

Abstract
Here we report the results of an analysis of variation at 128 EST-based microsatellites in wild Helianthus annuus, using populations from the species' typical plains habitat in Kansas and Colorado, as well as two arid desert and two distinct brackish marsh areas in Utah. The test statistics lnRV and lnRH were used to find regions of the genome that were significantly less variable in one population relative to the others and thus are likely to contain genes under selection. A small but detectable percentage (1.5–6%) of genes showed evidence for selection from both statistics in any particular environment, and a total of 17 loci showed evidence of selection in at least one environment. Distance-based measures provided additional evidence of selection for 15 of the 17 loci. Global FST-values were significantly higher for candidate loci, as expected under divergent selection. However, pairwise FST-values were lower for populations that shared a selective sweep. Moreover, while spatially separated populations undergoing similar selective pressures showed evidence of divergence at some loci, they evolved in concert at other loci. Thus, this study illustrates how selective sweeps might contribute both to the integration of conspecific populations and to the differentiation of races or species.
BECAUSE plants are sessile during a significant fraction of their life cycle, they are subject to strong selection for optimal performance under local environmental conditions. As a consequence, most plant populations are expected to exhibit significant local adaptation, an expectation that has been corroborated by a century of reciprocal transplant and common garden studies (Turesson 1922; Clausen et al. 1940, 1948; Schemske 1984; Stanton and Galen 1997; Donohue et al. 2000; Ramsey et al. 2003; Verhoeven et al. 2004; Angert and Schemske 2005). Significant progress has been made toward identifying phenotypic traits that contribute to adaptation and estimating the strength of selection on these traits (Arnqvist 1992; Dudley 1996; Dudley and Schmitt 1996; Nagy 1997; Petit and Thompson 1998; Weinig 2000; Kingsolver et al. 2001; Callahan and Pigliucci 2002; Ungerer et al. 2002; McKay et al. 2003; Etterson 2004; Gross et al. 2004; Griffith and Watson 2005). However, little is known about the genetic basis of locally adapted differences, particularly outside of the model plant, Arabidopsis thaliana (MitchellOlds 1996; Purugganan and Suddith 1998; Ungerer et al. 2002; Cork and Purugganan 2005; Stinchcombe et al. 2005). Important issues that remain to be addressed include the number, kinds, and effects of genes and mutations that contribute to local adaptation, the source of adaptive variants (new mutation, standing variation, or migration), the effects of genetic background and environment on allelic fitness, and the geographic extent of selective sweeps.
Providing answers to all of these questions, even within a single model system, is a formidable task. Nonetheless, progress is being made on several fronts. For example, QTL studies have been useful for estimating a minimum number of genes underlying fitness traits, either under laboratory (Sarigorla et al. 1992; MitchellOlds 1996; Bradshaw et al. 1998; Fry et al. 1998; Kim and Rieseberg 1999; Ungerer et al. 2003) or under field (Schemske and Bradshaw 1999; Lexer et al. 2003; Weinig et al. 2003b) conditions. QTL approaches also have been employed to estimate the fitness effects of particular genotypes across environments and have shown that different QTL may contribute to fitness when the environment changes (Weinig et al. 2003a). While these kinds of studies provide insight into the complex and conditional nature of adaptation to local environmental conditions, they rarely lead to a detailed understanding of the genetic basis of adaptation because a given QTL typically contains dozens or hundreds of genes, any one (or several) of which might be under selection. Only for major QTL is cloning and functional analysis likely to be feasible (although see (Kroymann and Mitchell-Olds 2005), yet these represent a biased subset of the genes and mutations contributing to adaptation.
Another strategy that has become increasingly popular is the search for correlations between sequence and expression variation at candidate genes and phenotypic variation at fitness-related traits (or fitness itself). This approach is likely to be most successful in model organisms (Pigliucci and Schmitt 1999; Kohn et al. 2000, 2003; Harr et al. 2002; Wayne and McIntyre 2002; Storz 2005), where the gene of interest has been functionally characterized. However, candidate gene lists are necessarily biased toward developmentally important or well-studied genes. These genes, while showing large phenotypic effects in the lab, may not be the most important sites involved in adaptation in nature.
While QTL and candidate gene genetic approaches provide insight into the number and kinds of genes that contribute to local adaptation, a new type of multilocus genetic screen may hold a more general route for the dissection of recent adaptation (Harr et al. 2002; Schlotterer 2002; Schlotterer and Dieringer 2005). Multiple individuals from two or more populations are genotyped, typically at microsatellite markers, and the test statics lnRV (Harr et al. 2002; Schlotterer 2002) and lnRH (Schlotterer and Dieringer 2005) are used to identify loci that are likely to have recently experienced strong selection. The rationale for this approach is that recent genetic changes driven by selection leave a record or “genetic signature” in the genome. These include a reduction of variation and/or increased differentiation at selected loci and tightly linked regions (Smith and Haigh 1974). This approach is particularly powerful when applied to the study of local adaptation, especially with multiple populations recently adapted to similar environmental conditions. The divergence of populations is recent and the cause of divergence from ancestral populations is clear––adaptation to different environmental conditions. This technique has the advantage of wide applicability to nonmodel systems, including those for which controlled crosses, and thus traditional genetics, are impossible. In addition, this type of forward-genetic screen may be somewhat less biased than an analysis based on obvious phenotypic differences between groups or known candidate genes. That is, selection may be detected for genes underlying traits that might otherwise not be measured.
Because this approach can be applied to natural populations, it is ideal for the study of genes that enable a species to adapt to new environments in the wild. This was used with some success in Drosophila (Schlotterer 2002; Schofl and Schlotterer 2004). A plant species that also has a remarkably diverse range of habitats is Helianthus annuus, the common sunflower. This species is among the most widespread and ubiquitous of the wild sunflower species in the United States (Heiser et al. 1969) and has populations adapted to numerous habitats, throughout North America from Mexico to Canada, and spanning both coasts. In some regions, hybridization with other native sunflower species may have allowed introgression of favorable alleles into the H. annuus background, thus facilitating the H. annuus invasion. This appears to have occurred in both Texas (Rieseberg et al. 1990) and California (Carney et al. 2000), but it is unclear if this mechanism is of widespread importance in H. annuus. In addition to introgression, adaptation in many areas could have involved either standing variation or new mutations in existing H. annuus alleles.
A particularly interesting region for examining local adaptation is the dry deserts of the American southwest. In much of the southwestern United States, H. annuus can grow only in more favorable years; sunflowers may have low or no germination if there is not enough moisture, and sunflower seeds may persist in the seed bank for many years (Alexander and Schrag 2003). However, sunflowers may instead obtain a more reliable source of water by living in or near a small stream or seep or living on the edge of a pond or marshy area. While plants in these environments escape desiccation, they are likely to encounter other, novel stresses, as water supplies may be contaminated by salts or other toxins. In addition, competition may be more important in habitats with a more reliable water supply.
The focus in this study is on a comparison of H. annuus from the fertile plains of Kansas and eastern Colorado with populations from the arid deserts of Utah. Additionally, multiple populations within Utah were compared, placing a special emphasis on the different adaptations to stresses specific to salt marshes surrounding the Great Salt Lake as compared to life in more arid habitats. For this study, we used microsatellites located within expressed sequence tags (ESTs), which have the advantage of being tightly linked to a known, expressed gene. Using a screen of 128 microsatellite markers, we looked for genes that may underlie adaptation to salt and drought stress in H. annuus. We also estimated gene flow (FST) between the different populations, overall and for individual loci, at both neutral and putatively selected loci. Several distance-based tests were used to look for additional evidence confirming the presence of selection and to infer its mode of action.
