

Abstract
Inferring population structure from genetic data sampled from some number of individuals is a formidable statistical problem. One widely used approach considers the number of populations to be fixed and calculates the posterior probability of assigning individuals to each population. More recently, the assignment of individuals to populations and the number of populations have both been considered random variables that follow a Dirichlet process prior. We examined the statistical behavior of assignment of individuals to populations under a Dirichlet process prior. First, we examined a best-case scenario, in which all of the assumptions of the Dirichlet process prior were satisfied, by generating data under a Dirichlet process prior. Second, we examined the performance of the method when the genetic data were generated under a population genetics model with symmetric migration between populations. We examined the accuracy of population assignment using a distance on partitions. The method can be quite accurate with a moderate number of loci. As expected, inferences on the number of populations are more accurate when θ = 4Neu is large and when the migration rate (4Nem) is low. We also examined the sensitivity of inferences of population structure to choice of the parameter of the Dirichlet process model. Although inferences could be sensitive to the choice of the prior on the number of populations, this sensitivity occurred when the number of loci sampled was small; inferences are more robust to the prior on the number of populations when the number of sampled loci is large. Finally, we discuss several methods for summarizing the results of a Bayesian Markov chain Monte Carlo (MCMC) analysis of population structure. We develop the notion of the mean population partition, which is the partition of individuals to populations that minimizes the squared partition distance to the partitions sampled by the MCMC algorithm.
MOST natural populations display some degree of population subdivision, either because they occupy a large geographic area and cannot act as a single randomly mating population or because geographical barriers reduce migration between different areas. The consequence is that subpopulations from different geographic regions occupied by a species show different allele frequencies. Population subdivision has profoundly important effects on the dynamics of alleles in populations and also on the statistical tests we might apply to genetic data sampled from individuals. It is well known that population subdivision affects the dynamics of alleles in a population under the influence of mutation, drift, and selection; hence, the eventual fate of an allele is affected by population subdivision (Wright 1940, 1943). Moreover, undetected population subdivision has an important (and usually negative) effect on statistical tests that are commonly applied to genetic data sampled from populations. For example, statistical tests of the presence of natural selection are adversely affected by undetected population subdivision, often having an inflated incidence of false positives (Andolfatto and Przeworski 2000; Nielsen 2001; Przeworski 2002; Hammer et al. 2003).
How can one identify the presence of population structure on the basis of genetic data sampled from some number of individuals? This is a long-standing problem in population genetics and has inspired a variety of approaches. One approach—F-statistics, developed by Wright (1951) and Malécot (1948)—attempts to characterize the effect of population subdivision by its inbreeding-like effect on excess homozygosity. These approaches can be quite sophisticated (e.g., Holsinger et al. 2002), but ultimately attempt to characterize the potentially complex pattern of population subdivision with a single statistic; FST provides a rather blunt tool for exploring population subdivision, with many different patterns of population subdivision, for example, producing a similar (positive) FST. Moreover, these approaches rely on preexisting labels; the assignment of individuals to populations is considered to be known before the analysis of the genetic data begins.
More recently, several authors have developed methods that do not rely upon a known assignment of individuals to populations, instead inferring the population structure. Perhaps the most widely used method is a Bayesian one developed by Pritchard et al. (2000). In its simplest form, the method of Pritchard et al. (2000) considers a fixed number of populations and, assuming linkage equilibrium and Hardy–Weinberg equilibrium of the alleles at the sampled loci, calculates the probability of assigning individuals to each of the populations. In its more fully developed form, the method has been modified to allow for admixture of individuals and linkage of the loci (Pritchard et al. 2000; Falush et al. 2003). Pritchard et al. (2000) use a variant of Markov chain Monte Carlo (MCMC) to approximate the probabilities of assigning individuals to populations. The Bayesian method of Pritchard et al. (2000) has the advantage that the uncertainty in the assignment of individuals to populations is easy to characterize. The method has also been of great practical importance, with uses in conservation genetics (Moodley and Harley 2005; Small et al. 2006), epidemiology (Leo et al. 2005; Michel et al. 2005; Nejsum et al. 2005; Ochsenreither et al. 2006), and studies of population demography (e.g., Rosenberg et al. 2002), and is often used as a first step in a genetic association study (Song and Elston 2006; Tsai et al. 2006; Yu et al. 2006).
The first step in many analyses of population structure is to decide how many populations are needed to explain the observations. Statistically, this is viewed as a clustering problem; the goal is to cluster individuals into populations. The number of mixture components in the clustering algorithm is usually considered fixed. The approach of Pritchard et al. (2000), for example, clusters individuals into one of a fixed number of populations. Pritchard et al. (2000) suggest a method based upon marginal likelihoods to determine the number of populations needed to explain the observations. Specifically, the method is applied several times to the data, with a varying number of populations for each treatment (say, one, two, three, etc., populations). The marginal likelihood can be calculated as the harmonic mean of the likelihoods sampled from the output of the MCMC method used to approximate the probability of assigning individuals to populations (Newton and Raftery 1994). Evanno et al. (2005) performed a simulation study investigating how well the method based on marginal likelihoods can identify the true number of populations. They found that the method performed poorly and suggested another statistic based upon the rate of change in the log probability of the data between successive analyses with increasing numbers of populations; this ad hoc statistic did a better job of correctly identifying the uppermost number of populations necessary to explain the data. Corander et al. (2003, 2004) take a different approach to clustering individuals into populations and allow the number of populations to vary to some degree. They start with the individuals assigned to prospective populations. For example, consider 100 individuals that were evenly sampled from 10 prospective populations. With the method of Corander et al. (2003, 2004), the minimum number of populations, in this example, is one (all of the prospective populations are merged together into one, sharing a common set of allele frequencies) and the maximum number of populations is 10 (none of the 10 prospective populations are merged). The method uses MCMC (see Green 1995) to explore possible patterns of merged and split prospective populations and has the advantage that the number of populations is allowed to vary within a limited range. However, with the method of Corander et al. (2003), it is impossible to escape the initial decision to place individuals together into the same prospective population.
More recently, Pella and Masuda (2006) applied a Dirichlet process prior to the problem of identifying population structure. Importantly, the Dirichlet process prior allows both the assignment of individuals to populations and the number of populations to be random variables; the number of populations, then, can in principle be estimated. The method of Pella and Masuda (2006) is similar to one proposed by Dawson and Belkhir (2001). Dawson and Belkhir (2001) propose both a maximum-likelihood and a Bayesian approach to infer the assignments of individuals to populations. Importantly, they also estimate the number of populations. Their method mostly differs in the prior that they place on the population assignments. Here, we examine the statistical behavior of the Dirichlet process prior as applied to the problem of inferring population structure. Besides performing simulations that probe the performance of the method, we describe a new way to summarize the results of a Bayesian MCMC analysis of population structure by using the mean partition.
MATERIALS AND METHODS
Our goal is to infer the assignment of individuals to populations on the basis of allele information for each individual. In this section, we describe a method for doing this, first described by Pella and Masuda (2006) that allows the number of populations to be a random variable. Specifically, the number of populations and the assignment of individuals to populations are treated as random variables with a Dirichlet process prior distribution (Ferguson 1973; Antoniak 1974). The description of the method entails considerable notation, and in Table 1 we provide a complete list of all the variables we consider.
TABLE 1.
Definitions of parameters used in this study
| Parameter | Description |
|---|---|
| K | No. of populations |
| L | No. of loci |
| Jl | No. of unique alleles observed at locus l |
| n | No. of individuals sampled |
| xilj | No. of copies of allele j at locus l in individual i |
| xil | No. of copies of alleles at locus l for individual i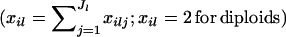
|
| xil | Allele information for individual i at locus l |
| Xi | Allele information for individual i |
| X | Allele information for all individuals |
| yklj | No. of copies of allele j at locus l in individuals assigned to population k |
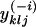 |
No. of copies of allele j at locus l in individuals assigned to population k, excluding information for individual i |
| ykl | No. of alleles observed for some locus l in individuals assigned to population k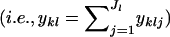
|
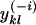 |
No. of alleles observed for some locus l in individuals assigned to population k, excluding information for individual i |
| pklj | Frequency of allele j at locus l in population k |
| pkl | Vector containing the allele frequencies for locus l in population k |
| λj | Dirichlet parameter for frequency of allele j |
| λ0 | Sum of the Dirichlet parameters for some locus and population 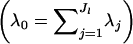
|
| zi | Assignment of individual i to a population [zi ∈ (1, …, K)] |
| z | Allocation vector containing the assignments of the n individuals to populations [i.e., z = (z1, z2, …, zn)] |
| α | Parameter of the Dirichlet process prior model that controls the “clumpiness” of the process |
| S(n, k) | Stirling no. of the second kind (no. of ways to partition n individuals into k populations) |
| Bn | Bell nos. (total no. of ways to assign n individuals to populations) |
| nak | Stirling nos. of the first kind |
| Γ(·) | Gamma function |
| I(·) | Indicator function, equaling 0 or 1 |
Data:
We assume that we have sampled the alleles for n individuals at L loci. At locus l, we observe Jl unique alleles. The number of copies of allele j at locus l in individual i is denoted xilj. Similarly, the number of copies of all alleles observed at locus l in individual i is denoted 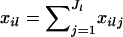 . The allelic information for individual i at locus l is contained in the vector,
. The allelic information for individual i at locus l is contained in the vector, 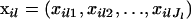 . For example, the information for individual i might look like
. For example, the information for individual i might look like
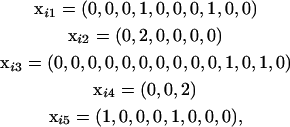 |
where there are L = 5 loci with the loci having J1 = 10, J2 = 6, J3 = 14, J4 = 3, J5 = 8 observed alleles. This example individual is homozygous at the second and fourth loci and heterozygous at the others. We denote the complete information for the ith individual as Xi = (xi1, xi2, …, xiL). Similarly, we denote the information for all of the n individuals as X = (X1, X2, …, Xn).
