

Summary
A Kinase Anchoring Proteins (AKAPs) target PKA to specific microdomains using an amphipathic helix that docks to N-terminal Dimerization/Docking (D/D) domains of PKA regulatory (R) subunits. To understand specificity, we solved the crystal structure of the helical motif from d-AKAP2, a dual-specific AKAP, bound to the RIIα D/D domain. The 1.6 Å structure reveals for the first time how this dynamic, hydrophobic docking site is assembled. A stable, hydrophobic docking groove is formed by the helical interface of two RIIα protomers. The flexible N-terminus of one protomer is then recruited to the site, anchored to the peptide through two essential isoleucines. The other N-terminus is disordered. This asymmetry provides greater possibilities for AKAP docking. While there is strong discrimination against RIα in the N-terminus of the AKAP helix, the hydrophobic groove discriminates against RIIα. RIα, with a cavity in the groove, can accept a bulky tryptophan, while RIIα requires valine.
Introduction
Signal transduction is mediated by protein:protein interactions frequently regulated dynamically by peptides docking to relatively small binding motifs. SH2, SH3, WW, and PDZ domains are examples of signaling modules that interact with short linear peptides where specificity is typically achieved by mostly hydrogen bonds and electrostatic interactions (Cohen et al., 1995). In contrast, the targeting of cAMP-dependent protein kinase (PKA) is mediated primarily by docking of an amphipathic helix where specificity is achieved not by backbone/side chain contacts but rather by the interactions of hydrophobic side chains. One of the most important mechanisms for targeting PKA is through A Kinase Anchoring Proteins (AKAPs) where the signature motif of the AKAP is an amphipathic helix that docks to the PKA regulatory (R) subunits (Carr et al., 1991). AKAPs thus serve as scaffolds to target PKA and other signaling molecules to microdomains in the cell in close proximity to their substrates (Wong and Scott, 2004). This targeting is an essential feature of PKA signaling and is an important mechanism for achieving specificity.
The PKA holoenzyme is comprised of a homodimer of R-subunits bound to two catalytic (C) subunits. Each protomer of R contains an N-terminal dimerization/docking (D/D) domain followed by a PKA inhibitor site and two tandem cAMP binding domains. In most cases, the D/D domain is thought to be functionally well segregated from the cAMP binding domains where the C-subunit binds (Heller et al., 2004). The D/D domain is an anti-parallel four-helix bundle that provides the docking surface for the AKAP helix (Banky et al., 2003; Newlon et al., 2001; Newlon et al., 1999). Although the fold of the D/D domain is conserved in all R subunits, sequence differences in RI and RII provide one mechanism for introducing specificity into the PKA signaling pathway (Hausken et al., 1994). Typically, AKAPs bind with high affinity (KD = 1–5 nM) to RII subunits (Carr et al., 1992; Herberg et al., 2000), whereas some AKAPs, termed dual-specificity AKAPs or d-AKAPs, bind to both RI and RII (Huang et al., 1997a; Huang et al., 1997b). RII subunits are typically localized to discrete sites in the cell, such as the plasma membrane, mitochondria, cytoskeleton, and centrosomes (Wong and Scott, 2004), while RI subunits tend to be more diffuse. Whereas docking of RII is more static, the RI subunits can be dynamically recruited to a specific site as seen in T-cells, where normally diffuse cytoplasmic RI is recruited to the cap site (Skalhegg et al., 1994).
d-AKAP2 is considered a dual-specific AKAP because it binds with high affinity to both RII and RI (KD= 2 nM and 48 nM, respectively) (Burns et al., 2003). The biological importance of RI vs. RII specificity was suggested by SNP analysis where a mutation in d-AKAP2 that reduces affinity only for RI has been associated with a shorter life span and possibly cardiac dysfunction (Kammerer et al., 2003). As summarized in Figure 1A, d-AKAP2 is a modular protein that contains two putative RGS domains followed by an A Kinase Binding (AKB) domain and a C-terminal PDZ binding motif (Gisler et al., 2003; Huang et al., 1997a). The biophysical properties of the AKB of dAKAP2 have been characterized extensively using a variety of solution methods. Protection of both the peptide and the D/D domains of RIα and RIIα was mapped by hydrogen/deuterium exchange coupled with mass spectrometry (H/DMS) (Burns-Hamuro et al., 2005), while the specificity requirements for binding RI vs RII were mapped using a peptide array (Burns-Hamuro et al., 2003). Although we can design AKAP peptides specific for RI and RII and can use them to disrupt cell function (Alto et al., 2003; Burns-Hamuro et al., 2003), we still cannot predict specificity a priori using an amphipathic helix as a template. How do we distinguish, for example, an amphipathic helix that has specificity for an AKAP from one that serves as a nuclear export signal (Wen et al., 1995)? Elucidating the stringent rules for AKAP specificity for PKA will require structures of complexes.
Figure 1. Amphipathic helices mediate D-AKAP2 interaction with RIIα D/D domain.
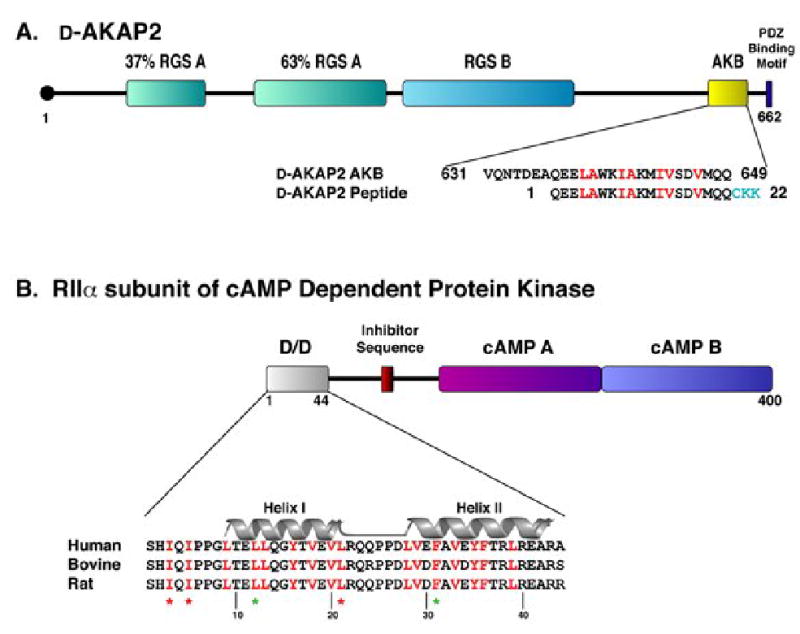
(A) Full length D-AKAP2 contains two putative RGS domains, a PDZ binding motif, and the A Kinase Binding (AKB) Domain, which forms an amphipathic helix for binding the D/D domain. This fragment corresponds to residues 623–649 (numbering according to Swiss-Prot entry O8845). For our studies, a 22-residue peptide was synthesized, including residues 631–649 of D-AKAP2 and three extra residues (shown in turquoise) on the C-terminus for functional purposes. For simplicity, we have numbered the peptide 1–22. Residues highlighted in red correspond to the surface of the helix involved in docking to the D/D domain of RIα and RIIα (Burns-Hamuro et al., 2005).
(B) The dimerization/ docking domain resides at the N-terminus of the RIIα subunit, which also includes an inhibitor sequence that binds the active site of the PKA catalytic subunit and two cAMP binding domains. Each protomer of the D/D domain forms a helix-turn-helix motif (Helix I and Helix II are labeled). The conserved hydrophobic, dimer interface is highlighted in red. Residues essential for dimerization are denoted by a green star and residues that are essential for AKAP binding, but do not effect dimerization are denoted by a red star (Hausken et al., 1994; Li and Rubin, 1995).