MATERIALS AND METHODS
Plant material:
In July 2003, fresh, young leaf tissue was collected from 48 plants in each of eight populations of H. annuus (Figure 1), including two plains populations (PLA1, PLA2) and two Utah desert populations (DES1, DES2) collected from typical habitats near roadsides, two brackish marsh populations from the northeast side of the Great Salt Lake (SMN1, SMN2), and two brackish marsh populations from the southwest side of the Great Salt Lake (SMS1, SMS2). Brackish marshes were identified by their proximity to visible salt flats. In addition, we identified any species within 10 m known to favor saline environments. Plants included in the survey were: Sarcobatus vermiculatus (greasewood), Atriplex sp. (saltbush), Salicornia utahensis (glasswort), and Allenrolfea occidentalis (iodine bush). All sites classified as saline had at least one of these four species growing nearby. DNA was extracted from 50 mg leaf tissue using DNeasy 96-well plate kits.
Figure 1.—

Map of populations. Shaded circles, plains populations; open circles, Utah desert populations; solid circles, brackish marsh populations.
Microsatellite analysis:
Screening was done using microsatellites derived from the sunflower Compositae Genome Project (CGP) database (http://compgenomics.ucdavis.edu/), which includes sequence information for ∼70,000 sunflower ESTs. A total of 288 microsatellite markers previously developed from the CGP database (http://cgpdb.ucdavis.edu/) were screened for reliability and the presence of variation in two populations (PLA1 and SMN1), yielding 102 usable markers. To add to these genes, the CGP database was searched using perl scripts for additional genes with useful microsatellites. Microsatellites were defined as any occurrence of five or more repeats of di-, tri, or tetramers. From this data set, 50 more microsatellites were chosen, and primers flanking the microsatellite were designed using Primer3, with an additional tail (GTCGTTTTACAACGTCGTG) added to the 5′ end of the forward primer for hybridization with fluorescent probes (Schuelke 2000), and synthesized by Integrated DNA Technologies (http://www.idtdna.com). As before, these 50 markers were screened for reliability and variability, and 26 primer pairs were chosen, yielding a total of 128 microsatellite markers analyzed on the full data set. It should be noted that this screening method is likely to remove loci that are consistently invariant in all populations examined. Thus, loci with extremely low mutation rates or consistently high levels of selection in all populations screened would not be included. However, invariant loci cannot yield meaningful comparisons between populations, so there is little that researchers can do to avoid this effect.
PCR:
During PCR, the 5′ end of the forward primer was labeled using fluorescent dye (Schuelke 2000; Wills et al. 2005). The PCR reactions were performed in volumes of 10 ml containing 2 ng DNA, 2 units Taq DNA polymerase, 0.2 μm fluorescent dye 6FAM, VIC, NED, or PET, 0.2 μm of each primer, 30 μm Tricine, 50 μm KCl, 2 μm MgCl2, and 100 μm each dNTP. Fragments were amplified using a “touchdown” PCR protocol, developed to reduce nonspecific primer binding and fragment amplification (Don et al. 1991). An initial denaturing cycle of 3 min at 95° was followed by 10 touchdown cycles, starting with a 58° annealing temperature but dropping 1° each cycle, of 30 sec at 94°, 30 sec at the annealing temperature, and 45 sec at 72°. These 10 cycles were followed by 29 cycles of 30 sec at 95°, 30 sec at 48°, and 45 sec at 72°, with a final elongation period at 72° for 20 min.
Genotyping:
Analysis of microsatellite fragment size was done on an ABI 3730 (Applied Biosystems, Foster City, CA). Multiple, nonoverlapping PCR fragments were pooled and diluted 1:60 with ddH2O. One microliter of the diluted PCR product was added to 8.9 μl ddH2O and 0.1 μl of the GenScan-500 Liz Size Standard (Applied Biosystems). Samples were denatured at 95°, snap cooled on ice, and then centrifuged to remove bubbles before loading onto the ABI 3730. For each marker, genotypes were scored using GeneMapper 3.7 (Applied Biosystems). Markers were scored automatically, with results manually corrected in cases of errors in the automatic scoring.
Selective sweeps:
For each population, observed heterozygosity, expected heterozygosity, and variance in microsatellite length were calculated using the program Microsatellite Analyzer (Dieringer and Schlotterer 2003). Global FST and all possible pairwise FST-values were calculated using the Weir and Cockerham method (Weir and Cockerham 1984) as implemented by GenePop 3.4 (Raymond and Rousset 1995). Additionally, global GST-values were calculated (Nei 1973) and yielded nearly identical results.
Populations were grouped into four pairs: the plains populations (PLA1, PLA2), the desert Utah populations (DES1, DES2), the southwestern Utah salt marsh populations (SMS1, SMS2), and the northeastern Utah salt marsh populations (SMN1, SMN2). These were verified as valid groupings using various algorithms including the neighbor-joining trees in PHILIP (Felsenstein 2005) and Bayesian clustering methods using the program Structure (Pritchard et al. 2000), as described below. Each of the four groups was then used as a single population for the lnRH and lnRV tests described below. Additionally, for some tests, all six of the Utah populations were grouped together for some analyses (see below).
To evaluate the significance of reductions in variance or heterozygosity, we used the statistics lnRH and lnRV, developed by Schlotterer and Dieringer (2005) and Schlotterer (2002), respectively. The two statistics lnRH and lnRV were calculated using the equations 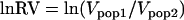 (Schlotterer 2002) and
(Schlotterer 2002) and 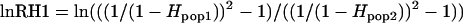 (Schlotterer and Dieringer 2005), respectively. These statistics are generally normally distributed, and simulations have confirmed that outliers are likely to be caused by selection (Schlotterer and Dieringer 2005). Additionally, independent, sequence-based measures of selection [e.g., HKA tests (Hudson et al. 1987)] have confirmed the existence of adaptive alleles within regions identified as candidates using these statistics (Harr et al. 2002; Vigouroux et al. 2002; DuMont and Aquadro 2005), as have SNP-based measures of reductions in nucleotide diversity (Ihle et al. 2006). It is important to note that these statistics are merely screens to emphasize candidate markers likely to be linked to genes under selection and do not indicate how close the marker is to the selected locus. Another caution in interpreting these tests is that they produce a low level of false positives (Schlotterer and Dieringer 2005). However, because the variance in repeat number and the heterozygosity of a population measure different aspects of the variation at a locus, using both statistics together lowers the rate of false positives by a factor of three (Schlotterer and Dieringer 2005). There is also a reduction in power, especially for detecting older sweeps. However, for detecting the most recent and strong selective sweeps, the combination of lnRH and lnRV statistics is as powerful as lnRV on its own, but with one-third the false-positive rate (Schlotterer and Dieringer 2005). Thus, for each locus, we calculated lnRH and lnRV statistics for several evolutionarily significant groupings. These groupings were based on the presumed relatedness among populations, as indicated by both neighbor-joining and Bayesian clustering algorithms (below). Specifically, we compared the plains to all Utah populations (desert and salt marsh) to test for differential selection associated with the invasion of the arid southwest. Within Utah, we compared the desert populations to each pair of salt marsh populations, pooled, to identify genes under selection, looking for potentially independent evolution of salt tolerance in each salt habitat. Significance of lnRH and lnRV for each comparison was calculated according to standard methods (Schlotterer 2002; Schlotterer and Dieringer 2005).