The combinatorics of assigning individuals to populations:
The n individuals are assigned to one of K populations. The information on the assignment of individuals to populations is contained in an assignment vector, z. Specifically, the assignment of individual i to population k is denoted zi = k, where zi ∈ (1, …, K). An example of an assignment vector for n = 5 individuals might look like
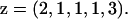 |
Here there are K = 3 populations with individuals 2, 3, and 4 being assigned to the same population. The number of individuals that are assigned to the ith population is denoted ηi.
The number of ways to assign n individuals to one of k populations is given by the Stirling numbers of the second kind,
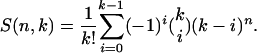 |
For our example of n = 5 individuals and K = 3 populations, there are a total of S(5, 3) = 25 ways to partition the individuals among populations. The total number of ways to partition n individuals among populations is given by the Bell numbers (Bell 1934). The Bell numbers, Bn, are calculated as the sum
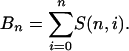 |
For example, n = 5 individuals can be partitioned among 1, 2, 3, 4, or 5 populations. The total number of ways to assign individuals to populations, then, is
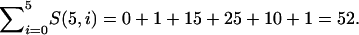 |
We label possible partitions of the individuals into populations using the restricted growth function notation of Stanton and White (1986), where elements are sequentially numbered with the constraint that the index numbers for two individuals are the same if they are found in the same population. For example, the allocation vector z = (2, 1, 1, 1, 3) is described using the restricted growth function notation as 1, 2, 2, 2, 3.
Assigning individuals to populations when K is fixed:
Pritchard et al. (2000) examined the case where the number of populations, K, is considered to be fixed, but the allocation vector, z, is treated as a random variable. They treated the problem in a Bayesian context. In a Bayesian framework, inferences are based upon the posterior probability of a parameter. For the problem of assigning individuals to populations, ideally one would calculate the posterior probability of an assignment vector f(z | X, K), which can be calculated using Bayes' theorem as
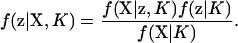 |
Probabilities of assignment vectors, however, are rarely calculated. The posterior probability of an assignment vector cannot be calculated analytically and must instead be approximated using a numerical technique such as MCMC. When the number of individuals is large, there are a vast number of possible assignment vectors and the posterior probability for even the most probable can be quite small and difficult to approximate accurately. For example, when n = 200 and K = 5 there are a total of S(200, 5) = 5.186 × 10137 possible ways to partition individuals among populations; in this case, even the best partitioning scheme might have a tiny probability that is difficult or impossible to approximate using a method like MCMC.
Instead of calculating the probability of an assignment vector, the more practical solution is to calculate the marginal posterior probability of assigning individual i to population k,
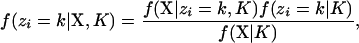 |
where f(X | zi = k, K) is the likelihood (the probability of the observations when individual i is assigned to population k) and f(zi = k | K) is the prior probability of assigning individual i to population k. This is the approach taken by Pritchard et al. (2000). They assume a uniform prior on populations, so that f(zi = k | K) = 1/K. For their model without admixture and assuming that the allele frequencies for each population are in linkage equilibrium and in Hardy–Weinberg equilibrium, the probability of the information for individual i is
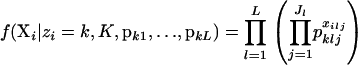 |
Here pklj is the frequency of allele j at locus l in population k; the vector pkl contains the allele frequencies for locus l in population k. The probability of the observations on all of the individuals is the product of the likelihoods for the individuals.
Pritchard et al. (2000), following the lead of Balding and Nichols (1995) and Rannala and Mountain (1997), assume that the allele frequencies have a flat Dirichlet prior probability distribution. The Dirichlet distribution has probability density
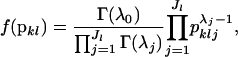 |
where 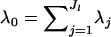 and Γ(·) is the Gamma function. The flat Dirichlet distribution has all λj = 1.
and Γ(·) is the Gamma function. The flat Dirichlet distribution has all λj = 1.
Pritchard et al. (2000) do not condition upon any particular combination of allele frequencies when calculating the likelihood, instead integrating the likelihood over all possible combinations of allele frequencies, each combination being weighted by its probability under a flat Dirichlet prior. They then approximate the posterior probability using MCMC. Specifically, the program Structure—a program that implements the method outlined above—uses Gibbs sampling to first sample allele frequencies for each locus conditioned on the assignment of individuals to populations. They then use another Gibbs sampling step to assign individuals to populations (assuming that the allele frequencies are fixed). Repetition of these two Gibbs sampling steps allows the program Structure to integrate over allele frequencies and assignment vectors. If one is interested in the marginal posterior probability that individual i is assigned to population k, one simply records the fraction of the time that the individual was assigned to that population; the fraction of the time the Markov chain has individual i in population k is a valid approximation of the probability that the individual is assigned to the population (Tierney 1996).
For the simple problem of assigning individuals to populations without admixture, one does not need to use this two-step Gibbs procedure; one can perform Gibbs sampling on the assignment of individuals to populations while analytically integrating over the possible allele frequencies (a point also made by Pella and Masuda 2006). The Gibbs sampling procedure works like this: Pick an individual denoted i. Reassign this individual to population k with probability
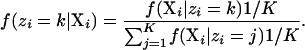 |
The likelihood, f(Xi | zi = k), is calculated by integrating over all possible combinations of allele frequencies:
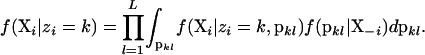 |
When individual i is placed into population k, the allele frequencies may not follow the prior probability distribution (e.g., a flat Dirichlet distribution) because other individuals may also be included in that population, thus modifying the probability density of different allele frequency combinations. Instead, the allele frequencies are drawn from the posterior probability distribution of allele frequencies for that population with individual i excluded. The notation X−i is read “all of the observations, excluding the observations made on individual i.” The likelihood is then
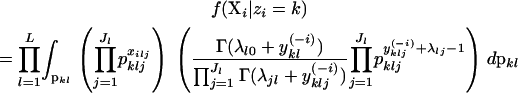 |
(1) |
 |
(2) |
Here, yklj is the number of copies of allele j at locus l in individuals that are assigned to population k and is calculated as
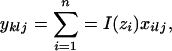 |
where I(zi) is an indicator function that equals one if individual i is assigned to population k (i.e., zi = k) and zero otherwise. The summation of yklj over all alleles is denoted 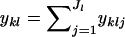 . Finally, the superscript “(−i)” indicates that the count excludes individual i.
. Finally, the superscript “(−i)” indicates that the count excludes individual i.
The integral in Equation 2 can be solved analytically. Remembering that
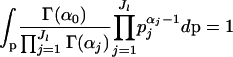 |
we have
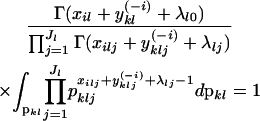 |
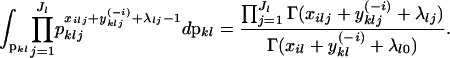 |
This means that
 |
(3) |
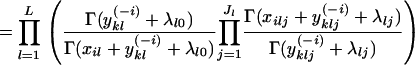 |
(4) |
This result was first derived by Rannala and Mountain (1997) and can be used to perform Gibbs sampling only with respect to the assignment of individuals to populations.
Assigning individuals to populations when K is a random variable:
Our goal is to assign individuals to populations while allowing both the allocation vector (z) and the number of populations (K) to both be random variables. To do this we assume that the joint prior probability of the allocation vector and the number of populations follows a Dirichlet process prior (Ferguson 1973; Antoniak 1974; Pella and Masuda 2006). Specifically, under the Dirichlet process prior the joint probability of z and K is
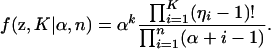 |
(5) |
The parameter α is a concentration parameter that determines the degree to which individuals are grouped together into the same population. In fact, the prior probability that two randomly chosen individuals (i and j) are grouped together in the same population is
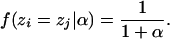 |
Clearly, when α is small, the prior probability of two individuals finding themselves in the same population is high. Moreover, the probability of having K populations is obtained by summing over all possible partitions for K populations,
 |
where nak is the absolute value of the Stirling numbers of the first kind. Finally, the expected number of populations is
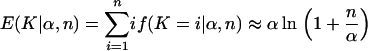 |
The Dirichlet process prior model, then, can be understood by considering the following procedure: First, randomly draw the number of populations (K) and the allocation vector (z) from the probability distribution described by Equation 5; then, for each population randomly draw the allele frequencies from the Dirichlet probability distribution prior (in this study, we use a flat Dirichlet). We use the word “Dirichlet” in two different senses: first, to describe the prior probability distribution on allele frequencies (this is the “Dirichlet probability distribution”) and, second, to describe the prior probability distribution on the allocation vector and on the number of populations (this is the “Dirichlet process prior”). Keep in mind that the Dirichlet probability distribution and the Dirichlet process prior are two different probability distributions.