Here we report the crystal structure of the D/D domain of rat RIIα bound to a 22-residue peptide from the AKB domain of d-AKAP2 (Figure 1A). Our structure reveals a unique docking mechanism and allows us to appreciate how hydrophobic side chains contribute to docking. In addition to a solvent-excluded hydrophobic interaction surface between the helical peptide and the groove formed by the antiparallel helices of the RIIα D/D domain described previously (Newlon et al., 2001), we see for the critical contributions of the dynamic N-terminus of RIIα, specifically Ile3R and Ile5R. The N-terminal tail of the second protomer within the dimer is disordered. Binding of the d-AKAP2 peptide thus introduces asymmetry into the D/D domain, and this asymmetry provides greater docking possibilities for the AKAPs. The crystal structure, correlated with extensive biochemical and biophysical data, defines the atomic determinants for RI and RII specificity of AKAP docking. While many mutations in the N-terminus of the AKAP peptide discriminate against RIα, the N-termini of the D/D domains for RIα and RIIα are flexible and also different. Thus, we cannot establish definitive isoform-specificity for this region until we have a structure of the RIα docked to the AKAP peptide. However, the hydrophobic groove formed by Helix I and Helix I’ is more rigid and here there is one isoform-specific site that discriminates strongly against RIIα. In the corresponding groove for RIα, there is a cavity that can accept bulky side chains, whereas RIIα has a flat hydrophobic surface. RIIα has a strong preference for Val at position 13 of the peptide and cannot accept larger residues, while the AKAP peptide with Trp at the same position binds to RIα. This site allows AKAP peptides to discriminate between RI and RII-isoforms, thus contributes to isoform-specificity as demonstrated in vitro and in cells.
Results and Discussion
Summary of the RIIα D/D: d-AKAP2 AKB complex structure
The fully refined structure of the D/D domain bound to a 22 residue AKAP peptide from dAKAP2 showed two dimers:AKAP complexes in the asymmetric unit (Dimer I and Dimer II) (Figure 2). Although each dimer:AKAP complex has a unique set of crystal contacts, they align with an RMSD of 0.32 Å for 64 equivalent Cα atoms. Crystallographic data and refinement statistics are included in the supplementary data. The backbone trace of the dimerization/docking domain of our model forms a four helix bundle and agree well with the previous NMR solution structures (Newlon et al., 2001), as discussed later. Each protomer contains two helices which pair to form an X-type, four-helix bundle. As previously reported, Helix I and II from one protomer interact in an antiparallel manner with their counterparts on the other protomer, Helix I’ and II’, respectively (Figure 2A). The two molecules in the asymmetric unit are joined through a stacking interaction (Figure 2D) between RIIα residues Pro25R’ and Pro26R’ of one molecule (Dimer I) and His2R of the other molecule (Dimer II). Gln24R’ of Dimer I also forms a hydrogen bond with Gln4R of Dimer II. Symmetrically related molecules cluster together by the N termini of the D/D domains as well as by contacts between several residues of the d-AKAP2 peptide with D/D domains of neighboring molecules. For example, Trp6P of the d-AKAP2 peptide forms a crystal contact with Pro25R and Pro26R of RIIα in a symmetrically related molecule.
Figure 2. High resolution crystal structure depicts RIIα D/D domain complexed with the D-AKAP2 peptide.
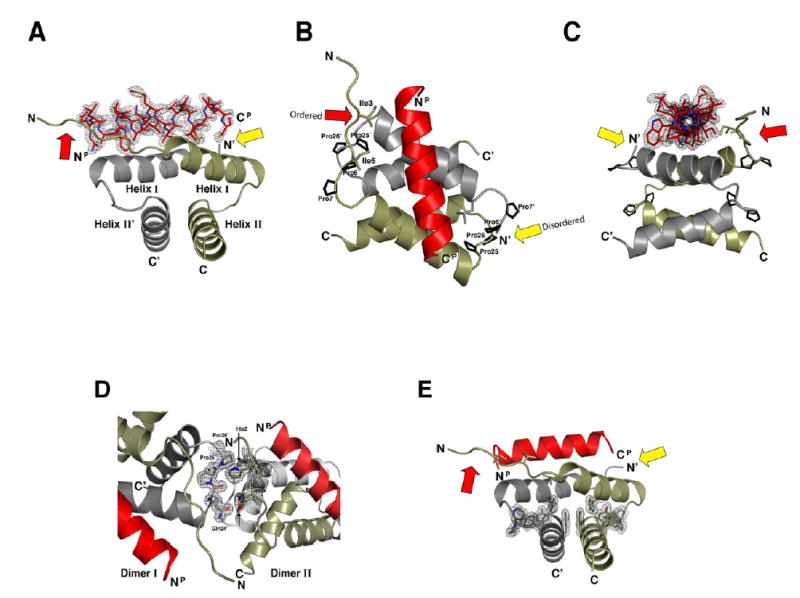
(A-C) The crystal structure of the RIIαD/D:D-AKAP2 complex is displayed at three different angles. The two protomers of the D/D domain, RIIαD/D and RIIαD/D’, are displayed in tan and grey, respectively. The D-AKAP2 peptide, shown in red, is bound to Helix I and Helix I’ of the RIIα D/D dimer. Prolines 6R, 7R, 25R, and 26R, which stablize the loops between helices, as well as, contribute to crystal packing, are highlighted.
(D) The two molecules in the asymmetric unit are associated by contacts involving Gln24R’, Pro25R’, and Pro25R’ of Dimer I and His2R and Gln4R of Dimer II.
(E) The hydrophobic core of the D/D domain is stabilized by several aromatic side chains extending from all four helices.
The dimer interface of each D/D domain is comprised of hydrophobic side chains from each helix (residues Leu12R, Leu13R, Tyr16R, Thr17R, Val20R, Leu21R, Leu28R, Val29R, Phe31R, Ala32R, Val33R, Tyr35R, Phe36R, Thr37R, and Leu39R from each protomer) forming a tight, solvent-excluded core. The structure includes over 200 water molecules, none of which are found within the dimer interface. The center of the dimer is especially enriched with phenylalanines and tyrosines which stack together to form the core (Figure 2E). Additionally, two motifs, characterized by two conserved adjacent prolines, are created by Pro6R, 7R and Pro25R, 26R (Figure 2B). Prolines 6R and 7R tether the N-terminal tail to Helix I, while prolines 25R and 26R stabilize the loop between Helices I and II. These two proline pairs (6R, 7R and 25R, 26R) are also in close proximity to each other on the exposed surface of the D/D domain. As indicated above, Pro25R and Pro26R also contribute to crystal packing, suggesting that this surface may have a propensity to bind other proteins.
Assembly of an asymmetric, hydrophobic binding pocket is revealed by the crystal structure
Two elements in the D/D domain, one stable and one flexible, create an asymmetric surface for the AKAP helix (Figure 3A). A preformed, stable hydrophobic groove is formed by the antiparallel packing of Helix I and Helix I’, while a flexible component derives from the N-terminal tail. Because only one tail is recruited to the site, binding of the AKAP peptide creates an inherent asymmetry. Side chains from the d-AKAP2 peptide form a hydrophobic ridge that nicely complements the surface displayed by RIIα. As mentioned, no water was found at the hydrophobic interface between the RIIα D/D domain and the d-AKAP2 peptide. The interface occludes an area of 360 Å2 (Lee and Richards, 1971). Figure 3C summarizes the multiple contacts between the d-AKAP2 peptide and the RIIα D/D domain.
