supplementary Figure S2 in the Supporting Information). Numerous hydrophobic interactions with the dimethylbenzene ring are also present, including a nearly perpendicular relationship between the aromatic ring of Phe401 and the re face of the FAD dimethybenzene ring (Figures 4 and 6A). Such a T-shaped configuration is known to stabilize benzene dimers and substituted benzene dimers by interactions between the electronegative π obitals of one aromatic ring and the electropositive edge of the second aromatic ring (61). Aromatic stacking between the si-face FAD and Trp169 also likely influences the two-electron reduction potential of the enzyme, which has been documented for some members of the ACAD family (62).
Figure 5.
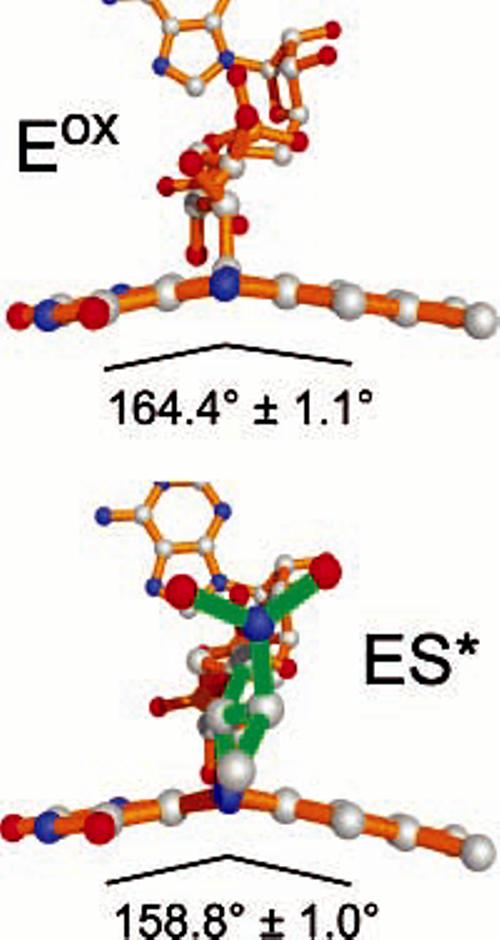
Isoalloxazine rings display deviations from planarity in both Eox and ES* structures. The angle shown is the average of all of the active sites in each asymmetric unit and depicts the C4–N5–C6 angle. Bonds for the FAD are shown in orange, and those of the nitrobutyl adduct are shown in green.
Figure 6.
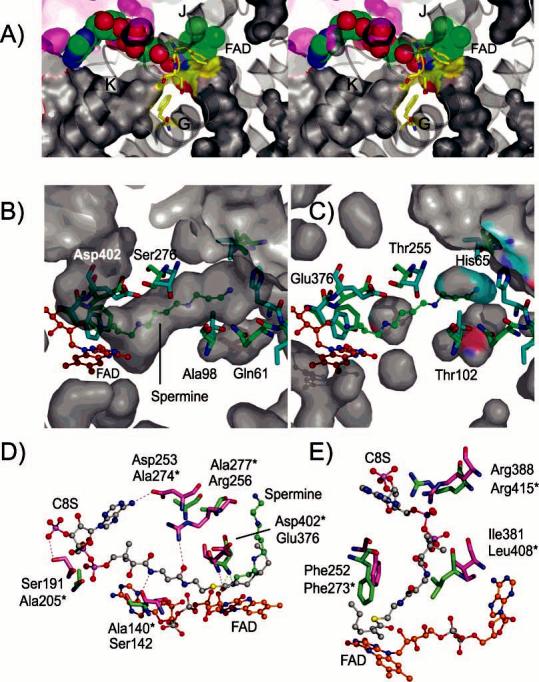
Comparison of substrate-binding channels in NAO and the MCAD–C8S complex. (A) Divergent stereoview of the solvent-accessible surfaces around the FAD in subunit A of the NAO Eox active site. The FAD is shown as CPK atoms with carbon, nitrogen, oxygen, and phosphorus atoms colored green, blue, red, and magenta, respectively. The active-site base, Asp402, as well as Phe401 and Phe273 are shown as sticks with carbon atoms colored yellow. The surfaces are colored according to the atoms that form the border and with gray surfaces from subunit A and purple surfaces from subunit B. The N5 channel extends from the bottom right to the right of α-helix G and terminates under the Phe401–Asp402 residues. The N1 channel extends from the bottom left, under the α-helix K, and terminates near Phe273 from α-helix G. (B) N5 substrate-access channel in NAO is occupied by a spermine molecule in subunit B of oxidized NAO (EI). For a comparison, several residues from the superimposed structure of the MCAD–C8S complex (PDB code 1udy) are shown in cyan. The NAO residues are labeled and shown in green, and the FAD residues are labeled and shown in orange. (C) Corresponding solvent-accessible surface areas in the MCAD–C8S complex. The MCAD residues are labeled and colored as in B. For a comparison, several residues from the superimposed structure of NAO–EI are shown in green. The MCAD–C8S cavities are interrupted by Thr255 and blocked from the exterior by His65. (D) Residues that form hydrogen bonds to the C8S molecule in the MCAD–C8S complex (magenta) and the corresponding residues from NAO (green, residue names with asterisks). (E) Some of the residues that constrict the N1 channel in NAO include Phe273, Leu408, and Arg415 and the corresponding residues in the MCAD–C8S complex.