We use Algorithm 3 from Neal (2000) to perform the numerical integration over the possible allocation vectors and number of populations. Neal (2000) describes a Gibbs sampling scheme for MCMC that works as follows. First, pick an individual, i, and remove it from the allocation vector z. If the individual was alone in a population, then the population is removed from computer memory, and the total number of populations is decreased by one. Otherwise, the count of the number of individuals in the population, η, is decreased by one. Place individual i into population k with probability
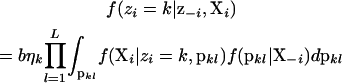 |
(see Equation 4) and into a new population k (all by itself) with probability
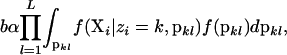 |
where f(pkl) is the Dirichlet prior density distribution on allele frequencies and b is a normalizing constant.
Computer program:
The methods described in this article have been implemented in a computer program called Structurama. The program, written in C++, implements a Gibbs MCMC sampling strategy for assigning individuals to populations when K is fixed and the strategy described by Neal (2000) when the number of populations is treated as a random variable with a Dirichlet process prior. The program is available for free download from http://www.structurama.org. Each MCMC cycle of the program involves a Gibbs scan of all of the individuals; hence, the total number of MCMC cycles for the analyses of this study is the product of the reported number of MCMC cycles and the number of individuals in the analysis.
The ability of the MCMC algorithm we implemented to work on any particular data set is, of course, unknown. However, we have found that the MCMC appears to work well for some problems that involve up to 1000 individuals. The MCMC we implement needs only to integrate over the placement of individuals into populations; unlike other implementations, we analytically integrate over allele frequencies. Moreover, we implemented (though did not use in this article) a variant of MCMC called Metropolis-coupled Markov chain Monte Carlo [MCMCMC or (MC)3 (Geyer 1991)]. This variant of MCMC better allows a Markov chain to explore the parameter space. We recommend not only that MCMCMC be used on difficult problems (those involving, say, ≥1000 individuals), but also that the results be checked carefully by comparing the inferences from multiple chains on the same data.
Summarizing the results of a MCMC analysis of population structure:
Considerable attention has been paid to the problem of how to summarize the results of a Bayesian analysis of population structure. The output of a Markov chain can be used to calculate the probabilities of assigning individuals to populations. For example, consider the following (arbitrarily constructed) output from a MCMC analysis of population structure:
Table 7.
| N | K | Arbitrary indexing | RGF indexing |
|---|---|---|---|
| 1 | 3 | 3 2 2 1 3 2 1 1 2 2 | 1 2 2 3 1 2 3 3 2 2 |
| 2 | 3 | 3 2 3 2 3 2 1 1 2 2 | 1 2 1 2 1 2 3 3 2 2 |
| 3 | 3 | 3 2 2 2 3 2 1 2 2 2 | 1 2 2 2 1 2 3 2 2 2 |
| 4 | 3 | 3 2 3 2 3 2 1 1 2 1 | 1 2 1 2 1 2 3 3 2 3 |
| 5 | 3 | 2 1 1 3 2 1 3 3 1 1 | 1 2 2 3 1 2 3 3 2 2 |
Here N is the sample number and K is the number of populations. There are a number of ways to describe the current state of the Markov chain. One way is to simply assign each population an index. The column labeled “Arbitrary indexing” shows this strategy. In fact, this is the strategy taken in the program Structure. The last column shows the labeling of the population partition using the restricted growth function (RGF) notation of Stanton and White (1986). The main problem with the arbitrary indexing scheme is that the labels are just that, arbitrary. In fact, if the Markov chain mixes properly, then it should be able to explore other, equivalent, indexing schemes for the partition. For example, the first and fifth samples shown in the example MCMC output, above, are equivalent in that they specify the same partition of individuals to populations. (Note that the first and fifth samples have the same description using the restricted growth function notation.) When there are K populations, there are K! equivalent arbitrary indexing schemes that can be applied to describe the same partition of individuals among populations. In other words, if the Markov chain mixes well (i.e., properly explores the parameter space), then the following six indexing schemes for the same partition should be visited by the Markov chain equally often: (1 1 1 2 2 2 3 3 3), (1 1 1 3 3 3 2 2 2), (2 2 2 1 1 1 3 3 3), (2 2 2 3 3 3 1 1 1), (3 3 3 1 1 1 2 2 2), (3 3 3 2 2 2 1 1 1). This problem was noted by Pritchard et al. (2000) and Stephens (2000), but their method for summarizing the results of the output of a MCMC analysis relies on the fact that in practice, only one of the equivalent labeling schemes for a partition is visited.
Here, we explore a number of different ways to summarize the results of a Bayesian MCMC analysis of population structure. The first method we explore simply summarizes the probability that each of the 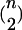 pairs of individuals are in the same population. The approximation of the probability that individual i and individual j are in the same population is simply the fraction of the time they were placed into the same population during the MCMC analysis and does not depend upon the labeling scheme for the partitions. The prior probability of any two individuals falling into the same population under the Dirichlet process prior is 1/(1 + α). This means that the Bayes factor in favor of individuals i and j being in the same population is
pairs of individuals are in the same population. The approximation of the probability that individual i and individual j are in the same population is simply the fraction of the time they were placed into the same population during the MCMC analysis and does not depend upon the labeling scheme for the partitions. The prior probability of any two individuals falling into the same population under the Dirichlet process prior is 1/(1 + α). This means that the Bayes factor in favor of individuals i and j being in the same population is
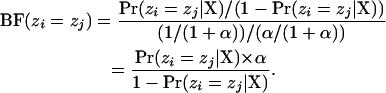 |
Lavine and Schervish (1999) argue that Bayes' factors should be interpreted as the degree by which an investigator changes his or her mind about a hypothesis after making some observations. In the context of inferring population structure, the Bayes factor would indicate how strongly two individuals are grouped into the same population.
Another strategy we explore for summarizing the results of a Bayesian analysis of population structure relies on a distance on partitions, described by Gusfield (2002). The basic idea is to find a partitioning of the individuals among populations that minimizes the squared distance to the partitions sampled by the MCMC algorithm (Figure 1). Here, we rely on the interpretation of the mean as minimizing the squared distance to a sample. For example, consider the usual interpretation of the mean of n real numbers, x1, x2, …, xn. If d(xi, μ) = (xi − μ), then the value of μ that minimizes  is
is 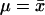 . We consider the partition distance,
. We consider the partition distance,  , and define the mean partition to be the partition,
, and define the mean partition to be the partition, 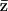 , that minimizes
, that minimizes 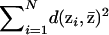 , where the summation is over all N of the partitions sampled by the MCMC algorithm. The partition distance,
, where the summation is over all N of the partitions sampled by the MCMC algorithm. The partition distance, 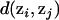 , is the minimum number of individuals that must be deleted from both zi and zj to make the two induced partitions the same. Equivalently, the partition distance of Gusfield (2002) can be thought of as the minimum number of elements that must be moved between populations in one of the partitions to make it identical to the other partition. We search for the mean partition using the following algorithm. First, we start with an arbitrarily chosen partition. Here, we start with one of the partitions sampled by the Markov chain. We then attempt to place each element (individual) of the mean partition into each of the other clusters (populations) and also into a cluster all by itself. At each step, we keep track of the mean squared distance to all of the sampled partitions and move to partitions that result in a smaller mean squared distance. The algorithm stops when a partition is found in which perturbation does not result in a smaller mean squared distance to sampled partitions. Our experience is that this algorithm works well in finding the mean partition, even when the starting partition is distant from the sampled partitions.
, is the minimum number of individuals that must be deleted from both zi and zj to make the two induced partitions the same. Equivalently, the partition distance of Gusfield (2002) can be thought of as the minimum number of elements that must be moved between populations in one of the partitions to make it identical to the other partition. We search for the mean partition using the following algorithm. First, we start with an arbitrarily chosen partition. Here, we start with one of the partitions sampled by the Markov chain. We then attempt to place each element (individual) of the mean partition into each of the other clusters (populations) and also into a cluster all by itself. At each step, we keep track of the mean squared distance to all of the sampled partitions and move to partitions that result in a smaller mean squared distance. The algorithm stops when a partition is found in which perturbation does not result in a smaller mean squared distance to sampled partitions. Our experience is that this algorithm works well in finding the mean partition, even when the starting partition is distant from the sampled partitions.