Figure 3. Complementary hydrophobic surfaces on RIIα D/D domain and D-AKAP2 mediate a tight interaction between helices.
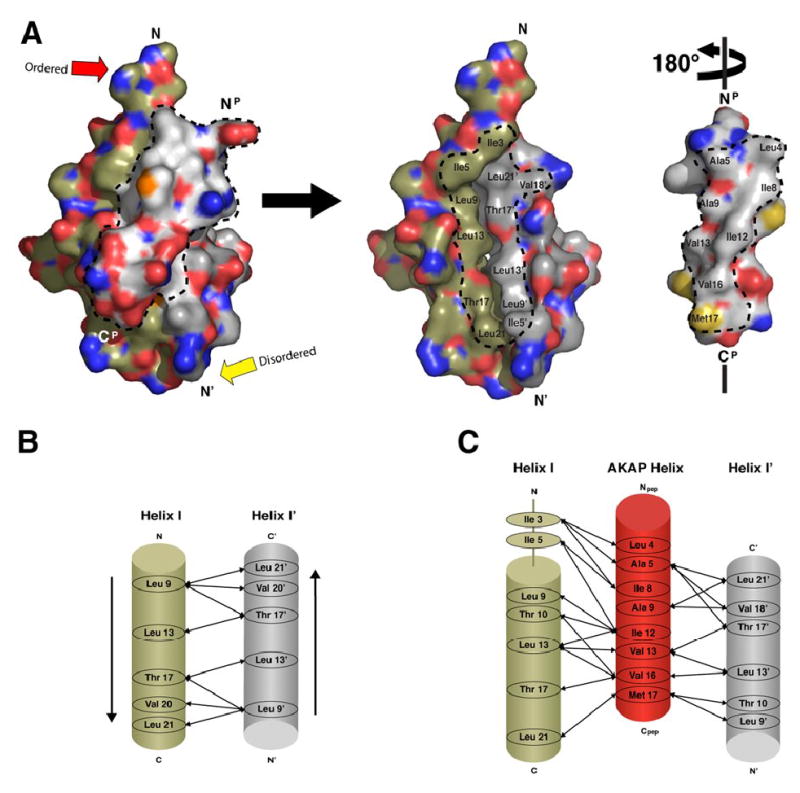
(A) The D-AKAP2 peptide is removed from the interface and rotated 180° in order to see the buried interacting surfaces displayed by the peptide and the RIIα D/D domain. The hydrophobic residues of the peptide form a ridge required for PKA binding. RIIα D/D domain displays a complementary hydrophobic surface, formed by Helix I and I’, that allows the peptide’s hydrophobic ridge to dock. Dotted lines outline the residues of both peptide and the D/D domain that pack together in the hydrophobic interface.
(B) Helix I and Helix I’ interact in an antiparallel manner to form the stable binding site for AKAP docking. Arrows represent the hydrophobic interactions between the two helices.
(C) Specific side chain interactions between residues of the D/D domain and peptide were elucidated and listed.
A cluster of hydrophobic residues from each protomer of RIIα D/D (Leu9R, Thr10R, Leu13R, Val16R, Thr17R, and Leu21R) create the preformed surface where the AKAP helix docks (Figure 3B). Many of these hydrophobic side chains also stabilize the dimer. Leu21R in this stable core is the only hydrophobic residue known to disrupt AKAP binding, but not dimerization, when mutated (Li and Rubin, 1995). As indicated in Figure 1B, mutations of other residues abolish dimerization and, as a secondary consequence, prevent AKAP binding (Li and Rubin, 1995). As discussed later, the position of the side chain of Leu13R differs in RIα and RIIα and is a specificity determinant for this hydrophobic groove.
Our structure reveals that the solvent-exposed and flexible N-terminal segment (residues 1–5) of RIIα is also an essential feature of the AKAP binding site. The side chains of Ile3R and Ile5R of one protomer extend in towards the peptide, making multiple hydrophobic contacts with d-AKAP2 (Figure 4A); the N-terminus of the other protomer is disordered (residues 1–3) and has higher temperature factors, suggesting that the N-terminus is flexible, especially the disordered end. Although Ile5 of the disordered end is seen, it is displaced X Å from its position compared to the ordered end and show a high temperature factor (Figure 5). Ile3R contacts Leu4P, Ala5P, and Ile8P of the d-AKAP2 peptide while Ile5R contacts Ile8P and Ile12P. Leu21R’ from the other protomer also extends upward into the docking surface to contact the d-AKAP2 peptide where it is juxtapositioned against Ile3R and Ile5R. As seen in Figure 4, Ile3R, Ile5R, and Leu21R’, previously identified by mutagenesis to be required for AKAP binding (Hausken et al., 1994; Li and Rubin, 1995), create a well-defined hydrophobic pocket. Our structure confirms that these residues are critical AKAP contact points. This binding module was not revealed by the previous NMR structures, as the N-termini were not well ordered. This essential hydrophobic recognition site requires the clustering of at least three residues: Leu21R’ is part of the stable core while Ile3R is recruited from the flexible tail. Ile5R is reoriented as well for binding. The side chain of Leu9R, which initiates Helix I, is also a stable part of this pocket; however, no mutants of Leu9R have been studied. The symmetry-related site in the same molecule is partially disordered, allowing one to appreciate the dynamic features of this binding element. Leu21R and Leu9R’, both part of the stable core, are in place, Ile5R’ is close, but Ile3R’ is not seen at all. Residues 4′-7′ are seen, but backbone and side chain positions are different. Residues 1′-3′ are not seen at all. Gly8R appears to be the clear hinge point for the N-terminal element. It is also important to note that we have two dimers in each asymmetric unit and in all cases the disordered N-terminus is not influenced or biased by crystal packing, thus serves as an internal control. This reinforces the argument that one N-terminus is disordered.
Figure 4. RIIα D/D domain forms an asymmetric pocket upon D-AKAP2 binding.
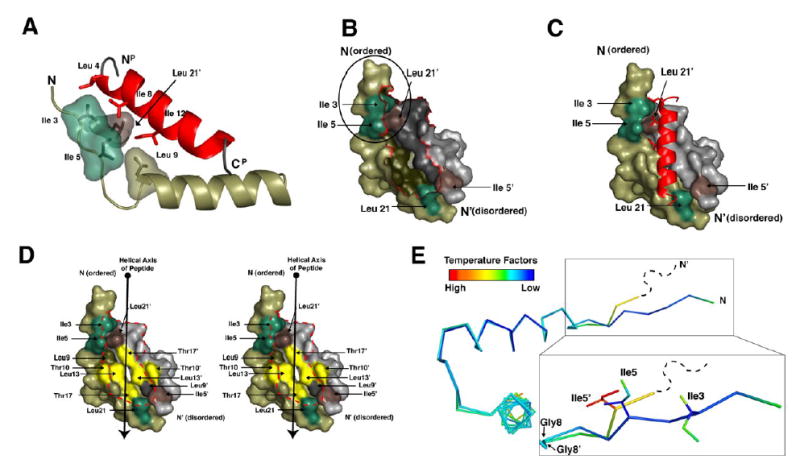
(A) Ile3R, Ile5R, Leu9R and Leu21R’ make critical contacts with peptide residues Leu4P, Ala5P, Ile8P, and Ile12P. These residues are highlighted to show their proximity to each other and the importance of the branching of the isoleucine side chains.