(Schlotterer and Dieringer 2005), respectively. These statistics are generally normally distributed, and simulations have confirmed that outliers are likely to be caused by selection (Schlotterer and Dieringer 2005). Additionally, independent, sequence-based measures of selection [e.g., HKA tests (Hudson et al. 1987)] have confirmed the existence of adaptive alleles within regions identified as candidates using these statistics (Harr et al. 2002; Vigouroux et al. 2002; DuMont and Aquadro 2005), as have SNP-based measures of reductions in nucleotide diversity (Ihle et al. 2006). It is important to note that these statistics are merely screens to emphasize candidate markers likely to be linked to genes under selection and do not indicate how close the marker is to the selected locus. Another caution in interpreting these tests is that they produce a low level of false positives (Schlotterer and Dieringer 2005). However, because the variance in repeat number and the heterozygosity of a population measure different aspects of the variation at a locus, using both statistics together lowers the rate of false positives by a factor of three (Schlotterer and Dieringer 2005). There is also a reduction in power, especially for detecting older sweeps. However, for detecting the most recent and strong selective sweeps, the combination of lnRH and lnRV statistics is as powerful as lnRV on its own, but with one-third the false-positive rate (Schlotterer and Dieringer 2005). Thus, for each locus, we calculated lnRH and lnRV statistics for several evolutionarily significant groupings. These groupings were based on the presumed relatedness among populations, as indicated by both neighbor-joining and Bayesian clustering algorithms (below). Specifically, we compared the plains to all Utah populations (desert and salt marsh) to test for differential selection associated with the invasion of the arid southwest. Within Utah, we compared the desert populations to each pair of salt marsh populations, pooled, to identify genes under selection, looking for potentially independent evolution of salt tolerance in each salt habitat. Significance of lnRH and lnRV for each comparison was calculated according to standard methods (Schlotterer 2002; Schlotterer and Dieringer 2005).
Distance-based tests of selection:
Several different methods use population divergence, rather than heterozygosity or variance differences, to test for selection. To test for additional evidence of selection, we used the coalescent-based simulation methods of Beaumont and Nichols (1996) and Vitalis et al. (2001), as well as a more robust Baysean test (Beaumont and Balding 2004).
For the Beaumont and Nichols (1996) method, we used the program FDIST2 (http://www.rubic.rdg.ac.uk/∼mab/software.html) to calculate P-values for each locus. We used the program to generate 100,000 simulated loci, providing an expected, neutral distribution of FST-values. To match our experimental design, each simulation had 48 individuals per population, eight populations, and 128 loci. This method provides evidence for divergent selection by looking for outliers with higher than expected FST-values, controlling for heterozygosity (Beaumont and Nichols 1996).
The Vitalis et al. (2001) coalescence-based approach was performed using the program DETSEL 1.0 (http://www.univ-montp2.fr/∼genetix/detsel.html). Null distributions were generated using the following parameters: population size before the split N0 = 50, 500; mutation rate μ = 0.01, 0.001, 0.0001; ancestral population size Ne = 500, 5000, 50,000; time since bottleneck T0 = 50, 500, 5000; and time since divergence t = 50, 500. This test defines an expected distribution of data points, on the basis of a defined P-value (in this case P = q − 1 = 0.01), and defines those loci outside of this expected region as showing evidence for recent selection. It has the advantage of being able to take into consideration a wide range of potential parameter values simultaneously and giving results that are robust to these starting assumptions.
Beaumont and Balding's (2004) hierarchical-Bayesian method was performed using the program newfst.c, which generates 2000 Markov chain Monte Carlo (MCMC) simulated loci on the basis of the distribution of FST given the data. Outliers from our data set were identified on the basis of this distribution following Beaumont and Balding (2004).
Gene function:
Genes for which a significant reduction in variability was found were investigated further to determine their predicted function and expression patterns. Blasts against GenBank using tBlastn on NCBI were used to find significant hits (Altschul et al. 1990, 1997). These genes were categorized according to known cellular and molecular functions, using gene ontology (GO) functional groups. Gene ontology is a standardized vocabulary used to describe the biological function of a gene and is an important tool for relating the putative functions of different genes in separate cells, tissues, or organisms (Ashburner et al. 2000; Harris et al. 2006). Genes were assigned to six broad functional groups: GO:0003824 (catalytic activity), GO:0030528 (transcription regulator activity), GO:0019538 (protein metabolism), GO:0005575 (cellular component), GO:0006281 (DNA repair), and GO:0005554 (molecular function unknown). In a separate analysis of biological function, genes were categorized according to whether or not they are known to be involved in response to stress (GO:0006950). On the basis of the GO functional groups, chi-square tests were used to determine whether the genes under selection have a different distribution of function than expected by chance.
Expression patterns, gene flow, and marker characteristics:
Because the sunflower EST libraries were not normalized, the number of redundant ESTs should be correlated with expression level. For each gene, the number of ESTs found and the tissues they were extracted from were calculated from the sunflower EST database (http://cgpdb.ucdavis.edu/). Using Statistical Package for Social Sciences, chi-square tests were used to determine whether the patterns of expression in the loci with significant selective sweeps were significantly different from those expected by chance given the distribution of expression patterns in all genes analyzed. Additionally, chi-square tests were used to examine whether microsatellite markers were in different parts of selected genes (5′-UTR, coding, or 3′-UTR) than in genes with neutral patterns of evolution. One-way ANOVAs were used to look at the association between selected loci and motif size, mean repeat number, chromosomal location, and global FST-values for each marker.
Population structure:
(δμ)2 (Goldstein et al. 1995; Slatkin 1995) was used as a measure of genetic distance because of its expected linear relationship to time of divergence (Goldstein et al. 1995). The (δμ)2 measure of divergence was calculated using the software Microsatellite Analyzer (Dieringer and Schlotterer 2003) for all population pairs, and the resulting genetic distance matrix was used to build a neighbor-joining tree using the program NEIGHBOR in PHYLIP (Felsenstein 2005). Additionally, we investigated the population structure in further detail, using the Bayesian clustering method of Pritchard et al. (2000), as implemented by the program Structure2.1. The number of populations (K) was estimated using the “no admixture” ancestral model with correlated alleles, allowing maximal population resolution, with K ranging from 2 to 8. Three independent runs of 100,000 MCMC generations and 20,000 generations of “burn in” were used for each value of K. The recent migration status of individuals within the populations was then assessed by incorporating the population information into the model.
RESULTS
Number of inferred selective sweeps:
In all three comparisons, the averages for both lnRH and lnRV were very close to zero and values were normally distributed, as expected for populations of roughly equal sizes. There were no significant genomewide reductions in variation or heterozygosity in any given population or region, although slightly higher diversity was seen in the plains populations than in Utah. Including all three comparisons, 19 genes were found with significant (P < 0.05) lnRH. Note that this is approximately the number expected by chance. A partially overlapping set of 19 genes showed significant levels of reduction in one or more comparisons (P < 0.05) using lnRV, and 17 of these loci were significant for both lnRH and lnRV. These loci thus had significant reductions in both allelic diversity and variance in allele size. For all of these loci, in the populations thought to be under selection, one allele was nearly completely fixed, with a few other alleles at very low frequency. Simulations have indicated that lnRH and lnRV measure different aspects of variation, despite high levels of correlation between the statistics, and that using both statistics together eliminates at least two-thirds of the false positives(Schlotterer and Dieringer 2005). Thus, of the 17 genes with both lnRV and lnRH significant (P < 0.05), 7 are expected to be false positive under conservative assumptions, given the sample size and number of comparisons (Schlotterer and Dieringer 2005). Using a higher threshold for significance (P < 0.01), for which only one false positive would be expected, 13 genes showed reduced variation indicating selection. Five genes showed such dramatic reductions in variation that they were significant under almost any threshold for both statistics (P < 0.001). In addition to the possibility of statistical false positives, these candidate genes may of course not be the targets of selection, but merely tightly linked to them. However, linkage disequilibrium is known to be extremely low in wild sunflowers (Liu and Burke 2006), so the region swept due to selection is likely to be quite small. Despite the possible presence of a few false positives, the full set of 17 significant genes was used in further analyses, as these are at the very least good candidate loci for further study.