Figure 1.—

Example of the calculation of the mean partition. The mean partition, 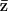 , minimizes the sum of the squared distances to the partitions sampled by the MCMC algorithm.
, minimizes the sum of the squared distances to the partitions sampled by the MCMC algorithm.
Analysis of real data:
We explored the method of Pella and Masuda (2006) on three empirical data sets:
The first data set we explored is of n = 216 Impala (Aepyceros melampus) individuals sampled at L = 8 microsatellite loci (Lorenzen et al. 2004, 2006).
We also examined a data set of n = 155 Taita thrush (Galbusera et al. 2000). Each bird was sampled at L = 7 microsatellite loci. The thrush data were also analyzed by Pritchard et al. (2000).
Finally, we examined Orth et al.'s (1998) data on n = 74 Mus musculus individuals sampled from Lake Casitas, California. The Mus data have L = 7 loci.
We performed 11 analyses for each of the three data sets (Impala, thrush, and Mus). Specifically, we ran analyses in which the number of populations was fixed (K = 1, …, 7) and in which the number of populations follows a Dirichlet process prior with a prior mean of E(K) = 2, E(K) = 5, E(K) = 10, and E(K) = 20. All MCMC analyses were run for a total of 1 million cycles.
Computer simulations:
We used computer simulation to explore the statistical behavior of population assignment under the Dirichlet process prior. Specifically, we explored two different models for generating genetic data. First, we simulated data under the Dirichlet process prior. We simulated up to 1000 data sets, each consisting of n = 100 individuals under the Dirichlet process with E(K) = 5. This means that each simulated data set might differ in the number of populations (and almost certainly differed in the allocation vector), but that the average number of populations over the replicates was close to five. We explored two scenarios here. First, we varied the number of loci. This allows us to investigate the effect of the number of observations on the performance of the method. We also explored the robustness of the method to misspecification of the “clumpiness” parameter (α) of the Dirichlet process prior. We did this by generating data with a prior mean of E(K) = 5, but analyzing the data with the prior mean set to E(K) = 5, E(K) = 10, or E(K) = 20. We designed the simulations to also allow us to check the validity of our implementation of the Dirichlet process prior. For example, when data are generated under the Dirichlet process prior and analyzed under the same model conditions [e.g., when we simulated and analyzed data with the prior mean of the number of populations set to E(K) = 5], a 95% credible set of the number of populations should contain the true number of populations with probability 0.95.
We also simulated data under a neutral coalescent model using the program ms (Hudson 2002). The model assumes an infinite-sites mutation model and no recombination so that each new mutation results in an unique allele (haplotype). To evaluate the type I error of the Dirichlet process prior method we simulated a single panmictic population with θ = 4Neu = (0.5, 1, 2, 4), where Ne is the effective population size and u is the mutation rate per generation. Our analyses of these simulations with Structurama suggest that the Dirichlet process prior method has low type I error (i.e., ≤5%) in the absence of population structure (see Table 3). While a full exploration of possible demographic models is beyond the scope of this study, a wide range of demographic models is straightforward to implement using the program ms (see the program documentation, http://home.uchicago.edu/rhudson1/source/mksamples.html). As a first-pass evaluation of Structurama's ability to detect population structure, we modeled a symmetric equilibrium island model with 2, 4, and 10 demes of equal size where θ = 4Neu = (0.5, 1.0, 2.0, 4.0), M = 4Nem = (0.5, 1.0, 2.0, 4.0), and m is the migration rate per generation. In each case, 100 diploid individuals were sampled in total, with equal numbers of individuals being sampled from each deme. Clearly, this demographic model and sampling scheme are highly simplistic and are intended only to be illustrative. The ms command lines we implemented are listed in the appendix. A perl script that digests the ms output into Structurama input is available on request to P. Andolfatto.
TABLE 3.
Results of simulations under a population genetics model when there is one population and the prior mean of the number of populations is 5, 10, or 20
|
E[E(K | X) − KT]
|
E[d(z, zT)]
|
Coverage
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KT | L | θ | 5 | 10 | 20 | 5 | 10 | 20 | 5 | 10 | 20 |
| 1 | 10 | 0.5 | 0.04 | 0.08 | 0.15 | 0.04 | 0.08 | 0.16 | 0.98 | 0.97 | 0.97 |
| 1 | 10 | 1.0 | 0.01 | 0.03 | 0.06 | 0.01 | 0.03 | 0.06 | 0.97 | 0.95 | 0.96 |
| 1 | 10 | 2.0 | 0.02 | 0.04 | 0.07 | 0.02 | 0.04 | 0.08 | 0.98 | 0.99 | 0.95 |
| 1 | 10 | 4.0 | 0.04 | 0.07 | 0.14 | 0.05 | 0.08 | 0.14 | 0.97 | 0.91 | 0.95 |
| 1 | 100 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.98 | 0.97 | 0.96 |
| 1 | 100 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.93 | 0.95 |
| 1 | 100 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.98 | 0.95 | 0.94 |
| 1 | 100 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.87 | 0.99 |
For each simulated data set, we ran a single Markov chain for a total of 100,000 cycles. We thinned the Markov chains by sampling every 25th sample and discarded samples taken during the first 12,500 cycles as the burn-in of the chain. The program Structurama, which can work in a batch-processing mode, was used for analysis of all simulated data.
RESULTS AND DISCUSSION
Figures 2–4 show the results of the analyses of the real data. Each figure shows the mean partition summarizing the MCMC samples for a particular analysis. Individuals are arrayed along columns, and each row represents a different analysis. Individuals sharing a color are grouped together into the same population. As expected, the partitions differ when the fixed number of populations varies. Interestingly, the mean partition is rather insensitive to the choice of the clumpiness parameter, α, when a Dirichlet process prior probability distribution is applied to the assignment vector and the number of populations; note that the same mean partition results when the prior mean of the number of populations is varied from E(K) = 5 to E(K) = 20.
Figure 2.—

The mean partitions for analyses of the Imapala Aegyceros melampus data (Lorenzen et al. 2004, 2006). Analyses were performed in which the number of populations was fixed (K = 1, K = 2, K = 3, K = 4, K = 5, K = 6, K = 7) or in which the number of populations was a random variable with a Dirichlet process prior [E(K) = 2, E(K) = 5, E(K) = 10, E(K) = 20]. The assignment of individuals (boxes) is indicated by color.
Figure 3.—

The mean partitions for analyses of the Taita thrush data (Galbusera et al. 2000). Analyses were performed in which the number of populations was fixed (K = 1, K = 2, K = 3, K = 4, K = 5, K = 6, K = 7) or in which the number of populations was a random variable with a Dirichlet process prior [E(K) = 2, E(K) = 5, E(K) = 10, E(K) = 20]. The assignment of individuals (boxes) is indicated by color.
Figure 4.—

The mean partitions for analyses of the Mus musculus data (Orth et al. 1998). Analyses were performed in which the number of populations was fixed (K = 1, K = 2, K = 3, K = 4, K = 5, K = 6, K = 7) or in which the number of populations was a random variable with a Dirichlet process prior [E(K) = 2, E(K) = 5, E(K) = 10, E(K) = 20]. The assignment of individuals (boxes) is indicated by color.
The problem of summarizing the results of an MCMC analysis of population structure is a difficult one, because the object of estimation—a partition representing the population structure—is not a standard statistical parameter. There is no natural way to order partitions, for example. In many ways, the problem of summarizing a collection of partitions (sampled, from say, a Markov chain) has parallels in the phylogenetics literature, where the object of inference is a phylogenetic tree. Trees, like partitions, cannot be naturally ordered. In the phylogenetics literature, a number of solutions have been considered, including consensus trees. Several methods have been suggested for summarizing the results of a MCMC analysis of population structure. As mentioned earlier, one solution is to arbitrarily assign indexes to populations. Although other choices of indexes will result in the same partition of individuals to populations, in practice the MCMC may fail to visit these alternative labeling schemes; the summary of the population index to which each individual is assigned, then, may be useful. Dawson and Belkhir (2001) suggest a clustering method for summarizing the results of an MCMC analysis of population structure. Their method applies a hierarchical clustering algorithm to identify well-supported clusters of individuals. Stephens (2000), on the other hand, takes a decision theoretic approach and attempts to find a partition that minimizes the posterior expected loss. Our method of constructing a mean partition best fits the decision theoretic approach, with a loss function that is the squared distance between a sampled partition and the true partition. The weakness of our approach is that it relies upon a distance on partitions (one described by Gusfield 2002), which is itself arbitrary. Unfortunately, only the Gusfield (2002) metric on partitions exists; other metrics on partitions are surely possible and could be usefully applied to the problem of summarizing the results of a MCMC analysis of population structure.