While the isoalloxazine ring in free oxidized flavins in solution is essentially flat (63-65), a survey of the structures of oxidized flavoproteins by Lennon et al. (66) found that isoalloxazine rings in many flavoproteins are significantly nonplanar, with a mean angle of 7 ± 5° for the angle between the pyrimidine and dimethylbenzene rings. The similarities of the bending of the flavin in the Eox and ES* systems in NAO would be expected to make the flavin in Eox more susceptible to nucleophilic attack at N5. Moreover, the structure of the flavin adduct in NAO–ES* is very similar to reduced flavin in solution (67). Distortions of the isoalloxazine ring systems have also been observed recently for N5 and C4a covalent adducts of monoamine and polyamine oxidases (68, 69), which have structures that are completely unrelated to NAO.
Substrate Access to the Active Site
Each of the structures reveals several solvent-accessible channels or excluded cavities associated with the FAD (Figure 6A). The largest solvent-accessible channel extends from the exterior of the protein and terminates at the FAD ribityl chain and N1 atom and at the side chain of Phe273. The location of Phe273 prevents the channel from extending to Asp402, the active-site base. We define this as the N1 channel and note that it is roughly analogous to the acyl-CoA substrate-binding site in the ACAD family members. The N1 channel in NAO is lined with several charged and hydrophilic residues and contains several solvent molecules. However, several residues that confer substrate affinity for acyl-CoA substrates in members of the ACAD family are not present in the N1 channel of NAO (see below).
The second channel also extends from the protein exterior and terminates at the FAD N5 atom and residues Phe401 and Asp402. We define this as the N5 channel and suggest that aliphatic substrates may enter the NAO active site through this channel. The 26 Å long channel is funnel-shaped and passes through the interface formed by the N- and C-terminal α-helical domains. The exterior opening of the N5 channel is approximately 15 Å wide and is surrounded by hydrophobic residues (Ile64, Pro65, Leu66, Ile105, Leu106, and Trp348) and charged or polar residues (Glu71, Ser72, Thr271, Glu342, Glu344, and Lys351). In the middle, an approximately 9 Å diameter constriction is formed by the side chains of residues Val95 and Val280. The interior walls of the channel are bordered exclusively with hydrophobic residues from α-helices C1, D, E, and G. The total solvent-accessible volume of the N5 channel is approximately 345 Å3. The spermine molecules in the EI active sites occupy approximately 72% of that volume. This correlates with the kinetic preference of NAO for hydrophobic nitroalkane substrates with each CH2 group contributing ∼2.6 kcal mol−1 of binding energy (10). The spermine molecule shields the C4a and N5 atoms of the re face from exposure to the solvent. The isoalloxazine ring in the covalent adduct of ES* is similarly excluded from the bulk solvent.
A third, solvent-excluded cavity is located adjacent to the FAD si face. It contains five ordered solvent molecules, but only the dimethylbenzene portion of the isoalloxazine ring borders the dumbbell-shaped cavity. The rest of the cavity boundary is comprised of the side chains of several residues as well as portions of the protein backbone (Phe43, Ile92, Thr93, Trp169, Ser171, Asn172, Glu231, Pro232, Leu234, Thr240, and Gly242). The cavity is very close to the A/B subunit interface and is only excluded from the bulk solvent by the side chains of Leu168, Trp169, and Pro232. The location of the cavity on the opposite face of the FAD with respect to the adduct suggests that it may not influence the reaction in a significant manner.
