

Abstract
Using the cinchonidine-derived phase-transfer catalyst de veloped by Park and Jew as a lead structure, we have prepared novel chiral ammonium salts and investigated their efficacy for the preparation of β-hydroxy α-amino acids via asymmetric aldol reactions. The modifications were performed at C3 of the cinchonidine nucleus and include dimers as well as catalysts possessing electron-deficient alkyne and alkene moieties. Some of the new catalysts yielded improvements relative to the Park–Jew catalyst in the aldol reaction.
Keywords: Cinchona alkaloids, β-hydroxy α-amino acids, Sonogashira coupling, Heck reaction, asymmetric aldol reaction
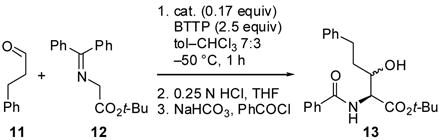
Quaternary ammonium salts derived from inexpensive and readily available Cinchona alkaloids have shown great utility as chiral phase-transfer catalysts, particularly in asymmetric alkylations.1 Recently, we examined the viability of these catalysts for the synthesis of β-hydroxy α-amino acids via aldol reactions, a topic first explored by Miller.2,3 We discovered that the N-2,3,4-trifluorobenzyl-substituted hydrocinchonidine derivative of Park and Jew4 afforded protected β-hydroxy α-amino acids in good yields under homogenous conditions in conjunction with the phosphazene base tert-butyliminotri(pyrrolid ino)-phosphorane (BTTP, Scheme 1).5 Although the syn diastereomers were obtained in good ee, the reaction exhib ited negligible diastereoselectivity. In an attempt to improve the selectivity of the aldol reaction, we designed novel cinchonidine-based monomeric and dimeric quaternary ammonium salts that retain the 2,3,4-trifluorobenzyl group of the Park–Jew catalyst. In this letter, we report the synthesis of these catalysts as well as their performance in the aldol reaction.
Scheme 1.
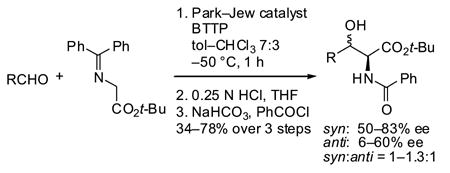
Asymmetric aldol reaction mediated by Park–Jew catalyst.
We viewed the C3-vinyl group of cinchonidine as a potential hand le for the construction of new catalysts. Modifications of this functional group such as dehydrogenation,6 hydroformylation,7 and oxidative cleavage8 are known; however, to the best of our knowledge, none of these protocols have been incorporated into the synthesis of quaternary ammonium salt catalysts. Hoffmann and Frackenpohl recently reported that C3-ethynyl Cinchona alkaloids are significantly more basic and polar than the parent compounds, thereby demonstrating that this functional group, although remote from the nitrogen, has a profound effect on its properties.9 Accordingly, we were optimistic that the catalytic abilities of cinchonidine-derived ammonium salts could be tuned via appropriate modifications of the C3-vinyl group. In an attempt to develop catalysts capable of forming tight ion pairs with enolates, we targeted ammonium salts with electron-deficient substituents at this site.
The preparation of C3-alkynyl ammonium salts 3a–d possessing fluorinated and trifluoromethylated aryl groups is summarized in Table 1. We employed 10,11-didehydrocinchonidine (1)6a as our starting material, and after considerable experimentation discovered that the aqueous Sonogashira coupling conditions of Bhattacharya and Sengupta10 were optimal for preparation of arylated compounds 2. Then, N-benzylation followed by O-allylation afforded the target compounds in good yield, with the exception of p-fluorophenyl-substituted salt 3b. In all cases, benzylation provided the quaternary ammonium salt in essentially quantitative yield without the need for further purification. Thus, the variability in the yields of 3 was a consequence of the O-allylation. Attempts to optimize the allylation in the synthesis of 3b were unsuccessful.
Table 1.