Figure 5 shows an alternative method for summarizing the results of the MCMC analysis of population structure. These plots—dubbed “plaid plots” by one of us—show the probabilities of all possible pairs of individuals being grouped together into the same population. The plots are rather complicated to read, at least on a first pass. The top left corner of each triangle shows the probability of individuals 1 and 2 being grouped into the same population. The top right corner of each graph shows the probability of individuals 1 and n finding themselves in the same population. Finally, the bottom right corner of each plot shows the probability of individuals n and n − 1 being grouped together in the same population. The color red indicates a probability >0.9 of two individuals being in the same population. One finds blocks of individuals all of whom share a high probability of being grouped together in the same population, but a low probability of being grouped with any other individuals (this is indicated by the “stair case”-like pattern of red coloration in the plots). Figure 5 also shows the Bayes factors for all pairs of individuals being grouped together in the same population. The Bayes factor measures the strength of the evidence grouping individuals together. Formally, it measures the degree by which an individual changes his or her mind about the grouping of a pair of individuals after observing the genetic data. Generally speaking, Bayes' factors >10 are considered “positive” or “strong” evidence in favor of a hypothesis (Jeffreys 1961), in this case the hypothesis being that individuals i and j are in the same population. Similarly, a Bayes factor <∼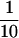 can be considered strong evidence against two individuals being in the same population. One advantage of the plaid plots is that they are invariant to the labeling of populations; all that is considered is whether two individuals are assigned to the same population or to different populations.
can be considered strong evidence against two individuals being in the same population. One advantage of the plaid plots is that they are invariant to the labeling of populations; all that is considered is whether two individuals are assigned to the same population or to different populations.
Figure 5.—

The probabilities and Bayes' factors of all pairs of individuals being grouped together into the same population. Each triangle shows all  pairs of individuals. The top left corner of a triangle shows the probability/Bayes' factor for individuals 1 and 2, the top right corner of the triangle shows the probability/Bayes' factor for individuals 1 and n, and the bottom corner of the triangle shows the probability/Bayes' factor for individuals n − 1 and n. Generally speaking, Bayes' factors >10 are strong evidence for two individuals being grouped together in the same population whereas Bayes' factors
pairs of individuals. The top left corner of a triangle shows the probability/Bayes' factor for individuals 1 and 2, the top right corner of the triangle shows the probability/Bayes' factor for individuals 1 and n, and the bottom corner of the triangle shows the probability/Bayes' factor for individuals n − 1 and n. Generally speaking, Bayes' factors >10 are strong evidence for two individuals being grouped together in the same population whereas Bayes' factors 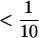 are strong evidence against grouping two individuals into the same population. All analyses were run assuming a Dirichlet process prior on the number of populations and a prior mean for the number of populations of E(K) = 5.
are strong evidence against grouping two individuals into the same population. All analyses were run assuming a Dirichlet process prior on the number of populations and a prior mean for the number of populations of E(K) = 5.
How many populations are required to explain the data? Figure 6 shows the approximate marginal likelihoods for varying numbers of populations for the three empirical data sets. The marginal likelihoods were calculated by taking the harmonic mean of the likelihoods for samples taken by the MCMC algorithm (Newton and Raftery 1994) when the number of populations is considered fixed. Note that it is difficult to explain the data with only a few populations (e.g., one or two), but that the marginal likelihoods are similar for larger numbers of populations. Typically, one picks the number of populations that is a compromise between the ability to explain the data (i.e., picking K such that the marginal likelihood is high) and parsimony (i.e., picking K as small as possible). One can do this using, for example, the Akaike information criterion (Akaike 1973) in which the marginal likelihood is penalized by the number of parameters (populations). Our approach involves sampling only assignments of individuals to populations and analytically integrates over uncertainty in allele frequencies; it is likely that our estimates of the marginal likelihood are more accurate than approaches that use MCMC to integrate over uncertainty in allele frequencies. Even so, Raftery et al. (2006) point out that while the harmonic mean is an unbiased estimator, it can have an infinite variance and is generally an unstable estimate of the marginal likelihood.
Figure 6.—

The marginal likelihoods Pr(X | K) when the number of populations (K) is fixed to different values (K = 1, 2, …, 7). (a) Impala data (Lorenzen et al. 2004, 2006); (b) Taita thrush data (Galbusera et al. 2000); (c) Mus musculus data (Orth et al. 1998).
Inference of the number of populations is automatic to the procedure when the number of populations and the assignment of individuals to populations are both considered random variables that follow a Dirichlet process prior. Figure 7 shows the prior and posterior probability distributions for K for four analyses of the Impala data set. The maximum posterior probability (MAP) estimate of the number of populations is K = 4 for three of the four analyses. When the prior mean of the number of populations is E(K) = 2, E(K) = 5, or E(K) = 10, most of the posterior probability is on K = 4. Only when the prior mean is E(K) = 20 does the MAP estimate of the number of populations shift to K = 5. In this study, we assumed a fixed value for the concentration parameter, α. For ease of interpretation, we set α using the prior mean on the number of populations; that is, we first chose a prior mean for K and then found the α that gives the desired prior mean. In some Bayesian analyses under a Dirichlet prior process, however, a probability distribution is put on the concentration parameter, α. Typically, a Gamma hyperprior is placed on α. We implemented the Gamma hyperprior in Structurama, but did not explore the effect of this approach on the inferences.
Figure 7.—

The prior, Pr(K), and posterior, Pr(K | X), probability distributions for the number of populations for the Impala data set when the prior mean of the number of populations varies.
How well can we expect to assign individuals to populations under the Dirichlet process model? We used simulation to explore the statistical behavior of the method. Table 2 summarizes the results for our first set of simulations. The first set of simulations was designed to explore the behavior of the method in a best-case scenario, in which all of the assumptions of the method are satisfied. We simulated data under a Dirichlet process prior in which E(K) = 5. We then analyzed the data under precisely the same model [i.e., with E(K) = 5] and under situations in which the prior is misspecified [i.e., with E(K) = 10 and E(K) = 20]. In the simulations, we varied the number of loci from L = 10 to L = 100. Note that the average distance of the partitions sampled by the MCMC algorithm to the true partition (zT) decreases as the number of loci increases. This result is to be expected; as the number of observations increases, inferences about the population assignment become more accurate. Here, we use the partition distance to measure the distance of the sampled partitions to the true partition in each simulation (Gusfield 2002). Also, as expected, inferences are less accurate when the concentration parameter, α, is misspecified. However, the effect of number of loci is much stronger than the effect of prior misspecification. We also noted the coverage probability for all the simulations. For each MCMC analysis of the simulated data, we constructed a 95% credible set for the number of populations. If the method is behaving correctly, a 95% credible set should contain the true number of populations 95% of the time. One can see that when the prior for α is not misspecified, the coverage (probability that the 95% credible set contains the true number of populations) is ∼0.95. However, when the prior model is misspecified, the 95% credible set contains the true number of populations with a probability <0.95. For example, in one of the simulations of L = 10 loci, data were simulated with a prior mean of 5, but analyzed assuming a prior mean for the number of populations of 20. In this set of simulations, the 95% credible set contained the true number of populations only 53% of the time. Importantly, the simulations suggest that misspecification of the prior on the number of populations becomes less important when the data have many loci. For instance, the coverage probability for the number of populations did not appear to be affected for the simulations of L = 100 loci. Finally, the bias in the estimates of the number of populations appears to be moderate in the simulations under the Dirichlet process prior. We measured bias as the difference between the average of the number of populations sampled by the MCMC algorithm and the true number of populations (E[E(K | X) − KT]). Generally, the number of populations was slightly overestimated when the prior mean chosen for the analysis was larger than the prior mean used to simulate the data.
TABLE 2.
Results of simulations under the Dirichlet process prior with E(K) = 5
| n | L | E(K) | E[E(K | X) − KT] | Coverage | E[d(z, zT)] | No. replicates |
|---|---|---|---|---|---|---|
| 100 | 10 | 5 | 0.056 | 0.954 | 5.08 | 1000 |
| 10 | 1.034 | 0.896 | 5.63 | |||
| 20 | 2.656 | 0.527 | 6.77 | |||
| 100 | 20 | 5 | 0.014 | 0.954 | 1.20 | 1000 |
| 10 | 0.400 | 0.940 | 1.36 | |||
| 20 | 0.955 | 0.817 | 1.70 | |||
| 100 | 100 | 5 | −0.002 | 0.950 | 0.016 | 500 |
| 20 | 0.014 | 0.958 | 0.021 |
In our second set of simulations, we simulated data under a model completely different from the Dirichlet process prior model under which the data were analyzed. Specifically, we simulated data under a population genetics model with K = 1, K = 2, K = 4, or K = 10 populations. We allowed θ = 4Nu to vary and also assumed a symmetric migration model between populations with varying migration rates (M = 4Nm). Tables 3–6 summarize the results of these simulations. Again, we measured the accuracy of the method as the average distance of the sampled partitions to the true partition. We also noted the bias (the degree by which the number of populations is over- or underestimated) and the coverage probability (the probability that a 95% credible set contains the true number of populations). Because there were only 100 simulations for each combination of parameter values, one should take the values for the coverage probabilities with a grain of salt; it is difficult to accurately determine coverage probabilities with only 100 replicates. Generally speaking, the simulations show that inferences of population structure are more accurate when (1) more loci are used, (2) migration rates are low, and (3) θ is large.
TABLE 4.