Of these 17 genes, 7 have been mapped in at least one study (S. Knapp, personal communication). These 7 microsatellites map to six different chromosomes. The 2 that map to the same linkage group (Figure 2) map to opposite ends of the linkage group. Moreover, in all seven cases, the nearest markers showed no reduction of lnRH (Figure 2) or lnRV (not shown). Thus, no evidence was found for linkage between markers under selection. It is thus likely that the 17 reductions in variation and heterozygosity represent 17 distinct selective sweeps.
Figure 2.—

Graphs of lnRH for mapped markers on three representative chromosomes, showing four examples of selective sweeps. Dashed line represents significance (P < 0.05).
Of the 17 candidate genes, 2 showed reduced variation only in the plains populations (Figure 3a). Four were found to be reduced significantly across all of the Utah populations examined (Figure 3a). Five and three loci had reductions in variation only in the northeastern (Figure 3b) and southwestern (Figure 3c) brackish marsh populations, respectively. Three loci, on the other hand, showed reductions in variation in both northeastern and southwestern salt populations, but not in desert populations. It should be emphasized that this result is not an artifact of spatial clustering of salt marsh populations; DES1 was closer to each of the salt marsh populations than they were to each other (Figure 1), but did not show the same selection patterns. Nor is this result likely to be due to a close relationship between the two salt populations; these populations do not cluster together when grouped by genetic distance [e.g., (δμ)2 (Figure 4) or fixation index (i.e., FST)]. Thus, spatially and genetically distinct populations thought to be undergoing similar selective pressures appear to have shared selective sweeps at several loci.
Figure 3.—

Graphs of lnRV and lnRH for all three comparisons. Gray areas contain points significant (P < 0.05) for lnRV and lnRH. (a) Plains populations vs. all Utah populations. (b) Utah desert vs. northeastern salt marsh populations (SMN). (c) Utah desert vs. southwestern salt marsh populations (SMS).
Figure 4.—

Unrooted neighbor-joining tree showing the relationships between the eight populations used, based on genetic distances, (δμ)2. Numbers represent bootstrap support, from 1000 iterations, using the Phylip 3.63 program “neighbor.”
Whether these represent independent, parallel sweeps or a single sweep that passed through multiple populations remains to be determined. In all three cases, the same size allele was apparently swept in both populations, which would apparently make a single mutation more likely. However, this could be due to two independent mutations in the same ancestral allele as well.
Gene function:
Candidate genes selected using the lnRH and lnRV statistics had a wide variety of predicted functions (Table 1). The 17 significant loci included genes with the following homology based on closest BLAST hits: four genes with catalytic or metabolic functions, five transcription factors, three cellular components, one DNA-repair gene, and four genes of unknown homology or function (Table 2). This was significantly different from the distribution expected by chance (χ2 = 15.28; 5 d.f.; P < 0.01), mainly due to fewer than expected genes with no known function or with kinase, ubiquitinase, or other protein metabolic function and more genes than expected with roles as transcription factors, cell structure, or catalytic functions. In addition to these molecular and structural functions, among these 17 candidate genes were 3 of the 4 known stress-response genes (GO: 0006950) from the 128 loci used, which was significantly different from that expected (χ2 = 4.25; 1 d.f.; P < 0.05). Of course, as stated above, it must be kept in mind that these genes are only candidate genes and may be tightly linked only to the actual locus under selection. However, the fact remains that their gene functions differ significantly from the expected distribution, indicating that they are not just a random set of genes.
TABLE 1.
Results of selection analyses lnRH, lnRV, fdist2, detSel, and newfst (see text), along with protein homology, putative function, and chromosomal locations for mapped markers
| Contig/EST | lnRHa | lnRVa | fdist2b | detSelc | newfstd | Populations | Function | Protein homology | Chromosome | cM |
|---|---|---|---|---|---|---|---|---|---|---|
| Contig 0291 | *** | ** | — | *** | — | SMS | Catalytic/metabolism | Secretory peroxidase | 15 | 36.2 |
| Contig 1731 | *** | *** | ***** | — | * | SMN | DNA binding | DNA-binding protein | 13 | 103 |
| Contig 1891 | *** | ** | ** | **** | * | Plains | DNA binding | DNA-binding protein | — | |
| Contig 2016 | *** | *** | — | — | * | SMN | DNA binding | Heat-shock protein | 16 | 36.8 |
| Contig 2135 | *** | ** | *** | **** | *** | SMS, SMN | DNA binding | Ovule development | — | |
| Contig 2281 | **** | *** | — | — | * | SMS, SMN | Catalytic/metabolism | Catalytic/metabolism | 11 | 39.1 |
| Contig 2293 | **** | **** | — | — | ** | Utah | Repair of DNA | Purple acid phosphatase | 8 | 79 |
| Contig 2727 | *** | *** | — | — | SMN | Unknown | — | 3 | 30 | |
| Contig 3403 | *** | **** | ** | *** | * | SMN | Unknown | None | — | — |
| Contig 3590 | *** | ** | — | — | — | Utah | Membrane | COPT5 | — | — |
| Contig 5468 | *** | *** | ** | — | ** | SMN | Catalytic/metabolism | Cellulose synthase like | — | — |
| QHA19L11 | *** | *** | *** | **** | ** | SMS | Unknown | None | — | — |
| QHA6L06 | ***** | ****** | — | — | *** | SMS | Membrane | Xyloglucan galactosyltransferase | — | — |
| QHB12L21 | *** | **** | *** | **** | ** | SMN | Cell wall | None | 10 | 77.4 |
| QHB33B17 | *** | ** | — | *** | **** | Utah | DNA binding | Squamosa promoter binding protein | 8 | 218 |
| QHG7N09 | **** | **** | — | **** | ** | Plains | Unknown | None | — | — |
| QHJ18D19 | ** | *** | — | *** | *** | SMS, SMN | Catalytic/metabolism | Putative dehydrogenase | — | — |
*P < 0.10, **P < 0.05, ***P < 0.01, ****P < 0.001, *****P < 0.0001.
Tests were performed following Schlotterer (2002) and Schlotterer and Dieringer (2005).
The results of the program fdist2, implementing the protocol from Beaumont and Nichols (1996).
The results from the program detSel, which performs the calculations outlined in Vitalis et al. (2001).
Summary of the results of the program newfst.c (Beaumont and Balding 2004).
TABLE 2.
Functional categories of genes analyzed, for all genes, for those found to be under selection, and the expected distribution
| GO category | Total | Observed selected | Expected selected |
|---|---|---|---|
| Catalytic/metabolism | 17 | 4 | 2 |
| Transcription | 15 | 5 | 2 |
| Protein metabolism | 18 | 0 | 2 |
| Cellular component | 6 | 3 | 1 |
| Repair of DNA | 2 | 1 | 1 |
| Unknown | 79 | 4 | 9 |
| Total | 128 | 17 | 17 |
Expression patterns, motifs:
The expression patterns of the significant genes were found to be not significantly different than expected by chance, although there were marginally significantly more genes from the seed hull tissues than expected when that tissue was analyzed alone (χ2 = 2.71; 1 d.f.; P < 0.1). There was no evidence for any effect of motif size, motif location, mean repeat number, chromosomal location, diversity of tissue expression, or level of expression on likelihood of detecting selective sweeps.