(B and C) The surface of RIIα D/D domain is displayed alone and with the AKAP. The Ile3R, Ile5R, Leu21R are shown in green. Ile5R’ and Leu21R’ are shown in brown. These residues are highlighted to illustrate the two potential docking sites created Ile3R, Ile5R and Leu21R’ and Ile5R’ and Leu21R. The N-terminus of RIIαD/D, including Ile3R and Ile5R (circled in B) is ordered in the structure and cluster with Leu21R’ to form a hydrophobic site that interacts with a number of side chains from the peptide. However, the N-terminal extension of RIIαD/D’ is disordered in our crystal, so it cannot be seen in our model.
(D) The hydrophobic groove of the RIIα D/D domain is illustrated with a stereo-representation of the D/D domain surface. Residues that line this groove and contribute to AKAP docking are highlighted. Residues colored in yellow represent the center of the groove, which are important for binding specificity.
(E) The two protomers from the D/D domain are aligned and colored according to B-factor to show the asymmetry more precisely. The RIIαD/D’ protomer is disordered at the N-terminus beyond residue 4 and residue 5 is relatively dynamic according to B-factors. However, this region is well ordered on the protomer that contacts the N-terminus of the peptide. The position of Ile5R and Ile5R’ is different due to changes around Gly8R and Gly8R’.
Figure 5. The B-factors of the crystal structure correlate with H/D exchange protection data.
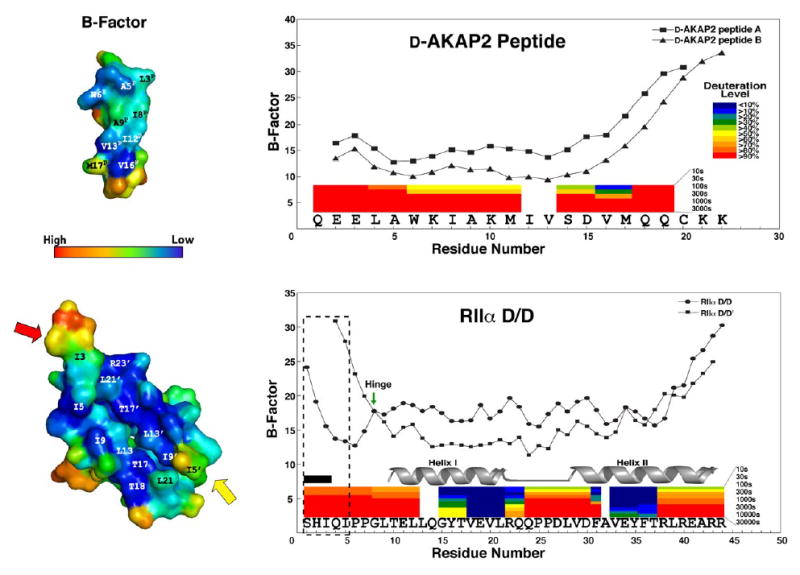
B-factors of both RIIα D/D domain and D-AKAP2 peptide are shown. We see a sharp rise in B-factor at the C-terminus of the peptide, which corresponds with a high deuteration rates demonstrated by previous H/DMS experiments (Burns-Hamuro et al., 2005). The colored bars represent the deuteration levels of peptides from D-AKAP2 AKB (631–649) in complex with RIIα D/D domain and the peptides of RIIαD/D domain in the presence of the peptide. In addition, we see elevation in B-factors at the termini of the RIIα D/D domain, which corresponds to high deuteration rates as well. The flexible N-terminus (1–5) is boxed to show the differences between the two protomers. RIIαD/D’ exhibits higher B-factors for residues 4 and 5 and residues 1–3 are disordered (labeled by the black box), suggesting great flexibility for this region. Gly 8 is marked with an arrow, because it may act as a hinge for the flexible N-terminus. The helix I and helix II are highly protected when bound to the AKAP.
Docking of the nonpalindromic d-AKAP2 peptide to the hydrophobic pocket of RIIα induces inherent asymmetry. Because the D/D domain is symmetrical, there are two possible binding modes for the AKAP, in principle. Thus, the chances of forming a productive complex are increased, since the AKAP can dock with multiple approaches and orientations. Leu21R (21R’), and Leu9R (9R’) are located at the C- and N-termini of Helix I (I’), and are all held in place by the packing of the four-helix bundle. However, only one N-terminus is ordered by AKAP binding; the other is disordered suggesting that the stable docking site for the AKAP is only assembled as a tight fit when the AKAP is bound. This induced fit, stimulated by AKAP docking, most likely accounts for the slow off-rate associated with the interaction (Burns et al., 2003) and may contribute to the high affinity binding of the RII subunit. It is most likely why mutations of either Ile3R and Ile5R can no longer pull down AKAPs (Hausken et al., 1994; Li and Rubin, 1995).
The crystal structure combined with biochemical and biophysical studies support the binding mechanism sugeested for D-AKAP2
Several biophysical methods have been used previously to characterize this same AKAP peptide, and this data is all consistent with the above model. Using peptide arrays, the AKB of d-AKAP2 was systematically mutated previously to identify the determinants for RI and RII specificity (Burns-Hamuro et al., 2003). The array data (Supplementary Figure 1) confirmed the helicity of the AKAP peptide, the specific importance of 2 turns of the helix (boxed in the figure), and the stringent requirement for Ile-specific sites. Based on the Ala scan, the most critical residues for RIIα binding on our peptide are Ile8P, Ala9P, Ile12P, and Val13P; RIIα binding was abolished for nearly all mutations at these sites. These residues define the center of the hydrophobic ridge on the peptide that docks to the hydrophobic surface of the D/D domain. They represent only two turns of the d-AKAP2 helix where the first turn complements the portion of the docking surface created by the N-terminal tail and the second turn goes mostly to the hydrophobic groove created by Helix I and I’. In the crystal structure, Ile8P of the peptide contacts Ile3R and Ile5R of the D/D domain, and only an isoleucine is uniquely able to make these multiple contacts. The branching of the side chain at Cβ is critical for bridging to both Ile3R and Ile5R. Ile12P also uses both branches to contact Ile5R and Leu13R of the D/D domain. Peptide residues Leu4P and Ala5P, which also participate in docking but can accept most hydrophobic substitutions, are not at all buried in the pocket. Moreover, the N-terminus of RIIα is flexible enough to accommodate multiple substitutions at these positions. The fourth essential residue,Val13P, does not bind to the tail; it binds to the center of the hydrophobic groove created by Helix I and I’, and bulkier side chains are not accepted. We will discuss this residue later as a determinant for specificity. Val16P, also located in the hydrophobic groove, is clearly preferred for its position, but the peptide can bind with reduced affinity when Val16P is replaced with other hydrophobic residues. This is also consistent with our structure.
Backbone amide H/D exchange followed by mass spectrometry (H/DMS) was also used to characterize the binding interfaces of d-AKAP2 and RI and RII D/D domains (24). The H/D protection data is consistent with our structure and also correlates nicely with the B-factors (Figure 5). Although the d-AKAP2 fragment (residues 623–662) alone was highly deuterated, indicating that it was fully exposed to solvent, increased amide protection was detected upon binding to the RIIα D/D domain. Based on higher deuteration levels at both ends, the binding surface was restricted to 634–647. Residues 1–19 of our peptide coincide with 631–649 of d-AKAP2 while residues 4–17 correspond to the protected regions. This region was shown to be involved in binding to the D/D in the structure. Furthermore, there is a rise in B-factors for residues 18–22 on the peptide consistent with the lack of protection. The H/D exchange profile was also characterized for the RIIα D/D domain in the presence and absence of the d-AKAP2 peptide. Protection of RIIα D/D in the absence of the AKAP was localized on helix I and helix II, and both of these helices showed increased protection upon d-AKAP2 binding. As reported previously, AKAP binding is sensed across the helix bundle, not just at the binding interface (Burns-Hamuro et al., 2005; Fayos et al., 2003; Newlon et al., 1999).