Comparisons of the Eox, EI, and ES* Active Sites
Each subunit in the asymmetric units of either oxidized NAO or the ES* complex overlays with root-mean-square deviation (rmsd) values of approximately 0.27 Å for 429 Cα atoms and 0.34 Å for 6876 backbone atoms. The structural overlay of homotetramers of oxidized NAO and the ES* complex reveals small shifts of the residues in the J–K loop and in several residues bordering the isoalloxazine ring si face. Perhaps the most important of these shifts are illustrated in Figure 3 and parts A and B of Supplementary Figure S3 in the Supporting Information. In subunit A of the oxidized enzyme (Eox, Figure 3C and Supplementary Figure S3A in the Supporting Information), the side-chain oxygen atoms of Asp402 form hydrogen bonds with the OH moiety of Ser276 and with the guanidino group of Arg409. There is also a hydrogen bond between the Asp402NH and a solvent molecule (Wat297), which is poised above the N5 of the FAD. Most of these interactions are maintained in the EI and ES* active-site complexes, with the exception of the solvent molecules because they are displaced by ligand binding. However, a comparison of subunit A in the Eox and ES* structures shows that the orientations differ for the side chains of Ser276 and Asp402 (parts A and C of Figure 3 and Supplementary Figure S3A in the Supporting Information). Indeed, the side chains of Asp402 and Ser271 are rotated into an alternative orientation in ES* with respect to the Eox. As a result, Asp402 in ES* interacts with the guanidino group of Arg409 and with the nitro group of the adduct (3.3 Å). This suggests that Asp402 in subunit A of ES* may be protonated, suggesting that the proton abstracted from the substrate by the active-site base does not exchange with the solvent until the products dissociate. In contrast, a comparison of the active sites within subunit B from the oxidized NAO (EI) and the ES* complex illustrates another set of conformations (parts B and D of Figure 3 and Supplementary Figure S3B in the Supporting Information). In the EI active sites, the protons from the terminal cationic nitrogen (N14 atom) in spermine likely form hydrogen bonds with the FAD 2′-OH ribityl chain. Moreover, the side chain of Glu76 is poised to interact equally with the cationic spermine N10 and N14 atoms, whereas Asp402 interacts with the cationic spermine N1 and N5 atoms. Together, these interactions stabilize the binding of the inhibitor directly above the FAD re face. However, the side chains of both Asp402 and Ser276 are in similar orientations within the B subunits, as was observed for the A subunit of oxidized NAO (Eox). Therefore, Asp402 appears to have at least two possible orientations, which are coupled to the orientation of Ser276 as well. In subunit B of ES*, the side-chain oxygen atom of Asp402 is about 0.5 Å further away from the nitro group (3.7 Å) than in subunit A.
Stability of the N5–FAD Adduct
Previous results have shown that the nitrobutyl–FAD adduct isolated from NAO is stable at low pH but not in basic solution where it converts to oxidized flavin (32). A similar conversion occurs within the enzyme but at a rate at least 160 times slower. Our structural analysis of ES* suggests that the remarkable stability of the adduct within the protein is likely the result of several factors (Figure 4). First, the analysis of solvent accessibility indicates that the adduct is excluded from the bulk solvent. Thus, the active-site environment almost perfectly sequesters the adduct from additional molecular influences. Second, Gadda et al. (32) have suggested that decay of the nitrobutyl flavin adduct in ES* requires that N1 be anionic. However, within the enzyme, the negative charge on N1 may be significantly stabilized and delocalized through resonance with the O2 atom of the pyrimidine ring. Our structures suggest that both the N1 and O2 atoms form hydrogen bonds with two main-chain amide protons and the hydroxyl group of Ser134, respectively (Figure 4). These influences would reduce the charge density on the N1 atom and, consequently, stabilize the adduct. Third, some of the active sites of the ES* structure show that the nitro group of the adduct interacts with the side chain of Asp402, which also interacts with Arg409. It is possible that the nitro–Asp402 interaction may reduce the capacity of NO2− to serve as a favorable leaving group (step 8 in Scheme 1).
Comparisons of NAO with ACAD Family Members
The structural homologues of NAO include family members of the mitochondrial acyl-CoA dehydrogenases (ACAD) (58, 70), acyl-CoA oxidase (ACO) I from Arabidopsis thaliana (71), ACO-II from rat liver peroxisomes (72), the acyl-ACP dehydrogenase from polyketide biosynthetic pathways (73), and the human glutaryl-CoA dehydrogenase/decarboxylase (74). NAO shares approximately 24% sequence identity (∼42% similarity) with various ACAD family members but only 13–16% sequence identity with the two ACOs (Table 2). The regions of homology are spread throughout the entire primary sequence, with the exception of the C-terminal region of the ACOs (Figure 2 and Supplementary Figure S1 in the Supporting Information). The structural homology of the subunits among the family members is evident from the rms differences (Figure 2 and Table 2) that range from 1.6 Å for the MCAD to 3.1 Å for the homologous core of the ACO-II. The Q scores (75) range between 0.64 (MCADs) and 0.2 (ACOs). Comparisons of the α4 holoenzymes yield the rms differences and Q scores that are all slightly worse than overlays of individual subunits. For example, the α4 structures of NAO–ES* and the MCAD complex with 3-thiaoctanoyl-CoA (MCAD–C8S, PDB code 1udy) (76) superimpose with an rmsd of 2.1 Å. This yields a Q score of 0.54 and includes 1465 atoms from residues from each protein. The largest degree of structural homology occurs in α-helices G and H, which are regions that form the core of each α4 holoenzyme (Figure 2). In contrast, the overlay in the regions at the points of the tetrahedra (the β-sheet domains) and at the C-terminal end of each subunit differ more significantly. These regions with very different degrees of structural homology are also present for other members of the ACAD family (58, 71-74).
Table 2.