Distance-based test of selection:
Distance-based tests of selection are prone to false positives because of sensitivity to demographic history (Whitlock and McCauley 1999) and heterogeneity among loci in mutation rate (Slatkin 1995; Balloux and Goudet 2002). Nonetheless, we expect the set of loci identified by distance-based tests to be enriched for true positives. This appears to be what we find here. Fifteen of the 17 “selected” loci from the lnRH and lnRV tests also showed evidence of selection in at least one of the three distance-based tests (Table 1). Because of the sensitivity of any FST-based tests to locus-specific phenomena not related to selection, we limit our discussion to those loci that show selection in the more robust lnRV and lnRH tests, which are specifically designed to control for locus-specific phenomena (Schlotterer and Dieringer 2005). Nevertheless, the additional evidence of selection from the FST-based tests is important in bolstering the results from the more robust tests. Since each of the five tests used relies on slightly different assumptions, loci that are repeatedly found to be outside of the range expected for neutral loci are extremely good candidates for genes under selection.
Consistent with the results of all five tests, global FST-values were significantly higher for selected loci (P < 0.05), as expected under divergent selection (Storz 2005), averaging 0.11 in the neutral loci and 0.15 in the selected loci. It should be noted that all global FST-values calculated were significantly >0 (thus indicating population structure).
Population comparisons:
Using the Bayesian methods implemented by the program Structure2.1 (Pritchard et al. 2000), we were able show that the eight populations collected from were indeed distinct genetic clusters and did not show evidence for substantial recent admixture or migration. With eight populations assumed, between 95 and 100% of individuals clustered together into their appropriate population groupings, with only three populations showing even one individual with substantial genetic material from other populations. The Structure analysis was not informative regarding the true number of populations [as the number of populations, K, in the model was increased the posterior probabilities Pr(X | K) increased progressively, with no clear discontinuities]. However, the results did reveal several values for K that captured important aspects of clustering within the data. When the model was set at lower numbers of assumed populations, the populations tended to cluster according to geographic regions. With only two populations assumed, individuals collected from all six Utah populations clustered together in all three iterations, while individuals from Kansas and Colorado (PLA1 and PLA2) clustered as a separate group. With more populations assumed, populations clustered into pairs. SMS1 and SMS2 clustered together in every analysis where more than three populations were assumed. Similarly, DES1 and DES2 clustered together in at least two of three of the runs for values of K from 3 to 7. SMN1 and SMN2 seemed to be more differentiated, but did cluster together in some analyses. The two pairs of salt marsh populations did not tend to cluster together; rather, SMS1 and SMS2 tended to cluster more closely with DES1 and DES2, particularly with the geographically closer DES2. On the basis of this analysis, we conclude that the four groupings used for the lnRH and lnRV tests are valid. Also, it appears that the two salt marsh regions show little genetic relationship or gene flow and show much closer genetic relationships to geographically proximal desert populations.
DISCUSSION
The geographic extent of selective sweeps:
The origin and spread of adaptive mutations has implications not only for understanding local adaptation and population-level phenomena, but also for interpreting the cohesions and divisions at the species level. For example, some authors argue that species are products of rather than units of evolution because gene flow within species often is too low to prevent differentiation through drift or local adaptation (Ehrlich and Raven 1969; Mishler 1999). However, even very low migration rates are sufficient for the spread of advantageous alleles (Morjan and Rieseberg 2004), leading Rieseberg and Burke (2001) to propose that selective sweeps associated with favorable mutations may allow species to be held together at some loci, while simultaneously differentiating at others.
The results presented here are broadly consistent with this proposal. While some genes sweep to fixation only within local populations, other genes sweep across a broader range of the species. Some selective sweeps define a single salt-tolerant population, three occur in spatially separated salt marsh populations, while others appear to have reduced variation across all of the Utah populations examined. Unfortunately, our design did not allow us to detect sweeps that have occurred across all populations of H. annuus, but another analysis utilizing comparisons with a hybridizing congener, H. petiolaris, indicates that specieswide selective sweeps are common as well (Y. Yatabe and L. H. Rieseberg, unpublished data).
There was a clear pattern of higher FST-values, and thus lower effective migration rates, for genes under selection than for other loci. This shows that selection may maintain higher levels of population differentiation at swept loci (FST = 0.15) than at other loci (FST = 0.11). However, for the four genes that were swept across all of Utah, there were dramatic increases in effective gene flow, such that pairwise FST-values were not significantly >0 between any Utah populations for these genes. Thus, selection has effectively obliterated all population structure within Utah at these loci.
Although spatially separated populations undergoing similar selective pressures show evidence of divergence at some loci, they evolve in concert at others. This is illustrated by three sweeps that are exclusive to the spatially separated salt marsh populations. In two of the cases, the same allele appears to be fixed in all salt populations examined. Those selective sweeps point to the importance of an allele that spreads through both clusters of salt marsh populations despite hundreds of miles of separation. In the third case, all salt populations examined show strong reductions in allelic diversity, but the northern salt populations are fixed for a different allele than the southern populations. This is likely due to two completely independent selective sweeps, each due to a separate adaptive mutation. For all three loci, whether the coincident patterns of variation occurred through long-distance dispersal of a new mutation or parallel evolution based on ancestral variation (Wood et al. 2005), strong selection at each locus is likely to be the cause of reduced variation in all populations. This is consistent with previous work looking at parallel evolution of edaphic races in Laesthenia californica (Rajakaruna et al. 2003).
In the present study, very few genes showed evidence for selection in the more benign and common plains populations, while relatively more genes appeared to be under selection in the more stressful and unusual environment of the salt marshes. The generality of this pattern of increased rates of molecular evolution in novel, unusual, or recently colonized environments remains to be determined. This may explain the widely reported pattern of increased phenotypic evolution in novel environments (Singer et al. 1993; Huey et al. 2000; Reznick and Ghalambor 2001; Schofl and Schlotterer 2004; Yeh 2004; Bonser and Geber 2005).
Caveats:
Because they are natural, not experimental, populations, the populations studied differ in many characteristics other than precipitation and salinity. Thus, some of the differentiation observed could have been due to some other selective force, such as herbivory, pathogens, or disturbance regime. Moreover, it is important to keep in mind that selection is not the only way that the distribution of variation can be changed at particular loci—reduced variation or increased differentiation can result from chance alone, e.g., genetic drift, bottlenecks, or founder events. As in this study, false positives can be minimized by using tests that account for population differences, looking for locus-specific effects, and by using multiple measures of variability, each of which measures different parameters and relies on different assumptions, e.g., heterozygosity and variance in allele size (Schlotterer and Dieringer 2005). Thus, loci with significant reductions in variability and/or increased differentiation are good candidates for further study and are quite likely to have been under recent selection (Schlotterer 2002; Vigouroux et al. 2002).
Of course, the microsatellites themselves are not likely to be the target of selection, but are merely tightly linked to it. Since each of the microsatellite loci used is located within ESTs, this gives us one good candidate gene that is known to be expressed and is tightly linked to each locus. In many of the cases so far examined, selective sweeps have affected only a very small region, potentially containing only one or a few genes (Clark et al. 2004; DuMont and Aquadro 2005; Storz 2005), except in the case of extremely strong selection (Kohn et al. 2000; Wootton et al. 2002). Moreover, recent estimates of linkage disequilibrium in wild H. annuus as negligible after 200–300 bp (Liu and Burke 2005) indicate that in most cases the region of reduced variability is likely to contain only a single gene or domain. Results for the domesticated H. annuus cultivars showed little linkage disequilibrium after 1800–1900 bp, which may be more reasonable for the more recently derived wild populations or for genes under strong selection. In any case, even if linkage disequilibrium is much higher, when a marker is found to be in a swept region, that marker is likely to be close to the actual locus under selection. Because of the small size of swept regions, however, even with the substantial number of loci we used, we are likely to have found only a very small fraction of the genes under selection in these H. annuus populations.