Results of simulations under a population genetics model when there are two populations and the prior mean of the number of populations is 5, 10, or 20
|
E[E(K | X) − KT]
|
E[d(z, zT)]
|
Coverage
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KT | L | θ | M | 5 | 10 | 20 | 5 | 10 | 20 | 5 | 10 | 20 |
| 2 | 10 | 0.5 | 0.5 | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.02 | 0.92 | 0.94 | 0.95 |
| 2 | 10 | 0.5 | 1.0 | −0.01 | −0.01 | 0.00 | 0.63 | 0.63 | 0.16 | 0.97 | 0.90 | 0.94 |
| 2 | 10 | 0.5 | 2.0 | −0.07 | −0.09 | −0.07 | 5.27 | 6.18 | 6.64 | 0.89 | 0.90 | 0.93 |
| 2 | 10 | 0.5 | 4.0 | −0.44 | −0.43 | −0.46 | 25.21 | 24.79 | 26.84 | 0.53 | 0.56 | 0.57 |
| 2 | 10 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.93 | 0.96 |
| 2 | 10 | 1.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.97 | 0.96 | 0.95 |
| 2 | 10 | 1.0 | 2.0 | −0.03 | −0.03 | −0.01 | 1.68 | 2.17 | 1.70 | 0.93 | 0.91 | 0.97 |
| 2 | 10 | 1.0 | 4.0 | −0.22 | −0.35 | −0.28 | 12.74 | 0.43 | 17.15 | 0.73 | 0.66 | 0.72 |
| 2 | 10 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.98 | 0.98 |
| 2 | 10 | 2.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.96 | 0.97 |
| 2 | 10 | 2.0 | 2.0 | 0.01 | 0.02 | 0.03 | 0.02 | 0.03 | 0.04 | 0.96 | 0.99 | 0.94 |
| 2 | 10 | 2.0 | 4.0 | −0.10 | −0.08 | −0.05 | 5.73 | 4.76 | 3.78 | 0.84 | 0.89 | 0.91 |
| 2 | 10 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.93 | 0.90 |
| 2 | 10 | 4.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.94 | 0.94 |
| 2 | 10 | 4.0 | 2.0 | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.01 | 0.95 | 0.97 | 0.95 |
| 2 | 10 | 4.0 | 4.0 | 0.00 | 0.01 | 0.02 | 0.04 | 0.05 | 0.56 | 0.97 | 0.96 | 0.96 |
| 2 | 100 | 0.5 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.91 | 0.96 | 0.96 |
| 2 | 100 | 0.5 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.88 | 0.92 | 0.97 |
| 2 | 100 | 0.5 | 2.0 | 0.00 | −0.02 | −0.01 | 0.00 | 1.00 | 0.50 | 0.95 | 0.92 | 0.92 |
| 2 | 100 | 0.5 | 4.0 | −0.82 | −0.79 | −0.77 | 41.00 | 39.50 | 38.50 | 0.18 | 0.19 | 0.23 |
| 2 | 100 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.98 | 0.91 |
| 2 | 100 | 1.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.95 | 0.97 |
| 2 | 100 | 1.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.95 | 0.95 |
| 2 | 100 | 1.0 | 4.0 | −0.25 | −0.30 | −0.24 | 12.50 | 15.00 | 12.00 | 0.73 | 0.69 | 0.73 |
| 2 | 100 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.93 | 0.97 |
| 2 | 100 | 2.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.91 | 0.94 |
| 2 | 100 | 2.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.99 | 0.93 | 0.98 |
| 2 | 100 | 2.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.93 | 0.96 |
| 2 | 100 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.96 | 0.93 |
| 2 | 100 | 4.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.97 | 0.94 |
| 2 | 100 | 4.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.95 | 0.95 |
| 2 | 100 | 4.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.94 | 0.96 |
TABLE 5.
Results of simulations under a population genetics model when there are four populations and the prior mean of the number of populations is 5, 10, or 20
|
E[E(K | X) − KT]
|
E[d(z, zT)]
|
Coverage
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KT | L | θ | M | 5 | 10 | 20 | 5 | 10 | 20 | 5 | 10 | 20 |
| 4 | 10 | 0.5 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.91 | 0.99 |
| 4 | 10 | 0.5 | 1.0 | 0.00 | 0.00 | −0.02 | 0.00 | 0.00 | 0.50 | 0.96 | 0.95 | 0.97 |
| 4 | 10 | 0.5 | 2.0 | −0.21 | −0.23 | −0.33 | 5.40 | 5.90 | 8.37 | 0.75 | 0.76 | 0.73 |
| 4 | 10 | 0.5 | 4.0 | −1.37 | −1.38 | −1.31 | 35.37 | 35.49 | 34.25 | 0.15 | 0.12 | 0.21 |
| 4 | 10 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.98 | 0.93 |
| 4 | 10 | 1.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.94 | 0.97 |
| 4 | 10 | 1.0 | 2.0 | −0.04 | −0.03 | −0.04 | 1.26 | 0.76 | 1.01 | 0.88 | 0.91 | 0.91 |
| 4 | 10 | 1.0 | 4.0 | −0.65 | −0.65 | −0.63 | 16.50 | 16.63 | 16.49 | 0.46 | 0.48 | 0.46 |
| 4 | 10 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.96 | 1.00 |
| 4 | 10 | 2.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.95 | 0.91 |
| 4 | 10 | 2.0 | 2.0 | −0.01 | −0.01 | 0.00 | 0.25 | 0.25 | 0.00 | 0.93 | 0.94 | 0.96 |
| 4 | 10 | 2.0 | 4.0 | −0.13 | −0.11 | −0.20 | 3.29 | 2.79 | 5.04 | 0.84 | 0.85 | 0.78 |
| 4 | 10 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.99 | 0.95 | 0.96 |
| 4 | 10 | 4.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.98 | 0.96 | 0.91 |
| 4 | 10 | 4.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.94 | 0.88 |
| 4 | 10 | 4.0 | 4.0 | −0.02 | −0.01 | −0.02 | 0.51 | 0.26 | 0.51 | 0.89 | 0.95 | 0.93 |
| 4 | 100 | 0.5 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.90 | 0.96 | 0.96 |
| 4 | 100 | 0.5 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.96 | 0.93 |
| 4 | 100 | 0.5 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.98 | 0.94 | 0.92 |
| 4 | 100 | 0.5 | 4.0 | −0.97 | −0.98 | −0.99 | 24.25 | 24.50 | 24.75 | 0.35 | 0.37 | 0.37 |
| 4 | 100 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.96 | 0.95 |
| 4 | 100 | 1.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.90 | 1.00 |
| 4 | 100 | 1.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.94 | 0.96 |
| 4 | 100 | 1.0 | 4.0 | −0.01 | −0.01 | −0.01 | 0.25 | 0.25 | 0.25 | 0.98 | 0.92 | 0.91 |
| 4 | 100 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.98 | 0.93 |
| 4 | 100 | 2.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.95 | 0.96 |
| 4 | 100 | 2.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.96 | 0.94 |
| 4 | 100 | 2.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.95 | 0.95 |
| 4 | 100 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.91 | 0.95 | 0.96 |
| 4 | 100 | 4.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.98 | 0.96 | 0.91 |
| 4 | 100 | 4.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.94 | 0.88 |
| 4 | 100 | 4.0 | 4.0 | −0.02 | −0.01 | −0.02 | 0.51 | 0.26 | 0.51 | 0.89 | 0.95 | 0.93 |
TABLE 6.