Sequence and Structural Homology between NAO–ES* and Selected Homologues
| subunit overlaya |
holoenzyme overlayb |
|||||||
|---|---|---|---|---|---|---|---|---|
| homologue | % identity (similarity) |
PDB code | Q score | rmsd (Å) | number of Cα atoms |
Q score | rmsd (Å) | number of Cα atoms |
| pMCADc | 23 (42) | 3mde | 0.63 | 1.67 | 369 | 0.31 | 1.76 | 745 |
| 1udy | 0.64 | 1.62 | 371 | 0.54 | 2.12 | 1465 | ||
| bMCADd | 25 (43) | 1ukw | 0.64 | 1.65 | 367 | |||
| hIVDHe | 18 (37) | 1ivh | 0.57 | 1.90 | 359 | 0.53 | 2.2 | 1476 |
| rSCADf | 22 (42) | 1jqi | 0.57 | 2.01 | 355 | 0.28 | 1.86 | 718 |
| bSCADg | 22 (41) | 1buc | 0.55 | 1.95 | 352 | 0.27 | 2.11 | 732 |
| GCADh | 16 (32) | 1siq | 0.47 | 2.09 | 343 | 0.39 | 2.63 | 1368 |
| 1sir | 0.51 | 1.96 | 349 | |||||
| ACPDi | 17 (33) | 1r2j | 0.48 | 2.15 | 327 | |||
| ACO-Ij | 16 (29) | 1w07 | 0.22 | 2.59 | 328 | 0.17 | 3.0 | 876 |
| ACO-IIk | 13 (26) | 1is2 | 0.21 | 3.11 | 331 | 0.14 | 3.3 | 837 |
Data from the secondary structure matching web server (75). The Q score is defined as NalgnNalgn/(1 + (rmsd/R0)2)Nres1Nres2, where Nalgn is the length of the alignment and Nres1 and Nres2 are the number of total residues in the NAO and the target PDB structure, respectively. Individual subunits for NAO–ES* and the target structure were superimposed.
The overlay was calculated for either an α2 or α4 holoenzyme.
Pig medium-chain acyl-CoA dehydrogenase (Sus scrofa).
Bacterial medium-chain acyl-CoA dehydrogenase (Thermus thermophilus Hb8).
Human isovaleryl-CoA dehydrogenase (Homo sapiens).
Rat short-chain acyl-CoA dehydrogenase (Rattus norvegicus).
Bacterial acyl-CoA dehydrogenase (Megasphaera elsdenii).
Glutaryl-CoA dehydrogenase decarboxylase (Homo sapiens).
Acyl-ACP dehydrogenase (Streptomyces hygroscopicus).
Acyl-CoA oxidase isoform I (Arabidopsis thaliana).
Acyl-CoA oxidase isoform II (Rattus norvegicus).
Members from both the ACAD and ACO families catalyze the oxidation of acyl-CoA thioesters to their corresponding trans-Δ2-enoyl-CoA. To our knowledge, none of these enzymes oxidize nitroalkanes. Moreover, NAO does not oxidize acyl-CoA substrates. Our structural analysis reveals important structural differences between NAO and the MCAD–C8S complex that may rationalize the lack of cross-reactivity. Perhaps the most important differences occur in the region of the subunit that provides binding interactions with the acyl-CoA substrate. For example, the Cα trace for NAO–ES* diverges quite significantly from MCAD–C8S in the corresponding regions that provide binding interactions with the adenine moiety of the acyl-CoA substrate (Figure 2B). Moreover, the side chains of Trp348 and Glu270 in NAO–ES* occupy the corresponding binding pocket for the adenine moiety of the acyl-CoA substrate in the MCAD–C8S complex. In addition, several substrate–protein hydrogen bonds in the MCAD–C8S complex are not possible for NAO. For example, Figure 6D shows that four residues that form hydrogen bonds with acyl-CoA substrates in the MCAD–C8S complex are substituted by alanine residues in NAO. As illustrated in Figure 2B, many of these features result in some of the largest differences in the N-terminal half of the MCAD–C8S and NAO–ES* structures.
The structure of the MCAD–C8S complex shows that the acyl-CoA substrate occupies the N1 channel adjacent to the ribityl portion of FAD (57, 76) (Figures 2B and 6D). In contrast, our structure of EI shows that spermine occupies the N5 channel on the opposite side of the FAD. Acyl-CoA substrates cannot bind productively to NAO via the N1 channel because it is interrupted by the side chains of Phe273, Leu408, and Arg415. In contrast to the N5 channel in NAO (Figure 6B), the N5 channel in MCAD does not extend to the exterior of the protein (Figure 6C). The side chain of His65 blocks the MCAD N5 channel, which is further interrupted on the interior by the side chains of Thr255 and Thr102. Consequently, the MCAD accommodates acyl-CoA substrates of limited size, but NAO binds a spermine molecule (14 atoms long) and oxidizes primary nitro substrates of various chain lengths.