Nature of selected genes:
The nature of this study precludes speculation on the type of genes that may sweep across an entire species range; for that a broader sampling of populations from multiple, closely related species would be necessary. However, this work provides insight into the percentage and types of genes involved in local and regional selective sweeps. In this study, we found that 1.5–6% of the loci examined, depending on the environment, showed significant reductions in heterozygosity and variance consistent with a recent selective sweep. Most of these loci showed additional evidence for selection on the basis of other statistical tests as well. Thus, while the large number of genes apparently under selection is somewhat surprising, our results are based on many independent tests of selection. Moreover, they are consistent with a growing body of evidence that a large percentage of a typical genome shows evidence for recent selection (Diller et al. 2002; Harr et al. 2002; Vigouroux et al. 2002, 2005; Kauer et al. 2003; Kayser et al. 2003; Schofl and Schlotterer 2004; Storz et al. 2004; Vasemagi et al. 2005; Wright et al. 2005).
The genes under selection exhibit a wide range of putative functions, including DNA binding, DNA repair, formation of cell walls, and catalyzing biosynthetic or metabolic reactions, as well as a number of unknown functions. The phenotypes associated with alternate alleles remain to be determined, but these results imply that adaptation to salt and drought is polygenic. Additionally, a substantial proportion of the selected genes (18%) show homology to known stress-response genes, from the GO classification, thus indicating that responses to abiotic stresses are indeed a major component of adaptation to the habitats examined. It should be emphasized, once again, that these genes are only candidate genes showing tight linkage to the markers identified as being under selection.
The three putative stress response genes apparently under selection have a variety of putative functions. One shows 85% identity to a secretory peroxidase thought to be involved in response to several different stresses (Tognolli et al. 2002) and that is highly homologous to a peroxidase upregulated in response to salt stress in cotton (Ritter et al. 1993). Another shows homology to a heat-shock protein from Arabidopsis. The third shows homology to a putative purple acid phosphatase, which is involved in response to phosphate starvation (Duff et al. 1994) and potentially other stresses (Olczak et al. 2003). In addition to the genes classified as “stress response” genes in GO, some of the selected genes have known or putative functions in adaptation to salinity or drought. For instance, one candidate locus shows homology to tocopherol cyclase, which can enhance tolerance of oxidative stress (Kanwischer et al. 2005).
In addition to these potential stress-response genes, a number of genes appear to be involved in determining the structure or function of the cell wall and cellular membrane. These include two ESTs with homology to membrane proteins (one of which is an ion channel) and one with homology to a cellulose-synthase-like protein. Other genes of interest include a squamosa promoter-binding protein, part of a family of DNA-binding proteins found only in plants (Klein et al. 1996), which regulates the expression of the MADS-box gene squamosa (Huijser et al. 1992), and AINTEGUMENTA (Elliott et al. 1996; Klucher et al. 1996), an APETALLA-2 like gene involved in floral organ development.
In addition to homology, the expression patterns of the genes in our database give us some idea of the types of traits potentially involved in adaptation. The prevalence of seed hull genes as candidates for genes under selection is interesting, as several aspects of seed morphology are extremely variable between populations, and, on the basis of our observations, are likely to be under selection. Seed coat coloration is one of the most variable traits between wild sunflower populations, with seeds colored to match the soil surface in every population examined (R. Randel, personal communication). Germination characteristics, which also vary highly between populations of H. annuus, are also likely to be affected by seed coat morphology and composition. Thus, these traits merit further study in these populations, with particular emphasis on the role of allelic variation at the selected loci.
Conclusions and future directions:
In summary, this study provides strong evidence that a large proportion of the genome is involved in adaptation and that selective sweeps play a major role in determining gene flow and genetic differences between populations. In terms of the types of genes potentially involved in adaptation, transcription factors appear to be quite important, but so do genes involved in catalyzing biosynthetic or metabolic reactions. In contrast, we found no evidence for selection on any of the protein kinases or other genes involved in protein synthesis, degradation, or regulation in our data set, despite their prevalence as the most common functional group.
Currently underway, an effort to sequence alleles of these candidate genes from these populations should provide insight into the differences between adaptive and ancestral sequences and may help us understand the origin of these adaptive alleles—either as new mutants or as part of the standing variation prior to selection. By gaining a greater understanding of the molecular basis for local adaptation, we hope to understand the contributions of standing variation, mutation, and migration in allowing species to colonize new habitats.
Acknowledgments
We thank Sarah Elmendorf, Briana Gross, Daniel Ortiz-Barrientos, Jared Strassburg, Yuval Sapir, Yoko Yatabe, and three anonymous reviewers for helpful advice and comments. This research was funded by National Science Foundation (NSF) (DBI0421630) and National Institutes of Health (GM059065) grants to L.H.R., as well as by NSF predoctoral and Integrative Graduate Education and Research Traineeship fellowships to N.C.K.
References
- Alexander, H. M., and A. M. Schrag, 2003. Role of soil seed banks and newly dispersed seeds in population dynamics of the annual sunflower, Helianthus annuus. J. Ecol. 91: 987–998. [Google Scholar]
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang et al., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angert, A. L., and D. W. Schemske, 2005. The evolution of species' distributions: reciprocal transplants across the elevation ranges of Mimulus cardinalis and M. lewisii. Evolution 59: 1671–1684. [PubMed] [Google Scholar]
- Arnqvist, G., 1992. Spatial variation in selective regimes—sexual selection in the water strider Gerris odontogaster. Evolution 46: 914–929. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., C. A. Ball, J. A. Blake, D. Botstein, H. Butler et al., 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balloux, F., and J. Goudet, 2002. Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Mol. Ecol. 11: 771–783. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and D. J. Balding, 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13: 969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and R. A. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. Ser. B Biol. Sci. 263: 1619–1626. [Google Scholar]
- Bonser, S. P., and M. A. Geber, 2005. Growth form evolution and shifting habitat specialization in annual plants. J. Evol. Biol. 18: 1009–1018. [DOI] [PubMed] [Google Scholar]
- Bradshaw, H. D., K. G. Otto, B. E. Frewen, J. K. McKay and D. W. Schemske, 1998. Quantitative trait loci affecting differences in floral morphology between two species of monkeyflower (Mimulus). Genetics 149: 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, H. S., and M. Pigliucci, 2002. Shade-induced plasticity and its ecological significance in wild populations of Arabidopsis thaliana. Ecology 83: 1965–1980. [Google Scholar]
- Carney, S. E., K. A. Gardner and L. H. Rieseberg, 2000. Evolutionary changes over the fifty-year history of a hybrid population of sunflowers (Helianthus). Evolution 54: 462–474. [DOI] [PubMed] [Google Scholar]
- Clark, R. M., E. Linton, J. Messing and J. F. Doebley, 2004. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Natl. Acad. Sci. USA 101: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen, J., D. D. Keck and W. M. Hiesey, 1940. Experimental studies on the nature of species. I. The effect of varied environments on western North American plants. Pub. 520, Carnegie Institution, Washington, DC.
- Clausen, J., D. D. Keck and W. M. Hiesey, 1948. Experimental studies on the nature of species. III. Environmental responses of climatic races of Achillea. Pub. 581, Carnegie Institution, Washington, DC.