Results of simulations under a population genetics model when there are 10 populations and the prior mean of the number of populations is 5, 10, or 20
|
E[E(K | X) − KT]
|
E[d(z, zT)]
|
Coverage
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KT | L | θ | M | 5 | 10 | 20 | 5 | 10 | 20 | 5 | 10 | 20 |
| 10 | 10 | 0.5 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.97 | 0.91 |
| 10 | 10 | 0.5 | 1.0 | −0.07 | −0.09 | −0.06 | 0.70 | 0.90 | 0.60 | 0.90 | 0.85 | 0.90 |
| 10 | 10 | 0.5 | 2.0 | −1.08 | −1.20 | −1.18 | 10.85 | 12.04 | 11.87 | 0.38 | 0.24 | 0.27 |
| 10 | 10 | 0.5 | 4.0 | −5.53 | −5.25 | −5.37 | 55.71 | 53.04 | 54.33 | 0.01 | 0.00 | 0.00 |
| 10 | 10 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.98 | 1.00 |
| 10 | 10 | 1.0 | 1.0 | −0.01 | −0.03 | −0.02 | 0.10 | 0.30 | 0.20 | 0.94 | 0.88 | 0.95 |
| 10 | 10 | 1.0 | 2.0 | −0.30 | −0.34 | −0.32 | 3.01 | 3.41 | 3.21 | 0.68 | 0.71 | 0.68 |
| 10 | 10 | 1.0 | 4.0 | −2.51 | −2.54 | −2.51 | 25.33 | 25.63 | 25.41 | 0.06 | 0.05 | 0.07 |
| 10 | 10 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.96 | 0.95 |
| 10 | 10 | 2.0 | 1.0 | 0.00 | −0.01 | 0.00 | 0.00 | 0.10 | 0.00 | 0.95 | 0.97 | 0.95 |
| 10 | 10 | 2.0 | 2.0 | −0.07 | −0.08 | −0.05 | 0.70 | 0.80 | 0.50 | 0.88 | 0.89 | 0.90 |
| 10 | 10 | 2.0 | 4.0 | −0.70 | −0.74 | −0.59 | 7.08 | 7.52 | 6.09 | 0.43 | 0.44 | 0.55 |
| 10 | 10 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.96 | 0.97 |
| 10 | 10 | 4.0 | 1.0 | 0.00 | −0.01 | −0.01 | 0.00 | 0.10 | 0.10 | 0.91 | 0.95 | 0.92 |
| 10 | 10 | 4.0 | 2.0 | −0.03 | −0.06 | −0.09 | 0.30 | 0.60 | 0.90 | 0.94 | 0.87 | 0.90 |
| 10 | 10 | 4.0 | 4.0 | −0.30 | −0.25 | −0.35 | 3.14 | 2.65 | 3.80 | 0.68 | 0.72 | 0.66 |
| 10 | 100 | 0.5 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.98 | 0.95 |
| 10 | 100 | 0.5 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.95 | 0.91 | 0.94 |
| 10 | 100 | 0.5 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.97 | 0.94 |
| 10 | 100 | 0.5 | 4.0 | −0.42 | −0.51 | −0.43 | 4.20 | 5.10 | 4.30 | 0.63 | 0.60 | 0.65 |
| 10 | 100 | 1.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.97 | 0.96 |
| 10 | 100 | 1.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | 0.93 | 0.93 |
| 10 | 100 | 1.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.91 | 0.94 |
| 10 | 100 | 1.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.91 | 0.96 | 0.93 |
| 10 | 100 | 2.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.91 | 0.95 |
| 10 | 100 | 2.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.91 | 0.95 | 0.96 |
| 10 | 100 | 2.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.94 | 0.96 |
| 10 | 100 | 2.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 0.95 | 0.92 |
| 10 | 100 | 4.0 | 0.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.96 | 0.91 | 0.97 |
| 10 | 100 | 4.0 | 1.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.99 | 0.93 | 0.99 |
| 10 | 100 | 4.0 | 2.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.94 | 0.93 | 0.95 |
| 10 | 100 | 4.0 | 4.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.97 | 0.94 | 0.93 |
Generally speaking, the results of the simulation study are in accord with what intuition would suggest: (1) Increasing the number of loci improves the ability of the method to correctly identify the partition, (2) estimation can suffer when the parameter of the Dirichlet process model, α, is misspecified, and (3) inference of population structure is more reliable when θ is large and M (migration rates) are low. The main result of the simulations, however, is not intuitive: One can accurately infer the number of populations in a sample using genetic data under a Dirichlet process model. Importantly, the number of loci necessary to accurately infer the number of populations is moderate in size and in the range of many published studies of population structure. Also, one can accurately infer the number of populations even when the parameter of the Dirichlet process model is misspecified (as long as the number of loci is moderately large).
Demographic models that are relevant to real populations are likely to be much more complex than the simple symmetric island model we have explored. In addition, markers used to infer population structure are often ascertained in a smaller sample of individuals and geographic sampling schemes can vary widely from study to study. Fortunately, Structurama is easy to use in conjunction with programs like ms (Hudson 2002; see materials and methods) and it is straightforward to evaluate the program's performance under virtually any arbitrary sampling scheme and demographic model. Future work could concentrate on the relative performance of different methods for inferring population structure. This work might benefit from consideration of the methods we devised in this article to measure the accuracy of population structure methods (such as the application of a distance on partitions).
Acknowledgments
This work was supported by National Science Foundation grant DEB-0445453 and National Institutes of Health grant GM-069801 awarded to J.P.H.
APPENDIX: COMMAND LINES USED FOR MS COALESCENT SIMULATIONS TO EVALUATE THE PERFORMANCE OF STRUCTURAMA
| One population (panmixia): |
| ./ms 200 10000 -t 0.5 >ms.t0.5_p1 |
| ./ms 200 10000 -t 1 >ms.t1_p1 |
| ./ms 200 10000 -t 2 >ms.t1_p2 |
| ./ms 200 10000 -t 4 >ms.t1_p4 |
| Two populations: |
| ./ms 200 10000 -t 0.5 -I 2 100 100 0.5 >ms.t0.5_p2_m0.5 |
| ./ms 200 10000 -t 0.5 -I 2 100 100 1 >ms.t0.5_p2_m1 |
| ./ms 200 10000 -t 0.5 -I 2 100 100 2 >ms.t0.5_p2_m2 |
| ./ms 200 10000 -t 0.5 -I 2 100 100 4 >ms.t0.5_p2_m4 |
| ./ms 200 10000 -t 1 -I 2 100 100 0.5 >ms.t1_p2_m0.5 |
| ./ms 200 10000 -t 1 -I 2 100 100 1 >ms.t1_p2_m1 |
| ./ms 200 10000 -t 1 -I 2 100 100 2 >ms.t1_p2_m2 |
| ./ms 200 10000 -t 1 -I 2 100 100 4 >ms.t1_p2_m4 |
| ./ms 200 10000 -t 2 -I 2 100 100 0.5 >ms.t2_p2_m0.5 |
| ./ms 200 10000 -t 2 -I 2 100 100 1 >ms.t2_p2_m1 |
| ./ms 200 10000 -t 2 -I 2 100 100 2 >ms.t2_p2_m2 |
| ./ms 200 10000 -t 2 -I 2 100 100 4 >ms.t2_p2_m4 |
| ./ms 200 10000 -t 4 -I 2 100 100 0.5 >ms.t4_p2_m0.5 |
| ./ms 200 10000 -t 4 -I 2 100 100 1 >ms.t4_p2_m1 |
| ./ms 200 10000 -t 4 -I 2 100 100 2 >ms.t4_p2_m2 |
| ./ms 200 10000 -t 4 -I 2 100 100 4 >ms.t4_p2_m4 |
| Four populations: |
| ./ms 200 10000 -t 0.5 -I 4 50 50 50 50 0.5 >ms.t0.5_p4_m0.5 |
| ./ms 200 10000 -t 0.5 -I 4 50 50 50 50 1 >ms.t0.5_p4_m1 |
| ./ms 200 10000 -t 0.5 -I 4 50 50 50 50 2 >ms.t0.5_p4_m2 |
| ./ms 200 10000 -t 0.5 -I 4 50 50 50 50 4 >ms.t0.5_p4_m4 |
| ./ms 200 10000 -t 1 -I 4 50 50 50 50 0.5 >ms.t1_p4_m0.5 |
| ./ms 200 10000 -t 1 -I 4 50 50 50 50 1 >ms.t1_p4_m1 |
| ./ms 200 10000 -t 1 -I 4 50 50 50 50 2 >ms.t1_p4_m2 |
| ./ms 200 10000 -t 1 -I 4 50 50 50 50 4 >ms.t1_p4_m4 |
| ./ms 200 10000 -t 2 -I 4 50 50 50 50 0.5 >ms.t2_p4_m0.5 |
| ./ms 200 10000 -t 2 -I 4 50 50 50 50 1 >ms.t2_p4_m1 |
| ./ms 200 10000 -t 2 -I 4 50 50 50 50 2 >ms.t2_p4_m2 |
| ./ms 200 10000 -t 2 -I 4 50 50 50 50 4 >ms.t2_p4_m4 |
| ./ms 200 10000 -t 4 -I 4 50 50 50 50 0.5 >ms.t4_p4_m0.5 |
| ./ms 200 10000 -t 4 -I 4 50 50 50 50 1 >ms.t4_p4_m1 |
| ./ms 200 10000 -t 4 -I 4 50 50 50 50 2 >ms.t4_p4_m2 |
| ./ms 200 10000 -t 4 -I 4 50 50 50 50 4 >ms.t4_p4_m4 |
| Ten populations: |
| ./ms 200 10000 -t 0.5 -I 10 20 20 20 20 20 20 20 20 20 20 0.5 >ms.t0.5_p10_m0.5 |
| ./ms 200 10000 -t 0.5 -I 10 20 20 20 20 20 20 20 20 20 20 1 >ms.t0.5_p10_m1 |
| ./ms 200 10000 -t 0.5 -I 10 20 20 20 20 20 20 20 20 20 20 2 >ms.t0.5_p10_m2 |
| ./ms 200 10000 -t 0.5 -I 10 20 20 20 20 20 20 20 20 20 20 4 >ms.t0.5_p10_m4 |
| ./ms 200 10000 -t 1 -I 10 20 20 20 20 20 20 20 20 20 20 0.5 >ms.t1_p10_m0.5 |
| ./ms 200 10000 -t 1 -I 10 20 20 20 20 20 20 20 20 20 20 1 >ms.t1_p10_m1 |
| ./ms 200 10000 -t 1 -I 10 20 20 20 20 20 20 20 20 20 20 2 >ms.t1_p10_m2 |
| ./ms 200 10000 -t 1 -I 10 20 20 20 20 20 20 20 20 20 20 4 >ms.t1_p10_m4 |
| ./ms 200 10000 -t 2 -I 10 20 20 20 20 20 20 20 20 20 20 0.5 >ms.t2_p10_m0.5 |
| ./ms 200 10000 -t 2 -I 10 20 20 20 20 20 20 20 20 20 20 1 >ms.t2_p10_m1 |
| ./ms 200 10000 -t 2 -I 10 20 20 20 20 20 20 20 20 20 20 2 >ms.t2_p10_m2 |
| ./ms 200 10000 -t 2 -I 10 20 20 20 20 20 20 20 20 20 20 4 >ms.t2_p10_m4 |
| ./ms 200 10000 -t 4 -I 10 20 20 20 20 20 20 20 20 20 20 0.5 >ms.t4_p10_m0.5 |
| ./ms 200 10000 -t 4 -I 10 20 20 20 20 20 20 20 20 20 20 1 >ms.t4_p10_m1 |
| ./ms 200 10000 -t 4 -I 10 20 20 20 20 20 20 20 20 20 20 2 >ms.t4_p10_m2 |
| ./ms 200 10000 -t 4 -I 10 20 20 20 20 20 20 20 20 20 20 4 >ms.t4_p10_m4 |
References
- Akaike, H., 1973. Information theory as an extension of the maximum likelihood principle, pp. 267–281 in Second International Symposium on Information Theory, edited by B. N. Petrov and F. Csaki. Akademiai Kiado, Budapest.