Comparison of the crystal structure of RIIαDD-d-AKAP2 and the NMR solution structures of RIIαD/D with Ht31 and AKAP79
The NMR solution structures revealed that the hydrophobic faces of the AKAP amphipathic helices of Ht31 and AKAP79 docked onto a well-ordered solvent-accessible hydrophobic groove on the surface of the D/D domain (Newlon et al., 2001). Additional solution studies revealed that the residues in the solvent-accessible groove experience increase in dynamics upon complex formation with an AKAP peptide (Fayos et al., 2003). In addition, protomer specific assignments revealed reduced dynamics for one protomer (including Ile3) and disorder in the other protomer upon binding to a different natural AKAP peptide (Ht31). We hypothesized that this increase in dynamics would facilitate binding to the diverse sequences within the AKAP family of proteins as the hydrophobic groove could accommodate multiple sequences by altering the contour of the hydrophobic groove once bound to an AKAP. The current structure of the d-AKAP-2 complex supports this prediction and allows us to expand upon this theme.
Despite the obvious similarities in global structure, the new crystallographic model reveals how the D/D domain surface can accommodate peptide binding. The importance of the N-terminal extended region of RIIα containing Ile3R and Ile5R was first revealed by mutagenesis (Hausken et al., 1994). Although mutagenesis first demonstrated the importance of Ile3R and Ile5R and interactions involving these side chains were suggested in the NMR analysis, the presence of two protonated histidines at pH 4.0, necessary for NMR analysis, likely deemphasized their hydrophobic interactions (Newlon et al., 2001). The crystal structure demonstrates clearly the importance of these two isoleucines and explains their stringent requirement for binding.
In addition, the conformational flexibility of the C-terminus of the AKAP peptide apparent in the NMR analysis (Figure 6A) correlates with the B-factors in the crystal structure and H/DMS (Figure 5). In Figure 6A the NMR structures of Ht31 and AKAP79 bound to the RIIα D/D domain are aligned with the crystal structure with the d-AKAP2 peptide. The inherent flexibility of the N- and C-termini of the D/D domain is most apparent from the NMR structures but also correlates with the temperature factors in the crystal (Figure 5). The peptides bind in nearly the same space; however, the crystal structure reveals a change in helical register between the AKAPs by as much as a quarter-turn. Despite this, the Ht31 and AKAP79 peptides display a hydrophobic ridge similar to what we see with d-AKAP2 (Figure 6C). This highlights that helix-helix interactions do not determine the precise position of the helix backbone, because only the side chains are involved in the recognition process. In the case of d-AKAP2, Ht31, and AKAP79, all naturally occurring AKAP peptides, the ridge comprised of hydrophobic side chains represents the recognition motif. We expect, given the diversity of AKAPs and lack of an absolute consensus sequence, that a variety of peptide orientations can be accommodated.
Figure 6. Crystal and NMR structures reveal conserved features for AKAP docking to RIIα and provide insight into RI binding.
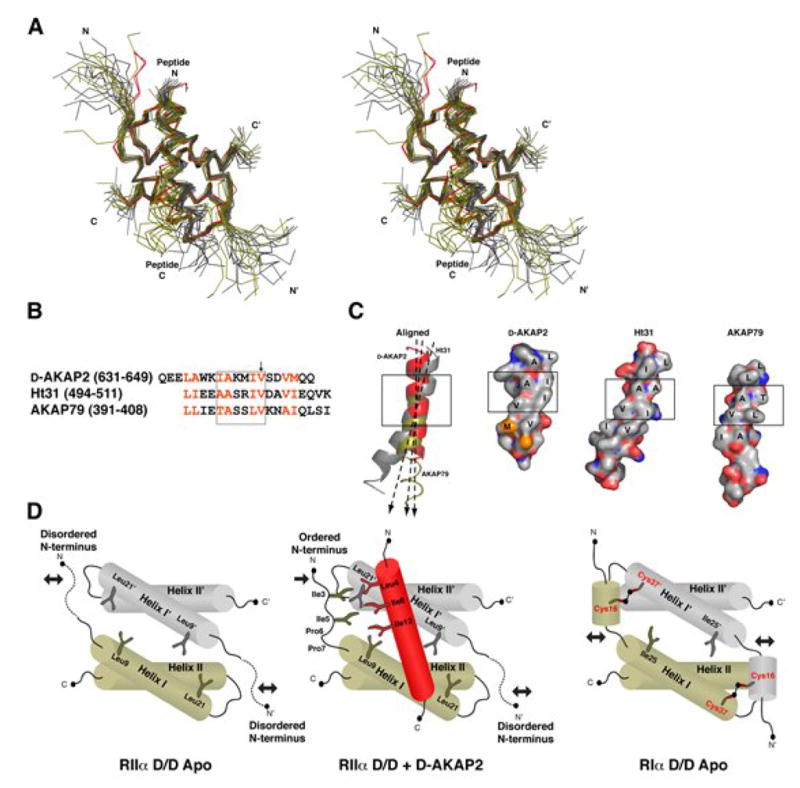
(A) Both Dimer I and Dimer II of the crystal structure (in red) are aligned with NMR structures of RIIα D/D bound to Ht31 (gray) and AKAP79 (olive) (Newlon et al., 2001). The structures are aligned based purely on Helix I and Helix I’. The AKAP peptides differ in helical register.
(B) The sequences of D-AKAP2, Ht31, AKAP79 involved in RIIα binding are aligned according to the structural alignment. The two turns of the helix that are critical for binding the D/D domain are shown in the box. The arrow points to Val13P, which is important for RI vs. RII specificity.
(C). Despite the diversity of sequence these AKAPs display a hydrophobic ridge that interacts with the D/D of RIIα. The two turns of the helices that are crucial for binding are boxed for each. Ht31 and AKAP79 peptides are each taken from one structure in an ensemble. The C-terminus of AKAP79 is not illustrated as a ribbon because it does not form a well ordered helix in the NMR structures. The axis of each peptide helix is shown to demonstrate that the D/D domain can accommodate different orientations of the AKAP helix.
(D) A cartoon representation of the D/D domain of RIIα subunits demonstrates the presence of a stable core and two flexible N-terminal tails. Upon binding to an AKAP, one of these tails is stabilized. RI subunits possess two conserved cysteines (Cys16R and Cys37R), which can stabilize the N-1 helix when disulfide bonded. Cys16R and Cys37R’ are located in analogous positions to Ile3R and Leu21R’ of RIIα (Banky et al., 2003), which are essential for binding. Crystal structures of RIα D/D may detail the specific involvement of these residues in AKAP docking.
AKAP binding specificity for RI vs. RII isoforms of PKA
While some AKAPs, such as d-AKAP2, have specificity for RI and RII subunits, most AKAPs do not bind well to RI. With the peptide array data, in combination with this structure, we can now define isoform specificity more precisely. As indicated in Supplementary Figure 1, the d-AKAP2 peptide can be divided into two segments: (1) the helical segment where side chains recognize the RIIα N-terminus and (2) the segment where the side chains dock to the hydrophobic groove. Clearly RII is much more tolerant of substitutions in the N-terminal site, as many point mutations in this region will abolish high affinity binding of RIα. Without a structure of this peptide bound to the RIα D/D domain, however, we cannot interpret those specificity differences more precisely. We can, however, address specificity differences in the hydrophobic groove, specifically the docking site for Val13P.