- Cork, J. M., and M. D. Purugganan, 2005. High-diversity genes in the Arabidopsis genome. Genetics 170: 1897–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieringer, D., and C. Schlotterer, 2003. MICROSATELLITE ANALYSER (MSA): a platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 3: 167–169. [Google Scholar]
- Diller, K. C., W. A. Gilbert and T. D. Kocher, 2002. Selective sweeps in the human genome: a starting point for identifying genetic differences between modern humans and chimpanzees. Mol. Biol. Evol. 19: 2342–2345. [DOI] [PubMed] [Google Scholar]
- Don, R. H., P. T. Cox, B. J. Wainwright, K. Baker and J. S. Mattick, 1991. Touchdown PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 19: 4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue, K., D. Messiqua, E. H. Pyle, M. S. Heschel and J. Schmitt, 2000. Evidence of adaptive divergence in plasticity: density- and site-dependent selection on shade-avoidance responses in Impatiens capensis. Evolution 54: 1956–1968. [DOI] [PubMed] [Google Scholar]
- Dudley, S. A., 1996. The response to differing selection on plant physiological traits: evidence for local adaptation. Evolution 50: 103–110. [DOI] [PubMed] [Google Scholar]
- Dudley, S. A., and J. Schmitt, 1996. Testing the adaptive plasticity hypothesis: density-dependent selection on manipulated stem length in Impatiens capensis. Am. Nat. 147: 445–465. [Google Scholar]
- Duff, S. M. G., G. Sarath and W. C. Plaxton, 1994. The role of acid phosphatases in plant phosphorus metabolism. Physiol. Plant. 90: 791–800. [Google Scholar]
- DuMont, V. B., and C. F. Aquadro, 2005. Multiple signatures of positive selection downstream of notch on the X chromosome in Drosophila melanogaster. Genetics 171: 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, P. R., and P. H. Raven, 1969. Differentiation of populations. Science 165: 1228–1232. [DOI] [PubMed] [Google Scholar]
- Elliott, R. C., A. S. Betzner, E. Huttner, M. P. Oakes, W. Tucker et al., 1996. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell 8: 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etterson, J. R., 2004. Evolutionary potential of Chamaecrista fasciculata in relation to climate change. 1. Clinal patterns of selection along an environmental gradient in the great plains. Evolution 58: 1446–1458. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J., 2005. Phylogeny inference package (Version 3.2). Cladistics 5: 164–166. [Google Scholar]
- Fry, J. D., S. V. Nuzhdin, E. G. Pasyukova and T. F. C. McKay, 1998. QTL mapping of genotype-environment interaction for fitness in Drosophila melanogaster. Genet. Res. 71: 133–141. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. B., A. R. Linares, L. L. Cavallisforza and M. W. Feldman, 1995. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc. Natl. Acad. Sci. USA 92: 6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith, T. M., and M. A. Watson, 2005. Stress avoidance in a common annual: reproductive timing is important for local adaptation and geographic distribution. J. Evol. Biol. 18: 1601–1612. [DOI] [PubMed] [Google Scholar]
- Gross, B. L., N. C. Kane, C. Lexer, F. Ludwig, D. M. Rosenthal et al., 2004. Reconstructing the origin of Helianthus deserticola: survival and selection on the desert floor. Am. Nat. 164: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., M. Kauer and C. Schlotterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99: 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M. A., J. I. Clark, A. Ireland, J. Lomax, M. Ashburner et al., 2006. The gene ontology (GO) project in 2006. Nucleic Acids Res. 34: D322–D326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser, C. B., D. M. Smith, S. Clevenger and W. C. Martin, 1969. The North American sunflowers (Helianthus). Mem. Torrey Bot. Club 22: 1–218. [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey, R. B., G. W. Gilchrist, M. L. Carlson, D. Berrigan and L. Serra, 2000. Rapid evolution of a geographic cline in size in an introduced fly. Science 287: 308–309. [DOI] [PubMed] [Google Scholar]
- Huijser, P., J. Klein, W. E. Lonnig, H. Meijer, H. Saedler et al., 1992. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene SQUAMOSA in Antirrhinum majus. EMBO J. 11: 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle, S., I. Ravaoarimanana, M. Thomas and D. Tautz, 2006. An analysis of signatures of selective sweeps in natural populations of the house mouse. Mol. Biol. Evol. 23: 790–797. [DOI] [PubMed] [Google Scholar]
- Kanwischer, M., S. Porfirova, E. Bergmuller and P. Dormann, 2005. Alterations in tocopherol cyclase activity in transgenic and mutant plants of Arabidopsis affect tocopherol content, tocopherol composition, and oxidative stress. Plant Physiol. 137: 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlotterer, 2003. A microsatellite variability screen for positive selection associated with the “Out of Africa” habitat expansion of Drosophila melanogaster. Genetics 165: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser, M., S. Brauer and M. Stoneking, 2003. A genome scan to detect candidate regions influenced by local natural selection in human populations. Mol. Biol. Evol. 20: 893–900. [DOI] [PubMed] [Google Scholar]
- Kim, S. C., and L. H. Rieseberg, 1999. Genetic architecture of species differences in annual sunflowers: implications for adaptive trait introgression. Genetics 153: 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsolver, J. G., H. E. Hoekstra, J. M. Hoekstra, D. Berrigan, S. N. Vignieri et al., 2001. The strength of phenotypic selection in natural populations. Am. Nat. 157: 245–261. [DOI] [PubMed] [Google Scholar]
- Klein, J., H. Saedler and P. Huijser, 1996. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol. Gen. Genet. 250: 7–16. [DOI] [PubMed] [Google Scholar]
- Klucher, K. M., H. Chow, L. Reiser and R. L. Fischer, 1996. The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell 8: 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, M. H., H. J. Pelz and R. K. Wayne, 2000. Natural selection mapping of the warfarin-resistance gene. Proc. Natl. Acad. Sci. USA 97: 7911–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, M. H., H. J. Pelz and R. K. Wayne, 2003. Locus-specific genetic differentiation at Rw among warfarin-resistant rat (Rattus norvegicus) populations. Genetics 164: 1055–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann, J., and T. Mitchell-Olds, 2005. Epistasis and balanced polymorphism influencing complex trait variation. Nature 435: 95–98. [DOI] [PubMed] [Google Scholar]
- Lexer, C., M. E. Welch, J. L. Durphy and L. H. Rieseberg, 2003. Natural selection for salt tolerance quantitative trait loci (QTLs) in wild sunflower hybrids: implications for the origin of Helianthus paradoxus, a diploid hybrid species. Mol. Ecol. 12: 1225–1235. [DOI] [PubMed] [Google Scholar]
- Liu, A., and J. M. Burke, 2006. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, J. K., J. H. Richards and T. Mitchell-Olds, 2003. Genetics of drought adaptation in Arabidopsis thaliana: I. Pleiotropy contributes to genetic correlations among ecological traits. Mol. Ecol. 12: 1137–1151. [DOI] [PubMed] [Google Scholar]
- Mishler, B. D., 1999. Getting rid of species?, pp. 307–315 in New Interdisciplinary Essays, edited by R. Wilson. MIT Press, Cambridge, MA.