Synthesis of C3-alkynyl ammonium salts 3a–d
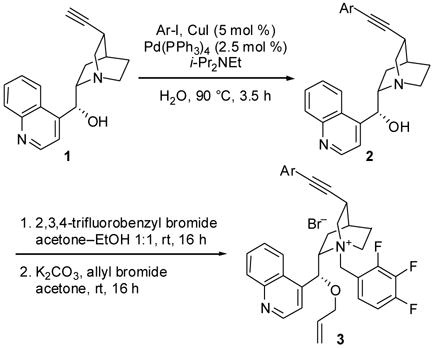
| Compound | Ar | Yield of 2 (%) | Yield of 3 (%) |
|---|---|---|---|
| 3a | p-CF3C6H4 | 96 | 52 |
| 3b | p-FC6H4 | 93 | 15 |
| 3c | 3,4-F2C6H3 | 92 | 64 |
| 3d | 2,4-F2C6H3 | 87 | 71 |
Unfortunately, despite examining numerous conditions, we were unable to perform the O-allylation on substrates possessing nitroaryl groups. Consequently, the order of steps was changed, and the Sonogashira coupling was performed last in the construction of nitro-containing catalysts 3e–g (Table 2). Benzylation and allylation of 1 proceeded under the same conditions shown in Table 1 for Sonogashira adduct 2, affording quaternary ammonium salt 4 in 79% yield. For the coupling of 4 with nitroaryl iodides, we found that the highest yields were obtained by employing TBAF as base11 under aqueous conditions. We were unable to obtain 3,4-dinitrophenyl-substituted salt 3g in good yield; nevertheless, enough of this compound was produced to examine its viability as a catalyst in the asymmetric aldol reaction.
Table 2.
Synthesis of C3-alkynyl ammonium salts 3e–g
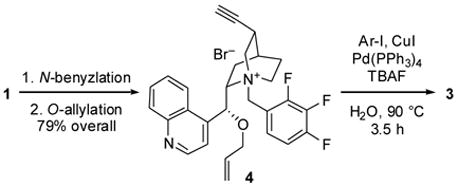
| Compound | Ar | Yield of 3 (%) |
|---|---|---|
| 3e | p-NO2C6H4 | 54 |
| 3f | 4-F-2-NO2C6H3 | 60 |
| 3g | 3,4-(NO2)2C6H3 | 22 |
C3-Alkenyl-substituted chiral ammonium salts 8 bearing fluorinated aryl groups were assembled as illustrated in Scheme 2, with a Heck reaction as the key step. Performing the Heck reaction after the N-benzylation and before the O-allylation delivered optimal results. The previously developed conditions were satisfactory for the benzylation and allylation steps, and PdCl2/Et3N was found to be the best system for the Heck coupling of ammonium salt 6 with fluorinated aryl iodides.
Scheme 2.
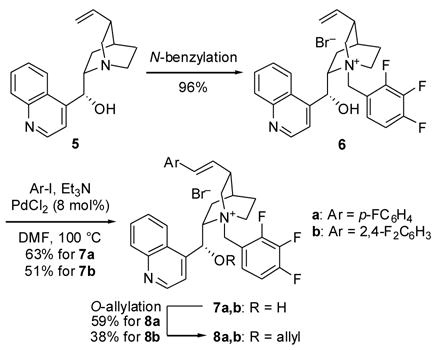
Synthesis of C3-alkenyl ammonium salts 8a and 8b.
In addition to the catalysts bearing electron-deficient groups at C3, we were interested in constructing dimeric catalysts linked through a C3 alkyne. Although dimeric cinchonidine-derived chiral phase-transfer catalysts are known, all previous examples of which we are aware involve dimerization via an N-benzyl or N-alkyl group.12 Thus, we decided to prepare a novel class of dimeric chiral ammonium salt catalysts. The synthesis of these catalysts is shown in Scheme 3. The previously constructed alkyne 4 could be dimerized in an Eglinton coupling,13 affording diyne 9 in good yield. We initially attempted to prepare phenyl-linked dimer 10a and biphenyl-linked dimer 10b in a single Sonogashira coupling employing two equivalents of 4 and the appropriate aryl d i-iodide. However, this approach was unsuccessful, so we performed two sequential couplings instead. The second reaction required increased loadings of the palladium and copper catalysts, and the products were obtained in modest yields. Each of the bis-ammonium salts 9 and 10 were extremely polar, requiring pure CH3OH for elution from a silica gel column.
Scheme 3.
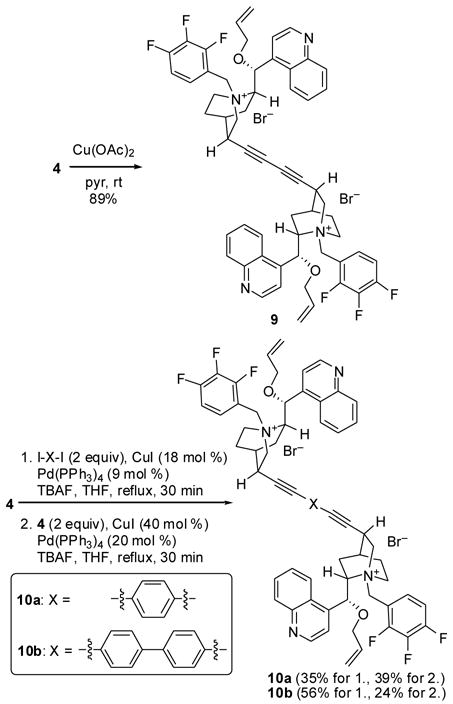
Synthesis of dimeric ammonium salts 9 and 10.