In contrast to the RIα subunit, the RIIα subunits are much more accommodating in terms of the AKAP sequences it can tolerate for binding. Many single point mutations will abolish RI binding, however, the peptide arrays revealed only one site in the d-AKAP2 peptide that strongly discriminates selectively against RIIα. This site is Val13R. RI subunits readily accept a Trp at this position, but replacing Val13P with Trp reduces RII affinity by 40-fold (Burns-Hamuro et al., 2003). Our crystal structure shows why. Trp is too bulky to fit in the hydrophobic groove of RIIα and would disrupt peptide docking by causing steric clashing with the surface created by Thr10R’, Leu13R’, Thr17R’ and Leu13R (Figure 7A,7B). Since Helix I and I’ align very well in RI and RII in the NMR structures, we aligned the NMR structure of the RIα D/D domain with our crystal structure. The homologous surface on RIα (created by Gln26R, Leu29R, Ile33R, and Leu29R’) contains a cavity sufficient for accommodating the bulky Trp side chain (Figure 7C) while the groove in RIIα is long and flat. This difference is caused by the orientation of the residue in the center of the groove, Leu13R and Leu29R in RIIα and RIα, respectively. Thus, while docking of the N-terminal tail is more restrictive for RI subunits, the hydrophobic groove corresponding to the docking site for one turn of the AKAP helix provides a surface that is actually more accommodating for RIα.
Figure 7. AKAP specificity for RII subunits of PKA can be toggled by Val13P mutations.
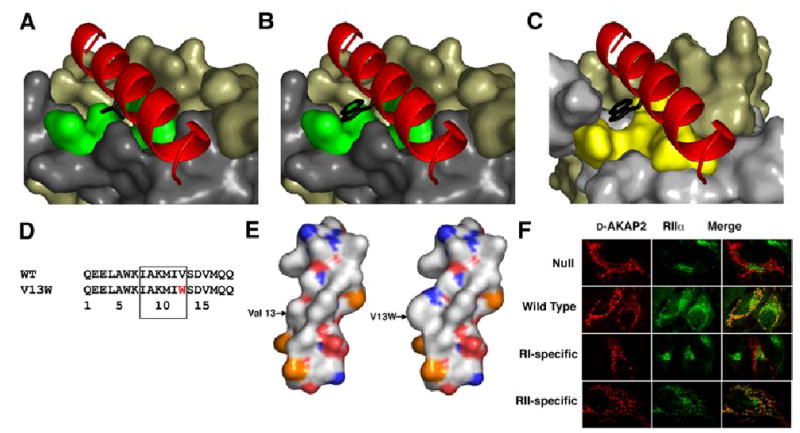
(A) Val13P, highlighted in black, binds to the surface of the D/D domain at the center of the hydrophobic groove comprised of Thr10R, Leu13R, Thr17R and Leu13R’, which are highlighted in green.
(B) Mutating Val13P to Trp eliminates binding to the RIIα D/D domain, as shown previously (Burns-Hamuro et al., 2003). Modeling this mutation into the D-AKAP2 in our structure causes steric clashes with the D/D domain; therefore, we believe this mutation disrupts the hydrophobic packing within the interface.
(C) By modeling our structure with the NMR structure of the RIα D/D domain (Banky et al., 2003), we observe a pocket in the RIα D/D domain that can accommodate the V13W mutation. This is consistent with peptide array data. Qln26R, Leu29R, Ile33R, and Leu29R’ form the surface highlighted in yellow can accommodate the Trp mutation in RIα.
(D) The sequence of the V13W mutation is shown above. This is located in the segment of the peptide that is critical for binding the D/D domains.
(E) The surfaces of the wild type and mutant peptides are quite different because the Trp protrudes from the peptide surface.
(F) The specificity of this mutation is demonstrated in vivo. Hela cells were transfected with wild-type, null, RI or RII specific versions of the D-AKAP2 peptide fused to a mitochondrial targeting sequence. Localization of these constructs is shown in the column on the left. RIIα was stained with fluorescent antibodies, which is shown in the middle column. Each AKAP construct was targeted to the mitochondria; however, only the wild type and RII-specific constructs successfully redirected the RIIα away from the golgi and to the mitochondria.
To test this in vivo, we transfected wild type and mutant versions of the d-AKAP2 peptide fused to a mitochondrial targeting sequence. With this vector we can not only disrupt targeting but also can redistribute endogenous R-subunits. In the mutant peptide, Val13 was replaced by Trp while two additional changes were engineered at Gln1 and Met17 for enhanced RI binding. The peptide has an affinity of 456 nM for RIIα, and we showed previously that this is sufficient to abolish targeting of a co-expressed RIIα subunit. We show here the effect of this mutation on the recruitment of endogenous RIIα. As seen in Figure 7, little RIIα is localized to the mitochondria with the null peptide whereas the wild type peptide recruits endogenous RIIα to the mitochondria. The introduction of a Trp at position 13, however, abolished the targeting of RIIα to the mitochondria as seen in Figure 7E. A single point mutation of Ala5P to Leu was sufficient to block the recruitment of endogenous RIα to the mitochondria but had no effect on the recruitment of endogenous RIIα. This mutation demonstrates the sensitivity of RI subunits to replacements in the N-terminus, but cannot be interpreted in molecular terms without a structure of the native peptide docked to RIα.
Plasticity of the D/D domain and the AKAP facilitates the diversity of cAMP signaling
More than 50 structurally diverse, but functionally similar, AKAP family members have been discovered to date (Michel and Scott, 2002). All AKAPs contain an amphipathic helix of 14–18 residues, which binds to the N-terminal dimerization/docking domain of the R subunits of PKA (Carr et al., 1991; Newlon et al., 1997; Newlon et al., 2001). The crystal structure of the RIIα D/D domain and d-AKAP2 provides insight into how RIIα binds such a wide array of AKAPs with high affinity. In the structure, the N-terminus of one protomer of RIIα is tightly bound to the AKAP, while the other is disordered (Figure 6D). The malleability of the N-terminus and the presence of two potential docking modes in the dimer enable the D/D domain to accommodate various sequences of AKAP peptides. The variability in helical register for bound AKAPs in the crystal and NMR structures confirms the plasticity and versatility of the dimerization/docking domain. In addition, peptide array data showed that only four residues on d-AKAP2 have strict sequence requirements for binding RIIα. The structure demonstrates how the RIIαD/D binding pocket can accommodate various side chains at the other positions of the AKAP peptide, especially at the N-terminus. This tight interaction can be summarized as a stable helix docking to a bipartite surface, where the surface created by Helix I and Helix I’ is rigid and the N-terminal tail is flexible (Figure 6D). The plasticity of the D/D domain, conveyed by the flexible N-termini, is likely to be crucial for facilitating PKA interactions with numerous AKAPs, allowing for amplified cAMP signaling at many cellular microdomains.
d-AKAP2 also has enhanced versatility compared to most AKAPs, because it can bind to both RI and RII isoforms with high affinity. Although the hydrophobic cores of RIα and RIIα are superimposable, the RI isoforms contain an abundance of charges on their AKAP binding surface (Banky et al., 2003). In addition, the N-terminus of the RI subunit contains a well-ordered helix, called the N-1 Helix. This helix, absent in RII subunits, replaces the N-terminal extended tail in RIIα. The flexible N-terminus of the RIIα subunit is positioned by two conserved prolines (Pro6 and Pro7) that are linked to the hydrophobic core by an intervening glycine. In contrast, the N-terminus of RIα is regulated by an intramolecular disulfide bond. The disulfide bond between Cys16R and Cys37R provides a stable anchor to the C-terminus of Helix I of the opposite protomer. Surprisingly, these two cysteines correlate spatially with Ile3R and Leu21R’ in the RIIα structure (Figure 6D). The N-terminus of RI is clearly more flexible when the cysteines are reduced. Moreover, the linker between the N-1 Helix and Helix I is dynamic, based on NMR structures (Banky et al., 2003). Crystal structures of RIαDD:AKAP complexes will reveal how d-AKAP2 can adapt to such differences. We expect that a network of both hydrogen bonds and salt bridges will contribute to the interface, in sharp contrast with the predominantly hydrophobic character of the RIIαD/d-AKAP2 complex.