- MitchellOlds, T., 1996. Genetic constraints on life-history evolution: quantitative-trait loci influencing growth and flowering in Arabidopsis thaliana. Evolution 50: 140–145. [DOI] [PubMed] [Google Scholar]
- Morjan, C. L., and L. H. Rieseberg, 2004. How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Mol. Ecol. 13: 1341–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, E. S., 1997. Selection for native characters in hybrids between two locally adapted plant subspecies. Evolution 51: 1469–1480. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak, M., B. Morawiecka and W. Watorek, 2003. Plant purple acid phosphatases—genes, structures and biological function. Acta Biochim. Pol. 50: 1245–1256. [PubMed] [Google Scholar]
- Petit, C., and J. D. Thompson, 1998. Phenotypic selection and population differentiation in relation to habitat heterogeneity in Arrhenatherum elatius (Poaceae). J. Ecol. 86: 829–840. [Google Scholar]
- Pigliucci, M., and J. Schmitt, 1999. Genes affecting phenotypic plasticity in Arabidopsis: pleiotropic effects and reproductive fitness of photomorphogenic mutants. J. Evol. Biol. 12: 551–562. [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure from mulitlocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purugganan, M. D., and J. I. Suddith, 1998. Molecular population genetics of the Arabidopsis CAULIFLOWER regulatory gene: nonneutral evolution and naturally occurring variation in floral homeotic function. Proc. Natl. Acad. Sci. USA 95: 8130–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakaruna, N., B. G. Baldwin, R. Chan, A. M. Desrochers, B. A. Bohm et al., 2003. Edaphic races and phylogenetic taxa in the Lasthenia californica complex (Asteraceae: Heliantheae): an hypothesis of parallel evolution. Mol. Ecol. 12: 1675–1679. [DOI] [PubMed] [Google Scholar]
- Ramsey, J., H. D. Bradshaw and D. W. Schemske, 2003. Components of reproductive isolation between the monkeyflowers Mimulus lewisii and M. cardinalis (Phrymaceae). Evolution 57: 1520–1534. [DOI] [PubMed] [Google Scholar]
- Raymond, M., and F. Rousset, 1995. Genepop(version-1.2). Population genetics softward for exact tests and ecumenicism. J. Hered. 86: 248–249. [Google Scholar]
- Reznick, D. N., and C. K. Ghalambor, 2001. The population ecology of contemporary adaptations: what empirical studies reveal about the conditions that promote adaptive evolution. Genetica 112: 183–198. [PubMed] [Google Scholar]
- Rieseberg, L. H., and J. M. Burke, 2001. The biological reality of species: gene flow, selection, and collective evolution. Taxon 50: 47–67. [Google Scholar]
- Rieseberg, L. H., S. Beckstromsternberg and K. Doan, 1990. Helianthus annuus ssp texanus has chloroplast DNA and nuclear ribosomal-RNA genes of Helianthus debilis ssp cucumerifolius. Proc. Natl. Acad. Sci. USA 87: 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter, D., R. D. Allen, N. Trolinder, D. W. Hughes and G. A. Galau, 1993. Cotton cotyledon cDNA encoding a peroxidase. Plant Physiol. 102: 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarigorla, M., M. E. Pe, D. L. Mulcahy and E. Ottaviano, 1992. Genetic dissection of pollen competitive ability in maize. Heredity 69: 423–430. [Google Scholar]
- Schemske, D. W., 1984. Population structure and local selection in Impatiens pallida (Balsaminaceae), a selfing annual. Evolution 38: 817–832. [DOI] [PubMed] [Google Scholar]
- Schemske, D. W., and H. D. Bradshaw, 1999. Pollinator preference and the evolution of floral traits in monkeyflowers (Mimulus). Proc. Natl. Acad. Sci. USA 96: 11910–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer, C., 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer, C., and D. Dieringer, 2005. A novel test statistic for the identification of local selective sweeps based on microsatellite gene diversity, pp. 55–64 in Selective Sweep, edited by D. Nurminsky. Landes Bioscience, Georgetown, TX.
- Schofl, G., and C. Schlotterer, 2004. Patterns of microsatellite variability among X chromosomes and autosomes indicate a high frequency of beneficial mutations in non-African D. simulans. Mol. Biol. Evol. 21: 1384–1390. [DOI] [PubMed] [Google Scholar]
- Schuelke, M., 2000. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18: 233–234. [DOI] [PubMed] [Google Scholar]
- Singer, M. C., C. D. Thomas and C. Parmesan, 1993. Rapid human-induced evolution of insect host associations. Nature 366: 681–683. [Google Scholar]
- Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. M., and J. Haigh, 1974. Hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- Stanton, M. L., and C. Galen, 1997. Life on the edge: adaptation versus environmentally mediated gene flow in the snow buttercup, Ranunculus adoneus. Am. Nat. 150: 143–178. [DOI] [PubMed] [Google Scholar]
- Stinchcombe, J. R., A. L. Caicedo, R. Hopkins, C. Mays, E. W. Boyd et al., 2005. Vernalization sensitivity in Arabidopsis thaliana (brassicaceae): the effects of latitude and FLC variation. Am. J. Bot. 92: 1701–1707. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., 2005. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 14: 671–688. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., B. A. Payseur and M. W. Nachman, 2004. Genome scans of DNA variability in humans reveal evidence for selective sweeps outside of Africa. Mol. Biol. Evol. 21: 1800–1811. [DOI] [PubMed] [Google Scholar]
- Tognolli, M., C. Penel, H. Greppin and P. Simon, 2002. Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 288: 129–138. [DOI] [PubMed] [Google Scholar]
- Turesson, G., 1922. The genotypical response of the plant species to the habitat. Hereditas 3: 211–350. [Google Scholar]
- Ungerer, M. C., S. S. Halldorsdottir, J. L. Modliszewski, T. F. C. Mackay and M. D. Purugganan, 2002. Quantitative trait loci for inflorescence development in Arabidopsis thaliana. Genetics 160: 1133–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerer, M. C., C. R. Linder and L. H. Rieseberg, 2003. Effects of genetic background on response to selection in experimental populations of Arabidopsis thaliana. Genetics 163: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasemagi, A., J. Nilsson and C. R. Primmer, 2005. Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon (Salmo salar L.). Mol. Biol. Evol. 22: 1067–1076. [DOI] [PubMed] [Google Scholar]
- Verhoeven, K. J. F., T. K. Vanhala, A. Biere, E. Nevo and J. M. M. Van Damme, 2004. The genetic basis of adaptive population differentiation: a quantitative trait locus analysis of fitness traits in two wild barley populations from contrasting habitats. Evolution 58: 270–283. [PubMed] [Google Scholar]
- Vigouroux, Y., M. McMullen, C. T. Hittinger, K. Houchins, L. Schulz et al., 2002. Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proc. Natl. Acad. Sci. USA 99: 9650–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux, Y., S. Mitchell, Y. Matsuoka, M. Hamblin, S. Kresovich et al., 2005. An analysis of genetic diversity across the maize genome using microsatellites. Genetics 169: 1617–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis, R., K. Dawson and P. Boursot, 2001. Interpretation of variation across marker loci as evidence of selection. Genetics 158: 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne, M. L., and L. M. McIntyre, 2002. Combining mapping and arraying: an approach to candidate gene identification. Proc. Natl. Acad. Sci. USA 99: 14903–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinig, C., 2000. Differing selection in alternative competitive environments: shade-avoidance responses and germination timing. Evolution 54: 124–136. [DOI] [PubMed] [Google Scholar]
- Weinig, C., L. A. Dorn, N. C. Kane, Z. M. German, S. S. Hahdorsdottir et al., 2003. a Heterogeneous selection at specific loci in natural environments in Arabidopsis thaliana. Genetics 165: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinig, C., J. R. Stinchcombe and J. Schmitt, 2003. b QTL architecture of resistance and tolerance traits in Arabidopsis thaliana in natural environments. Mol. Ecol. 12: 1153–1163. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., and D. E. McCauley, 1999. Indirect measures of gene flow and migration. Heredity 82: 117–125. [DOI] [PubMed] [Google Scholar]
- Wills, D. M., M. L. Hester, A. Z. Liu and J. M. Burke, 2005. Chloroplast SSR polymorphisms in the Compositae and the mode of organellar inheritance in Helianthus annuus. Theor. Appl. Genet. 110: 941–947. [DOI] [PubMed] [Google Scholar]
- Wood, T. E., J. M. Burke and L. H. Rieseberg, 2005. Parallel genotypic adaptation: when evolution repeats itself. Genetica 123: 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton, J. C., X. R. Feng, M. T. Ferdig, R. A. Cooper, J. B. Mu et al., 2002. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418: 320–323. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley et al., 2005. The effects of artificial selection of the maize genome. Science 308: 1310–1314. [DOI] [PubMed] [Google Scholar]
- Yeh, P. J., 2004. Rapid evolution of a sexually selected trait following population establishment in a novel habitat. Evolution 58: 166–174. [DOI] [PubMed] [Google Scholar]