- Andolfatto, P., and M. Przeworski, 2000. A genome-wide departure from the standard neutral model in natural populations of Drosophila. Genetics 156: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniak, C. E., 1974. Mixtures of Dirichlet processes with applications to non-parametric problems. Ann. Stat. 2: 1152–1174. [Google Scholar]
- Balding, D. J., and R. A. Nichols, 1995. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96: 3–12. [DOI] [PubMed] [Google Scholar]
- Bell, E. T., 1934. Exponential numbers. Am. Math. Mon. 41: 411–419. [Google Scholar]
- Corander, J., P. Waldmann and M. J. Sillanpää, 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander, J., P. Waldmann, P. Marttinen and M. J. Sillanpää, 2004. BAPS2: enhanced possibilities for the analysis of population structure. Bioinformatics 20: 2363–2369. [DOI] [PubMed] [Google Scholar]
- Dawson, K. J., and K. Belkhir, 2001. A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet. Res. 78: 59–77. [DOI] [PubMed] [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software Structure: a simulation study. Mol. Ecol. 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, T. S., 1973. A Bayesian analysis of some nonparametric problems. Ann. Stat. 1: 209–230. [Google Scholar]
- Galbusera, P., L. Lens, E. Waiyaki, T. Schenck and E. Mattysen, 2000. Effective population size and gene flow in the globally, critically endangered Taita thrush, Turdus helleri. Conserv. Genet. 1: 45–55. [Google Scholar]
- Geyer, C. J., 1991. Markov chain Monte Carlo maximum likelihood, pp. 156–163 in Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface, edited by E. M. Keramidas. Interface Foundation, Fairfax Station, VA.
- Green, P. J., 1995. Reversible jump MCMC computation and Bayesian model determination. Biometrika 82: 711–732. [Google Scholar]
- Gusfield, D., 2002. Partition-distance: a problem and class of perfect graphs arising in clustering. Inform. Process. Lett. 82: 159–164. [Google Scholar]
- Hammer, M. F., F. Blackmer, D. Garrigan, M. W. Nachman and J. A. Wilder, 2003. Human population structure and its effects on sampling Y chromosome sequence variation. Genetics 164: 1495–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger, K. E., P. O. Lewis and D. K. Dey, 2002. A Bayesian approach to inferring population structure from dominant markers. Mol. Ecol. 11: 1157–1164. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Jeffreys, H., 1961. Theory of Probability, Ed. 3. Oxford University Press, Oxford.
- Lavine, M., and M. J. Schervish, 1999. Bayes factors: what they are and what they are not. Am. Stat. 53: 119–122. [Google Scholar]
- Leo, N. P., J. M. Hughes, X. Yang, S. K. S. Poudel, W. G. Brogdon et al., 2005. The head and body lice of humans are genetically distinct (Insecta: Phthiraptera, Pediculidae): evidence from double infestations. Heredity 95: 34–40. [DOI] [PubMed] [Google Scholar]
- Lorenzen, E. D., and H. R. Siegismund, 2004. No suggestion of hybridization between the vulnerable black-faced impala (Aepyceros melampus petersi) and the common impala (A. m. melampus) in Etosha NP, Namibia. Mol. Ecol. 13: 3007–3019. [DOI] [PubMed] [Google Scholar]
- Lorenzen, E. D., P. Arctander and H. R. Siegismund, 2006. Regional genetic structuring and evolutionary history of the impala Aepyceros melampus. J. Hered. 97: 119–132. [DOI] [PubMed] [Google Scholar]
- Malécot, G., 1948. Les Mathématiques D l' Hérédité. Masson et Cie, Paris.
- Michel, A. P., M. J. Ingrasci, B. J. Schemerhorn, M. Kern, G. Le Goff et al., 2005. Rangewide population genetic structure of the African malaria vector Anopheles funestus. Mol. Ecol. 14: 4235–4248. [DOI] [PubMed] [Google Scholar]
- Moodley, Y., and E. H. Harley, 2005. Population structuring in mountain zebras (Equus zebra): the molecular consequences of divergent demographic histories. Conserv. Genet. 6: 953–968. [Google Scholar]
- Neal, R. M., 2000. Markov chain sampling methods for Dirichlet process mixture models. J. Comput. Graph. Stat. 9: 249–265. [Google Scholar]
- Nejsum, P., E. D. Parker, J. Frydenberg, A. Roepstorff, J. Boes et al., 2005. Ascariasis is a zoonosis in Denmark. J. Clin. Microbiol. 43: 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, M. A., and A. E. Raftery, 1994. Approximate Bayesian inference with the weighted likelihood bootstrap. J. R. Stat. Soc. B 56: 3–48. [Google Scholar]
- Nielsen, R., 2001. Statistical tests of neutrality in the age of genomics. Heredity 86: 641–647. [DOI] [PubMed] [Google Scholar]
- Ochsenreither, S., K. Kuhls, M. Schaar, W. Presber and G. Schonian, 2006. Multilocus microsatellite typing as a new tool for discrimination of Leishmania infantum MON-1 strains. J. Clin. Microbiol. 44: 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth, A., T. Adama, W. Din and F. Bonhomme, 1998. Hybridation naturelle entre deux sous espéces de souris domestique Mus musculus domesticus et Mus musculus castaneus prés de Lake Casitas (Californie). Genome 41: 104–110. [DOI] [PubMed] [Google Scholar]
- Pella, J., and M. Masuda, 2006. The Gibbs and split-merge sampler for population mixture analysis from genetic data with incomplete baselines. Can. J. Fish. Aquat. Sci. 63: 576–596. [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery, A. E., M. A. Newton, J. M. Satagopan and P. N. Krivitsky, 2006. Estimating the Integrated Likelihood via Posterior Simulation Using the Harmonic Mean Identity (Memorial Sloan-Kettering Cancer Center Department of Epidemiology and Biostatistics Working Paper Series, Working Paper 6).
- Rannala, B., and J. L. Mountain, 1997. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 94: 9197–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2981–2985. [DOI] [PubMed] [Google Scholar]
- Small, M. P., A. E. Frye, J. F. Von Bargen and S. F. Young, 2006. Genetic structure of chum salmon (Oncorhynchus keta) populations in the lower Columbia River: Are chum salmon in Cascade tributaries remnant populations? Conserv. Genet. 7: 65–78. [Google Scholar]
- Song, K. J., and R. C. Elston, 2006. A powerful method of combining measures of association and Hardy-Weinberg disequilibrium for fine-mapping in case-control studies. Stat. Med. 25: 105–126. [DOI] [PubMed] [Google Scholar]
- Stanton, D., and D. White, 1986. Constructive Combinatorics. Springer-Verlag, New York.
- Stephens, M., 2000. Dealing with label-switching in mixture models. J. R. Stat. Soc. Ser. B 62: 795–809. [Google Scholar]
- Tierney, L., 1996. Introduction to general state-space Markov chain theory, pp. 59–74 in Markov Chain Monte Carlo in Practice. Chapman & Hall, London.
- Tsai, H. J., J. Y. Kho, N. Shaikh, S. Choudhry, M. Naqvi et al., 2006. Admixture-matched case-control study: a practical approach for genetic association studies in admixed populations. Hum. Genet. 118: 626–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1940. Breeding structure of populations in relation to speciation. Am. Nat. 74: 232–248. [Google Scholar]
- Wright, S., 1943. Isolation by distance. Genetics 28: 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetical structure of populatons. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]
- Yu, J. M., G. Pressoir, W. H. Briggs, I. V. Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38: 203–208. [DOI] [PubMed] [Google Scholar]