Concurrent with our studies, a crystal structure of the RIIα D/D domain complexed with AKAP-IS, an AKAP peptide engineered for enhanced affinity and specificity for binding RII subunits was solved (Gold et al, submitted). As seen in Supplementary Figure 2, the D/D domain of this structure aligns closely with ours. Based on the alignment of the D/D domains, the AKAP peptides align in register such that the critical residues of d-AKAP2 for R-binding (Ile8P, Ala9P, Ile12P and Val13P) are located in the same space as Leu8P, Ala9P, Ile12P, and Val13P of AKAP-IS. This structure provides independent confirmation that these positions are structurally conserved, including Val13P, which is crucial for RII specificity. In addition, the AKAP-IS structure reveals ordering of Ile3 in only one protomer of the RIIα which contacts the N-terminus of the AKAP-IS, and thus independently confirms our model for binding. The AKAP-IS structure, however, is anchored by additional engineered hydrogen bonds and hydrophobic contacts that are not present in our structure.
Experimental Procedures
Protein expression and purification
Rattus norvegicus RIIα D/D (1–44) (Figure 1B) was cloned into a pET15b vector (Novagen) downstream of the his-tag and thrombin cleavage sequence, using NdeI and BamHI restriction sites. The clones were expressed in BL21 (DE3) competent E.coli (Stratagene) and grown in YT media containing 10 μg/ml ampicillin. Cells were grown to O.D.600 = 0.8, then induced with 0.5 mM IPTG for 6 hours. Cells were resuspended in 20mM Tris, 100mM NaCl and ruptured in a French pressure cell. The his-tagged RIIα D/D was purified using ProBond resin (Invitrogen) and eluted with 10 mM EDTA. The his-tag was then cleaved with thrombin for 36 hours. The protein was purified further using a Mono S cation exchange column in 20mM potassium phosphate (pH 5.0) with a gradient of 10–500 mM KCl followed by gel filtration on an S75 column (Pharmacia). Gel filtration was performed in the final buffer consisting of 10mM Na acetate (pH 5.0), 75 mM NaCl, 1 mM TCEP, and 10% glycerol. The protein was quantified based on the extinction coefficient 5960 M−1 cm−1 at 280 nm.
Crystallization of the RIIαD/D:peptide complex
The d-AKAP2 peptide QEELAWKIAKMIVSDVMQQCKK (Figure 1A) was synthesized and HPLC-purified to purity greater than 95% by Anaspec (San Jose). Residues 1–19 of this peptide correspond to residues 631–649 in d-AKAP2. Here we use the numbering indicated in Figure1A. The C-terminal residues are not part of d-AKAP2, but do not interfere with binding. Cys20P was added as a site for covalent labeling with a fluorophore (Burns-Hamuro et al., 2003), while the Lys21P and Lys22P improve the peptides solubility. Peptide was added to the purified RIIαD/D in a 2-fold molar excess. The complex was concentrated with a 3 kDa cutoff Amicon Ultra (Millipore) to 50mg/ml and subjected to crystallization trials using the hanging drop method. The complex crystallized in 100mM HEPES (pH 7.5), 20% PEG 8000 at 277 K after 1 week.
Crystallographic data collection and refinement
Data was collected at cryogenic temperature (100 K) using 20% glycerol as a cryoprotectant. Using the same single crystal, two complete data sets were collected, first on a rotating anode X-ray generator home source at UCSD and on synchrotron beamline 5.0.3 of the Advance Light Source, Lawrence Berkeley National Labs (Berkeley, California) (Table 1). Data was processed using Denzo/HKL2000 (Otwinowski et al., 1997). Due to dense ice rings at 3.75 Å, 2.24 Å, and 1.92 Å resolution, data bins with corresponding to these Bragg spacings showed 63.5%, 60.8 %, and 56.4 % completeness respectively The final data, however, showed a completeness of 87.9% for the resolution range of 35.0–1.60 Å. The initial model for the molecular replacement was constructed using the least varied region of the RIIα dimer from the NMR ensemble (residue 10R – 44R) (PDB access code:2DRN) (Newlon et al., 2001). Molecular replacement using MOLREP (Vagin and Teplyakov, 1997) of the CCP4 package (CCP4 1994) found two dimers in the asymmetric unit using all data between 25.0 and 2.5 Å resolution. The resulting electron density map showed clear density for each dimer, but not d-AKAP2 peptides. After a few rounds of manual model building followed by refinement with REFMAC5 (CCP4 1994), the improved phases clearly showed additional difference density for the peptides, at the periphery of each dimer which was used to model the peptides . After the addition of 214 waters using ARP/WARP (Lamzin et al., 1993), the final refinement implementing TLS refinement (Winn et al., 2001) for each chain converged to R and Rfree values of 0.208 and 0.238, respectively with excellent geometry (Table 1). The final model includes residues 1 to 43 of one protomer and residues 5 to 43 of the other protomer. Both molecules in the asymmetric unit are nearly identical with an RMSD of 0.17Å for the 96 equivalent Cα atoms. The shorter protomer is denoted with an (‘) for the sake of distinguishing between the two . Residues 2 to 22 and 2 to 20 were included for the AKAP peptides. The coordinates have been deposited in the Protein Data Bank under the accession code 2HWN [PDB]. Molecular representations in Figures 2, 3, 4, 6, and 7 were made using PyMol (DeLano Scientific) (DeLano, 2002).
Table 1.
Structure determination and statistics.
| Data set | Home source (Cu Kα) | ALS beamline 5.0.3 |
|---|---|---|
| Space group | C2 | C2 |
| Cell constants (Å, °) | a=98.9, b=44.3, c=72.9, β =124.0 | a=98.9, b=44.3, c=72.9, β = 124.0 |
| Number of crystals | 1 | 1 |
| Wavelength (Å) | 1.542 | 1.000 |
| Dmin (Å) | 2.67 | 1.6 |
| Mosaicity (°) | 0.6 | 0.6 |
| Unique reflections | 14461 | 40139 |
| Average redundancy | 2.6 (2.4) | 23.5 |
| Rsym⋄(%) | 5.4 (19.1)⊥ | 4.5 (41.1) |
| Completeness (%) | 97.9 (98.1) | 87.9§ (99.9) |
| <I>/<σI> | 22.2 (2.93) | 28.3 (2.8) |
| Resolution range for refinement (Å) | 20 to 1.6 | |
| Total reflections used | 30976 | |
| Number of protein atoms | 1663 | |
| Number of water molecules | 214 | |
| RMSD bond lengths (Å) | 0.007 | |
| RMSD bond angles (°) | 1.04 | |
| Rwork|| (%) | 20.8 | |
| Rfree¶ (%) | 23.8 | |
| Average B factor (Å2) | 38.4 |
Rsym = ∑h∑i|I(h) − I(h)i|I∑h∑i I(h)i, where I(h) is the mean intensity after rejections.