Catalysts 3a–g, 8a–b, 9, and 10a–b were evaluated in the aldol reaction between hydrocinnamaldehyde (11) and tert-butyl glycinate benzophenone imine (12). The results of this investigation are contained in Table 3. For purposes of comparison, the Park–Jew catalyst mediates the reaction between 11 and 12 to afford a 64% yield of 13 with a syn/anti ratio of 1.3:1. The syn and anti isomers are obtained with 80% and 33% ee, respectively.5 Ammonium salt 3d possessing a 2,4-difluorophenyl-substituted alkyne afforded the best ee’s of any of our new catalysts; unfortunately, the diastereoselectivity was both modest and reversed, with the major anti isomer having low ee. In fact, anti-13 was produced in low ee by all of the catalysts examined. p-Fluorophenyl-substituted alkyne catalyst 3b was able to improve the diastereoselectivity of the reaction (2.6:1 dr in favor of the syn isomer), but no enhancement of ee’s was observed. Comparison of the results obtained with 3b and 3d shows that the presence or absence of an ortho fluoro substituent exerts a significant effect on the aldol reaction. While the underlying reasons for this effect are unclear, it is possible that aromatic F···H interactions14 could be involved. Catalyst 3a, which bears a p-trifluoromethyl-substituted phenyl group on the alkyne, afforded the aldol products in very good yield (86%); however, the dr and ee’s were similar to those delivered by the Park–Jew catalyst. Z-alkene-containing catalysts 8a and 8b performed more poorly than did their alkyne analogues 3b and 3d, indicating that the geometry of the C3-substituent plays an important role in catalyst efficiency.
Table 3.
Survey of catalysts in asymmetric aldol reaction
| Catalyst | Yield (%) | Syn/Anti | Syn ee (%)b | Anti ee (%)b |
|---|---|---|---|---|
| 3a | 86 | 1.2:1 | 82 | 39 |
| 3b | 54 | 2.6:1 | 82 | 36 |
| 3c | 40 | 1.5:1 | 79 | 30 |
| 3d | 50 | 1:1.8 | 91 | 44 |
| 3e | 76 | 1.3:1 | 83 | 34 |
| 3f | 57 | 1:4.7 | 20 | 5 |
| 3fa | 40 | 1:1.1 | 61 | 8 |
| 3g | 76 | 1:3.7 | 13 | 3 |
| 3ga | 50 | 1:2.2 | 25 | 9 |
| 8a | 38 | 1:1.5 | 51 | 3 |
| 8b | 38 | 1.1:1 | 65 | 14 |
| 9a | 57 | 1:1.3 | 43 | 2 |
| 10aa | 57 | 1.1:1 | 85 | 38 |
| 10ba | 50 | 1:1.2 | 74 | 23 |
CH2Cl2 was used as the solvent.
Measured by HPLC (Chiralcel OD-H, 98:2 hexanes/i-PrOH, 1 mL/min).
The dimeric catalysts 9 and 10a–b were insoluble in the toluene–chloroform solvent system used for most of the aldol reactions.15 Thus, reactions employing these compounds were performed in CH2Cl2. Inspection of the results obtained with the dimers reveals that the phenyl group serves as the optimal spacer between the two catalyst units (cf. 10a vs. 9 and 10b). Nevertheless, none of the dimeric ammonium salts performed better than the Park–Jew catalyst in the aldol reaction.
In conclusion, we have synthesized novel monomeric and dimeric cinchonidine-derived chiral ammonium salts and examined their efficiency in the construction of β-hydroxy α-amino acids via asymmetric aldol reactions.16 We have discovered individual catalysts which improve upon the performance of the Park–Jew catalyst in terms of yield (3a), syn/anti ratio (3b), and ee (3d). We have yet to find a single catalyst which performs well in all three areas. Currently, the biggest limitation of this method is the modest diastereoselectivity. Since asymmetric alkylations of 12 mediated by cinchonidine-derived phase-transfer catalysts typically proceed with excellent ee, the problem is likely due to low facial selectivity of the aldehyde. The methods developed in this study for functionalization of the Cinchona alkaloid scaffold (Sonogashira coupling, Heck reaction) should prove useful to others interested in modifying this class of alkalo ids.
Supplementary Material
Experimental procedures and characterization data for compounds 3 and 8–10 are available.