The structural comparisons of NAO with ACADs also highlight differences in the hydrogen bonding associated with the active-site base, the FAD, and the substrate. The structural, mechanistic, mutagenic, and spectroscopic analyses of the MCAD support detailed proposals for the structural contributions of active-site residues to catalysis (58, 70). For example, the substrate binds to the MCAD along the N1 channel. The redox-active Cα-Cβ bonds of the substrate are situated between the isoalloxazine ring, the J–K active-site loop, and the active-site base (Glu376). This yields a set of hydrogen-bonding interactions that are proposed to not only orient the substrate and Glu376 but also increase the pKa of Glu376 and decrease the pKa of the substrate α proton (58, 70). Interestingly, the side chain of Arg256 in MCAD occupies an approximately analogous position as Arg409 in NAO. However, the guanidino group of MCAD–Arg256 interacts with the side chain of MCAD–Ser330 and the pantothenoic acid carbonoyl oxygen atoms of the acyl-CoA substrate. Consequently, the Cα–Cδ atoms of MCAD–Arg256 pack against the side-chain atoms of MCAD–Glu376, which contrasts with the hydrogen-bonding and ionic interactions between the side chains of NAO–Arg409 and NAO–Asp402. We anticipate that substrate binding to NAO may resemble the spermine orientation observed in the EI active sites and/or the 2-nitrobutyl portion of the ES* adduct. In the structures of NAO, the 2′- and 4′-hydroxyl groups of the FAD ribityl chain are hydrogen-bonded to the amide nitrogen of the active-site base, Asp402. The ribityl 4′-OH group also forms hydrogen bonds with the amide nitrogen of Gly403 and the carbonyl oxygen of Leu400. These interactions contrast with the MCAD–C8S complex, in which the substrate forms analogous hydrogen bonds. Consequently, the FAD in NAO is closer to the J–K active-site loop than in the ACAD family members. The protein–FAD interactions also terminate the N5 channel in NAO before it reaches Asp402.
CONCLUSIONS
Our crystal structures of NAO show that it has a similar structure to other members of the ACAD family. However, important differences include the substrate-access channel, the FAD microenvironment, and the hydrogen-bonding pattern of the active-site base with substrate molecules. Consequently, even though NAO and ACAD belong to the same family, they do not exhibit cross-reactivity with substrates for their reductive half-reactions. The stability of the N5–FAD adduct in NAO–ES* can be rationalized by structural features that include steric occlusion and hydrogen bonding that delocalizes the negative charge on N1 of the isoalloxazine ring.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by funds from the Georgia Tech Research Corporation, the Georgia Institute of Technology Office of the Vice Provost for Research, an American Chemical Society Petroleum Research Fund type G Grant (40310-G4), an American Heart Association Grant in Aid (0555286B) to A.M.O., and NIH Grant GM58698 to P.F.F. Portions of this research were carried out at (a) beamline X26C of the National Synchrotron Light Source, Brookhaven National Laboratory (supported by the U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences, under Contract Number DE-AC02-98CH10886), (b) with general user proposal time allotted at IMCA-CAT beamline 17-ID at the Advanced Photon Source (APS, IMCA-CAT is supported by the Industrial Macro-molecular Crystallography Association through a contract with Illinois Institute of Technology), (c) at BIOCARS beamline 14-BMC of the APS (supported by the National Institutes of Health, National Center for Research Resources, under Grant RR07707), and (d) at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the APS (SER-CAT supporting institutions may be found at www.ser-cat.org/members.html). We are grateful for many insightful discussions with Professors Giovanni Gadda and Dale E. Edmondson.
Footnotes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the corresponding file names: oxidized NAO, 2C12 and R2C12sf; NAO–ES*, 2C0U and R2C0Usf.
Abbreviations: NAO, nitroalkane oxidase; NAO–ES* or ES*, NAO trapped during the turnover of nitroethane as an N5-(2-nitrobutyl)-1,5-dihydro-FAD adduct; Eox, the subunits of oxidized NAO that do not contain spermine in the active site; EI, the subunits of oxidized NAO with spermine bound in the active site; ACAD, acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; MCAD–C8S, the MCAD complex with 3-thiaoctanoyl-CoA; ACO, acyl-CoA oxidase; Se-Met, selenomethionine.