Numbers in parentheses correspond to the highest resolution shell of data, which were 2.8 to 2.7 Å for the Cu Kα data set and 1.66 to 1.6 Å for the ALS data set.
The diffraction images from ALS showed three ice rings at 3.75 Å, 2.24 Å, and 1.92 Å. Each data bins including corresponding ice rings showed 63.5%, 60.8 %, and 56.4 % completeness respectively.
Rwork = ∑h||Fobs(h)|−|Fcalc(h)||I∑h|Fobs(h)|; no I/σ cutoff was used during refinement.
5.1% of the truncated data set was excluded from refinement to calculate Rfree.
Amide H/D Exchange Coupled with Mass Spectrometry (H/DMS)
The H/D exchange experiments performed on d-AKAP2 and RIIα D/D domain were previously described (Burns-Hamuro et al., 2005).
Peptide Array Binding Studies
Peptide Array experiments were performed using the C-terminus of d-AKAP2 as were previously described (Burns-Hamuro et al., 2003).
Immunofluorescence studies
The targeting constructs containing the C-terminal 156 residues of d-AKAP2 with different mutations have a 30-residue mitochondrial targeting sequence from d-AKAP1a on the N-terminal and a Flag tag on the C-terminal as described previously (Burns-Hamuro et al., 2003). These constructs were used to transfect Hela cells using Polyfect agent (Qiagen) according to the manufacturer’s instructions. The cells were fixed with 4% paraformaldehyde for 10 min, 15h after transfection. The cells were then treated with 0.2% Triton-100 in PBS for 5 min before staining with antibodies sequentially in 1% BSA in PBS. A rabbit antibody against RIIα (Santa Cruz) and a fluorescein-conjugated secondary antibody both at a 1:100 dilution were used to stain endogenous RIIα. A cy3-conjugated anti-Flag antibody (Sigma) at a 1:400 dilution was used to stain the transfected targeting constructs. The images were taken with a confocal microscope (BioRad MRC1000).
Supplementary Material
Acknowledgments
We would like to thank N. Nguyen at UCSD x-ray facility for his assistance with data collection. We thank C. Ralston, A. Dauz, and the Advanced Light Source (ALS) staffs for their assistance with data collection at ALS beamline 5.0.1 and 8.3.1 (Lawrence Berkeley National Laboratory). We especially thank L. Ten Eyck for his insight on the final data processing and G. Sarma for his comments on refinement. We thank L. Burns and G. Anand for helpful comments and E. Radzio-Andzelm for her help with the figure 4. This work was funded in part by NIH DK 54441 to SST. FSK is supported by a NIH doctoral fellowship (1 F31 GM 67578-01). CK is supported by the American Cancer Fellowship Grant# PF-05-238-01-GMC. The Advanced light source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division of the U.S. Department of Energy under Contract DE-AC02-05CH11231 at Lawrence Berkeley National Laboratory. None of the authors have a conflict of interest related to the work discussed in this manuscript.
References
- Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, Scott JD. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A. 2003;100:4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banky P, Roy M, Newlon MG, Morikis D, Haste NM, Taylor SS, Jennings PA. Related protein-protein interaction modules present drastically different surface topographies despite a conserved helical platform. J Mol Biol. 2003;330:1117–1129. doi: 10.1016/s0022-2836(03)00552-7. [DOI] [PubMed] [Google Scholar]
- Burns LL, Canaves JM, Pennypacker JK, Blumenthal DK, Taylor SS. Isoform specific differences in binding of a dual-specificity A-kinase anchoring protein to type I and type II regulatory subunits of PKA. Biochemistry. 2003;42:5754–5763. doi: 10.1021/bi0265729. [DOI] [PubMed] [Google Scholar]
- Burns-Hamuro LL, Hamuro Y, Kim JS, Sigala P, Fayos R, Stranz DD, Jennings PA, Taylor SS, Woods VL., Jr Distinct interaction modes of an AKAP bound to two regulatory subunit isoforms of protein kinase A revealed by amide hydrogen/deuterium exchange. Protein Sci. 2005;14:2982–2992. doi: 10.1110/ps.051687305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson GL, Cantor CR, Braun A, Taylor SS. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci U S A. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- Cohen GB, Ren R, Baltimore D. Modular binding domains in signal transduction proteins. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- Fayos R, Melacini G, Newlon MG, Burns L, Scott JD, Jennings PA. Induction of flexibility through protein-protein interactions. J Biol Chem. 2003;278:18581–18587. doi: 10.1074/jbc.M300866200. [DOI] [PubMed] [Google Scholar]
- Gisler SM, Madjdpour C, Bacic D, Pribanic S, Taylor SS, Biber J, Murer H. PDZK1: II. an anchoring site for the PKA-binding protein D-AKAP2 in renal proximal tubular cells. Kidney Int. 2003;64:1746–1754. doi: 10.1046/j.1523-1755.2003.00267.x. [DOI] [PubMed] [Google Scholar]
- Hausken ZE, Coghlan VM, Hastings CA, Reimann EM, Scott JD. Type II regulatory subunit (RII) of the cAMP-dependent protein kinase interaction with A-kinase anchor proteins requires isoleucines 3 and 5. J Biol Chem. 1994;269:24245–24251. [PubMed] [Google Scholar]
- Heller WT, Vigil D, Brown S, Blumenthal DK, Taylor SS, Trewhella J. C subunits binding to the protein kinase A RI alpha dimer induce a large conformational change. J Biol Chem. 2004;279:19084–19090. doi: 10.1074/jbc.M313405200. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol. 2000;298:329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci U S A. 1997a;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997b;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- Kammerer S, Burns-Hamuro LL, Ma Y, Hamon SC, Canaves JM, Shi MM, Nelson MR, Sing CF, Cantor CR, Taylor SS, Braun A. Amino acid variant in the kinase binding domain of dual-specific A kinase-anchoring protein 2: a disease susceptibility polymorphism. Proc Natl Acad Sci U S A. 2003;100:4066–4071. doi: 10.1073/pnas.2628028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr. 1993;D49:129–149. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Li Y, Rubin CS. Mutagenesis of the regulatory subunit (RII beta) of cAMP-dependent protein kinase II beta reveals hydrophobic amino acids that are essential for RII beta dimerization and/or anchoring RII beta to the cytoskeleton. J Biol Chem. 1995;270:1935–1944. [PubMed] [Google Scholar]
- Michel JJ, Scott JD. AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol. 2002;42:235–257. doi: 10.1146/annurev.pharmtox.42.083101.135801. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Hausken ZE, Scott JD, Jennings PA. The A-kinase anchoring domain of type IIalpha cAMP-dependent protein kinase is highly helical. J Biol Chem. 1997;272:23637–23644. doi: 10.1074/jbc.272.38.23637. [DOI] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. Embo J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat Struct Biol. 1999;6:222–227. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Skalhegg BS, Tasken K, Hansson V, Huitfeldt HS, Jahnsen T, Lea T. Location of cAMP-dependent protein kinase type I with the TCR-CD3 complex. Science. 1994;263:84–87. doi: 10.1126/science.8272870. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. 2001;D57:122 – 133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.