Acknowledgments
We thank the National Institutes of Health (GM70483) and Brigham Young University (Graduate Studies Fellowship to B.M., Undergraduate Research Awards to J.L.P.) for financial support of this work. We also thank Dr. Jens Frackenpohl for helpful comments concerning the synthesis and purification of 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Vachon J, Lacour J. Chimia. 2006;60:266. [Google Scholar]; (b) O’Donnell MJ. Acc Chem Res. 2004;37:506. doi: 10.1021/ar0300625. [DOI] [PubMed] [Google Scholar]; (c) Lygo B, Andrews BI. Acc Chem Res. 2004;37:518. doi: 10.1021/ar030058t. [DOI] [PubMed] [Google Scholar]; (d) Kacprzak K, Gawroski J. Synthesis. 2001:961. [Google Scholar]
- 2.Gasparski CM, Miller MJ. Tetrahedron. 1991;47:5367. [Google Scholar]
- 3.For an effective BINOL-based phase-transfer catalyst, see: Ooi T, Kameda M, Taniguchi M, Maruoka K. J Am Chem Soc. 2004;126:9685. doi: 10.1021/ja048865q.; Ooi T, Taniguchi M, Kameda M, Maruoka K. Angew Chem Int Ed. 2002;41:4542. doi: 10.1002/1521-3773(20021202)41:23<4542::AID-ANIE4542>3.0.CO;2-3. However, the high cost of BINOL and the lengthy synthesis required for this catalyst makes the development of cinchonidine-derived catalysts accessible via short routes a worthy goal. [DOI] [PubMed] [Google Scholar]
- 4.Jew S-s, Yoo M-S, Jeong B-S, Park I-Y, Park H-g. Org Lett. 2002;4:4245. doi: 10.1021/ol0267679. [DOI] [PubMed] [Google Scholar]
- 5.Mettath S, Srikanth GSC, Dangerfield BS, Castle SL. J Org Chem. 2004;69:6489. doi: 10.1021/jo049283u. [DOI] [PubMed] [Google Scholar]
- 6.(a) Braje WM, Frackenpohl J, Schrake O, Wartchow R, Beil W, Hoffmann HMR. Helv Chim Acta. 2000;83:777. [Google Scholar]; (b) Frackenpohl J, Braje WM, Hoffmann HMR. J Chem Soc, Perkin Trans 1. 2001:47. [Google Scholar]
- 7.Lambers M, Beijer FH, Padron JM, Toth I, de Vries JG. J Org Chem. 2002;67:5022. doi: 10.1021/jo0255173. [DOI] [PubMed] [Google Scholar]
- 8.Merschaert A, Delbeke P, Daloze D, Dive G. Tetrahedron Lett. 2004;45:4697. [Google Scholar]
- 9.Hoffman HMR, Frackenpohl J. Eur J Org Chem. 2004:4293. [Google Scholar]
- 10.Bhattacharya S, Sengupta S. Tetrahedron Lett. 2004;45:8733. [Google Scholar]
- 11.Liang Y, Xie YX, Li JH. J Org Chem. 2006;71:379. doi: 10.1021/jo051882t. [DOI] [PubMed] [Google Scholar]
- 12.(a) Chinchilla R, Mazón P, Nájera C. Tetrahedron: Asymmetry. 2002;13:927. [Google Scholar]; (b) Park H-g, Jeong B-S, Yoo M-S, Lee J-H, Park M-k, Lee Y-J, Kim M-J, Jew S-s. Angew Chem Int Ed. 2002;41:3036. doi: 10.1002/1521-3773(20020816)41:16<3036::AID-ANIE3036>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (c) Siva A, Murugan E. J Mol Catal A: Chem. 2005;241:111. [Google Scholar]
- 13.Tobe Y, Utsumi N, Nagano A, Sonoda M, Naemura K. Tetrahedron. 2001;57:8075. [Google Scholar]
- 14.Doyon JB, Jain A. Org Lett. 1999;1:183. doi: 10.1021/ol9905250. [DOI] [PubMed] [Google Scholar]
- 15.For the original application of this solvent mixture in phase-transfer-catalyzed alkylations, see refs 4 and 12b.
- 16.For recent developments in the synthesis of β-hydroxy α-amino acids, see: Makino K, Iwasaki M, Hamada Y. Org Lett. 2006;8:4573. doi: 10.1021/ol061796v.; Crich D, Banerjee A. J Org Chem. 2006;71:7106. doi: 10.1021/jo061159i. Please see the latter paper or ref 5 for a summary of the strategies employed in the asymmetric synthesis of these compounds. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for compounds 3 and 8–10 are available.