SUPPORTING INFORMATION AVAILABLE
(Figure S1) Amino acid sequence and 2° structural alignments of NAO compared to several homologues. (Figure S2) LIGPLOT schematic representation of the protein–ligand interactions. (Figure S3) (A, top) Divergent stereoview comparing Eox (subunit A) with ES* (subunit A) and (B, bottom) divergent stereoview comparing EI (subunit B) with ES* (subunit B). (Table S1) Angles between the dimethylbenzene and pyrimidine rings of the FADs in the NAO structures. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Venulet J, van Etten RL. Biochemistry and Pharmacology of the Nitro and Nitroso Group. Vol. 2. Interscience Press; New York: 1970. [Google Scholar]
- 2.Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol. Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- 3.Kamisaki Y, Wada K, Bian K, Balabanli B, Davis K, Martin E, Behbod F, Lee YC, Murad F. An activity in rat tissues that modifies nitrotyrosine-containing proteins. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11584–11589. doi: 10.1073/pnas.95.20.11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DJ, Bright HJ. Propionate-3-nitronate oxidase from Penicillium atrovenetum is a flavoprotein which initiates the autoxidation of its substrate by O2. J. Biol. Chem. 1987;262:14428–14434. [PubMed] [Google Scholar]
- 5.Hipkin CR, Salem MA, Simpson D, Wainwright SJ. 3-nitropropionic acid oxidase from horseshoe vetch (Hippocrepis comosa): A novel plant enzyme. Biochem. J. 1999;340:491–495. [PMC free article] [PubMed] [Google Scholar]
- 6.Alston TA, Mela L, Bright HJ. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spain JC. Biodegradation of nitroaromatic compounds. Annu. Rev. Microbiol. 1995;49:523–555. doi: 10.1146/annurev.mi.49.100195.002515. [DOI] [PubMed] [Google Scholar]
- 8.Spain JC, Hughes JB, Knackmuss HJ. Biodegradation of Nitroaromatic Compounds and Explosives. CRC Press LLC; Boca Raton, FL: 2000. [Google Scholar]
- 9.Kido T, Hashizume K, Soda K. Purification and properties of nitroalkane oxidase from Fusarium oxysporum. J. Bacteriol. 1978;133:53–58. doi: 10.1128/jb.133.1.53-58.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadda G, Fitzpatrick PF. Substrate specificity of a nitroalkane oxidizing enzyme. Arch. Biochem. Biophys. 1999;363:309–313. doi: 10.1006/abbi.1998.1081. [DOI] [PubMed] [Google Scholar]
- 11.Singh SB, Kulshrestha G. Microbial degradation of pendimethalin. J. Environ. Sci. Health B. 1991;26:309–321. doi: 10.1080/03601239109372737. [DOI] [PubMed] [Google Scholar]
- 12.Zayed SM, Mostafa IY, Farghaly MM, Attaby HS, Adam YM, Mahdy FM. Microbial degradation of trifluralin by Aspergillus carneus, Fusarium oxysporum, and Trichoderma viride. J. Environ. Sci. Health B. 1983;18:253–267. doi: 10.1080/03601238309372367. [DOI] [PubMed] [Google Scholar]
- 13.Kido T, Tanizawa K, Inagaki K, Yoshimura T, Ishida M, Hashizume K, Soda K. 2-Nitropropane dioxygenase from Hansenula mrakii: Re-characterization of the enzyme and oxidation of anionic nitroalkanes. Agric. Biol. Chem. 1984;48:2549–2554. [Google Scholar]
- 14.Gorlatova N, Tchorzewski M, Kurihara T, Soda K, Esaki N. Purification, characterization, and mechanism of a flavin mononucleotide-dependent 2-nitropropane dioxygenase from Neurospora crassa. Appl. Environ. Microbiol. 1998;64:1029–1033. doi: 10.1128/aem.64.3.1029-1033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Tan H. Cloning, expression, and characterization of a gene encoding nitroalkane-oxidizing enzyme from Streptomyces ansochromogenes. Eur. J. Biochem. 2002;269:6302–6307. doi: 10.1046/j.1432-1033.2002.03350.x. [DOI] [PubMed] [Google Scholar]
- 16.Porter DJ, Bright HJ. Mechanism of oxidation of nitroethane by glucose oxidase. J. Biol. Chem. 1977;252:4361–4370. [PubMed] [Google Scholar]
- 17.Swoboda BE, Massey V. Purification and properties of the glucose oxidase from Asperillus niger. J. Biol. Chem. 1965;240:2209–2215. [PubMed] [Google Scholar]
- 18.Porter DJ, Voet JG, Bright HJ. Direct evidence for carbanions and covalent N5-flavin-carbanion adducts as catalytic intermediates in the oxidation of nitroethane by d-amino acid oxidase. J. Biol. Chem. 1973;248:4400–4416. [PubMed] [Google Scholar]
- 19.Heasley CJ, Fitzpatrick PF. Kinetic mechanism and substrate specificity of nitroalkane oxidase. Biochem. Biophys. Res. Commun. 1996;225:6–10. doi: 10.1006/bbrc.1996.1122. [DOI] [PubMed] [Google Scholar]
- 20.Fitzpatrick PF, Orville AM, Nagpal A, Valley MP. Nitroalkane oxidase, a carbanion-forming flavoprotein homologous to acyl-CoA dehydrogenase. Arch. Biochem. Biophys. 2005;433:157–165. doi: 10.1016/j.abb.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Lee JY, Moon SS, Hwang BK. Isolation and antifungal and antioomycete activities of aerugine produced by Pseudomonas fluorescens strain MM-B16. Appl. Environ. Microbiol. 2003;69:2023–2031. doi: 10.1128/AEM.69.4.2023-2031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gadda G, Fitzpatrick PF. Biochemical and physical characterization of the active FAD-containing form of nitroalkane oxidase from Fusarium oxysporum. Biochemistry. 1998;37:6154–6164. doi: 10.1021/bi973085y. [DOI] [PubMed] [Google Scholar]
- 23.Daubner SC, Gadda G, Valley MP, Fitzpatrick PF. Cloning of nitroalkane oxidase from Fusarium oxysporum identifies a new member of the acyl-CoA dehydrogenase super-family. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2702–2707. doi: 10.1073/pnas.052527799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J. Mol. Biol. 2003;328:581–592. doi: 10.1016/s0022-2836(03)00319-x. [DOI] [PubMed] [Google Scholar]
- 25.Miyazawa S, Osumi T, Hashimoto T, Ohno K, Miura S, Fujiki Y. Peroxisome targeting signal of rat liver acylcoenzyme A oxidase resides at the carboxy terminus. Mol. Cell Biol. 1989;9:83–91. doi: 10.1128/mcb.9.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koller A, Spong AP, Luers GH, Subramani S. Analysis of the peroxisomal acyl-CoA oxidase gene product from Pichia pastoris and determination of its targeting signal. Yeast. 1999;15:1035–1044. doi: 10.1002/(SICI)1097-0061(199908)15:11<1035::AID-YEA432>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 27.Olivier LM, Kovacs W, Masuda K, Keller GA, Krisans SK. Identification of peroxisomal targeting signals in cholesterol biosynthetic enzymes. AA-CoA thiolase, HMG-coA synthase, MPPD, and FPP synthase. J. Lipid Res. 2000;41:1921–1935. [PubMed] [Google Scholar]
- 28.Gadda G, Choe DY, Fitzpatrick PF. Use of pH and kinetic isotope effects to dissect the effects of substrate size on binding and catalysis by nitroalkane oxidase. Arch. Biochem. Biophys. 2000;382:138–144. doi: 10.1006/abbi.2000.2009. [DOI] [PubMed] [Google Scholar]
- 29.Gadda G, Fitzpatrick PF. Mechanism of nitroalkane oxidase: 2. pH and kinetic isotope effects. Biochemistry. 2000;39:1406–1410. doi: 10.1021/bi992255z. [DOI] [PubMed] [Google Scholar]
- 30.Valley MP, Fitzpatrick PF. Inactivation of nitroalkane oxidase upon mutation of the active site base and rescue with a deprotonated substrate. J. Am. Chem. Soc. 2003;125:8738–8739. doi: 10.1021/ja036045s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valley MP, Tichy SE, Fitzpatrick PF. Establishing the kinetic competency of the cationic imine intermediate in nitroalkane oxidase. J. Am. Chem. Soc. 2005;127:2062–2066. doi: 10.1021/ja043542f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gadda G, Edmondson RD, Russell DH, Fitzpatrick PF. Identification of the naturally occurring flavin of nitroalkane oxidase from fusarium oxysporum as a 5-nitrobutyl-FAD and conversion of the enzyme to the active FAD-containing form. J. Biol. Chem. 1997;272:5563–5570. doi: 10.1074/jbc.272.9.5563. [DOI] [PubMed] [Google Scholar]
- 33.Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 34.Hendrickson WA, Ogata CM. Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol. 1997;276:494–523. doi: 10.1016/S0076-6879(97)76074-9. [DOI] [PubMed] [Google Scholar]
- 35.Valley MP, Fitzpatrick PF. Reductive half-reaction of nitroalkane oxidase: Effect of mutation of the active site aspartate to glutamate. Biochemistry. 2003;42:5850–5856. doi: 10.1021/bi034061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagpal A, Valley MP, Fitzpatrick PF, Orville AM. Crystallization and preliminary analysis of active nitroalkane oxidase in three crystal forms. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:1456–1460. doi: 10.1107/S0907444904013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeelen JP, Hiltunen JK, Ceska TA, Wierenga RK. Crystallization experiments with 2-enoyl-CoA hydratase, using an automated “fast-screening” crystallization protocol. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:443–447. doi: 10.1107/S0907444994001277. [DOI] [PubMed] [Google Scholar]
- 38.Powell HR. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999;55:1690–1695. doi: 10.1107/s0907444999009506. [DOI] [PubMed] [Google Scholar]
- 39.Evans PR. Proceedings of CCP4 Study Weekend. 1997 [Google Scholar]
- 40.Kraut J, Sieker LC, High DF, Freer ST. Chymotrypsinogen: A three-dimensional Fourier synthesis at 5 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 1962;48:1417–1424. doi: 10.1073/pnas.48.8.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howell L, Smith D. Identification of heavy-atom derivatives by normal probability methods. J. Appl. Crystallogr. 1992;25:81–86. [Google Scholar]
- 42.Collaborative Computational Project The CCP4 suite: Programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de la Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 45.Abrahams JP, Leslie AGW. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 46.Terwilliger TC. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr., Sect. A: Found. Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 48.Schuettelkopf AW, van Aalten DMF. PRODRG—A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 49.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 50.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 51.Barton GJ. ALSCRIPT—A tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 52.DeLano WL. DeLano Scientific; San Carlos, CA: 2001. [Google Scholar]
- 53.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 54.Kantardjieff KA, Rupp B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein–nucleic acid complex crystals. Protein Sci. 2003;12:1865–1871. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthews BW. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 56.Kim JJ, Wu J. Structure of the medium-chain acyl-CoA dehydrogenase from pig liver mitochondria at 3 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 1988;85:6677–6681. doi: 10.1073/pnas.85.18.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JJ, Wang M, Paschke R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7523–7527. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JJ, Miura R. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur. J. Biochem. 2004;271:483–493. doi: 10.1046/j.1432-1033.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- 59.Gadda G, Banerjee A, Dangott LJ, Fitzpatrick PF. Identification of a cysteine residue in the active site of nitroalkane oxidase by modification with N-ethylmaleimide. J. Biol. Chem. 2000;275:31891–31895. doi: 10.1074/jbc.M003679200. [DOI] [PubMed] [Google Scholar]
- 60.Gadda G, Banerjee A, Fitzpatrick PF. Identification of an essential tyrosine residue in nitroalkane oxidase by modification with tetranitromethane. Biochemistry. 2000;39:1162–1168. doi: 10.1021/bi9921743. [DOI] [PubMed] [Google Scholar]
- 61.Sinnokrot MO, Valeev EF, Sherrill CD. Estimates of the ab initio limit for π–π interactions: The benzene dimer. J. Am. Chem. Soc. 2002;124:10887–10893. doi: 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- 62.Pellett JD, Becker DF, Saenger AK, Fuchs JA, Stankovich MT. Role of aromatic stacking interactions in the modulation of the two-electron reduction potentials of flavin and substrate/product in Megasphaera elsdenii short-chain acylcoenzyme A dehydrogenase. Biochemistry. 2001;40:7720–7728. doi: 10.1021/bi010206s. [DOI] [PubMed] [Google Scholar]
- 63.Leijonmarck M, Werner P-E. Studies on flavin derivatives: The crystal structure of 1,10-ethylene-7,8-dimethylisoalloxazinium iodide monohydrate. Acta Chem. Scand. 1970;24:2916–2928. [Google Scholar]
- 64.Moonen CT, Vervoort J, Müller F. Reinvestigation of the structure of oxidized and reduced flavin: Carbon-13 and nitrogen-15 nuclear magnetic resonance study. Biochemistry. 1984;23:4859–4867. doi: 10.1021/bi00316a007. [DOI] [PubMed] [Google Scholar]
- 65.Moonen CT, Vervoort J, Müller F. Carbon-13 nuclear magnetic resonance study on the dynamics of the conformation of reduced flavin. Biochemistry. 1984;23:4868–4872. doi: 10.1021/bi00316a008. [DOI] [PubMed] [Google Scholar]
- 66.Lennon BW, Williams CH, Jr., Ludwig ML. Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 1999;8:2366–2379. doi: 10.1110/ps.8.11.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Werner P-E, Linnros B, Leijonmarck M. Studies on flavin derivatives: The crystal structure of 5-diethyl-3,7,8,-10-tetramethyl-1,5-dihydroisoalloxazine trihydrate. Acta Chem. Scand. 1971;25:1297–1312. [Google Scholar]
- 68.Binda C, Angelini R, Federico R, Ascenzi P, Mattevi A. Structural bases for inhibitor binding and catalysis in polyamine oxidase. Biochemistry. 2001;40:2766–2776. doi: 10.1021/bi002751j. [DOI] [PubMed] [Google Scholar]
- 69.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 2004;271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 71.Pedersen L, Henriksen A. Acyl-CoA oxidase 1 from Arabidopsis thaliana. Structure of a key enzyme in plant lipid metabolism. J. Mol. Biol. 2005;345:487–500. doi: 10.1016/j.jmb.2004.10.062. [DOI] [PubMed] [Google Scholar]
- 72.Nakajima Y, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Tamaoki H, Miura R. Three-dimensional structure of the flavoenzyme acyl-CoA oxidase-II from rat liver, the peroxisomal counterpart of mitochondrial acyl-CoA dehydrogenase. J. Biochem. 2002;131:365–374. doi: 10.1093/oxfordjournals.jbchem.a003111. [DOI] [PubMed] [Google Scholar]
- 73.Watanabe K, Khosla C, Stroud RM, Tsai SC. Crystal structure of an acyl-ACP dehydrogenase from the FK520 polyketide biosynthetic pathway: Insights into extender unit biosynthesis. J. Mol. Biol. 2003;334:435–444. doi: 10.1016/j.jmb.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 74.Fu Z, Wang M, Paschke R, Rao KS, Frerman FE, Kim JJ. Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: Structural bases of dehydrogenation and decarboxylation reactions. Biochemistry. 2004;43:9674–9684. doi: 10.1021/bi049290c. [DOI] [PubMed] [Google Scholar]
- 75.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 76.Satoh A, Nakajima Y, Miyahara I, Hirotsu K, Tanaka T, Nishina Y, Shiga K, Tamaoki H, Setoyama C, Miura R. Structure of the transition state analog of medium-chain acyl-CoA dehydrogenase. Crystallographic and molecular orbital studies on the charge-transfer complex of medium-chain acyl-CoA dehydrogenase with 3-thiaoctanoyl-CoA. J. Biochem. 2003;134:297–304. doi: 10.1093/jb/mvg143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.