

Abstract
Linkage disequilibrium (LD) analysis in outbred populations uses historical recombinations to detect and fine map quantitative trait loci (QTL). Our objective was to evaluate the effect of various factors on power and precision of QTL detection and to compare LD mapping methods on the basis of regression and identity by descent (IBD) in populations of limited effective population size (Ne). An 11-cM region with 6–38 segregating single-nucleotide polymorphisms (SNPs) and a central QTL was simulated. After 100 generations of random mating with Ne of 50, 100, or 200, SNP genotypes and phenotypes were generated on 200, 500, or 1000 individuals with the QTL explaining 2 or 5% of phenotypic variance. To detect and map the QTL, phenotypes were regressed on genotypes or (assumed known) haplotypes, in comparison with the IBD method. Power and precision to detect QTL increased with sample size, marker density, and QTL effect. Power decreased with Ne, but precision was affected little by Ne. Single-marker regression had similar or greater power and precision than other regression models, and was comparable to the IBD method. Thus, for rapid initial screening of samples of adequate size in populations in which drift is the primary force that has created LD, QTL can be detected and mapped by regression on SNP genotypes without recovering haplotypes.
RECENT advances in technology, such as high-density single-nucleotide polymorphism (SNP) genotyping, have increased the feasibility of quantitative trait loci (QTL) detection and fine mapping in outbred populations using historical populationwide linkage disequilibrium (LD). Goals for LD mapping include both QTL detection and fine mapping of a previously detected QTL, although most studies and methods developed for LD mapping may deal with only one of these (Zöllner and Pritchard 2005). LD mapping has been used extensively to identify genes for monogenic diseases in humans (Peltonen 2000). Contrary to the situation in humans, extensive LD over a long range was observed in dairy cattle, sheep, and pigs (Farnir et al. 2000; Mcrae et al. 2002; Tenesa et al. 2003; Nsengimana et al. 2004; Harmegnies et al. 2006) because of limited effective population sizes. Thus, LD mapping in livestock, and in other populations with (historically) limited effective population sizes, might be effective using marker maps of more limited density than what is required for most human populations because of the extensive LD that is created by drift in populations of limited effective sizes (Terwilliger et al. 1998; Farnir et al. 2002). Also, because LD mapping can be implemented in commercial breeding populations, resulting QTL can immediately be implemented for marker-assisted selection (Dekkers and Hospital 2002).
Several statistical methods for LD mapping have been developed, including random-effects models based on identity by descent (IBD) (Meuwissen and Goddard 2000) and least-squares methods based on regression of phenotype on marker genotypes or haplotypes (Long and Langley 1999; Grapes et al. 2004, 2006). IBD methods model covariances between individuals by deriving IBD probabilities for QTL alleles carried by alternate marker haplotypes under some assumptions about population history (Meuwissen and Goddard 2000). By combining IBD-based LD mapping methods and linkage analysis, Meuwissen et al. (2002) mapped a QTL within a region <1 cM for twinning rate in large half-sib cattle families.
Grapes et al. (2004) showed that regression methods for LD mapping can be competitive with the IBD method in terms of accuracy of fine mapping within a previously identified QTL region. For the IBD approach, Grapes et al. (2006) found that derivation of IBD probabilities based on haplotypes of 4–6 markers around the postulated QTL position resulted in greater mapping precision than IBD derived using all 10 markers, because the latter resulted in a flatter likelihood curve that did not discriminate between alternate QTL positions.
Grapes et al. (2004, 2006) did not compare IBD vs. regression methods in terms of power to detect QTL. In addition, they simulated 10 or 20 evenly spaced SNPs in the base population and used them for QTL fine mapping in the final generation although some of them were fixed after 100 generations of random mating. In practice, SNPs that are not informative will not be used for analysis. Our objective was to use well-spaced and segregating SNPs to evaluate the effect of various factors, including SNP density and effective population size, on power and precision to detect QTL using regression- and IBD-based LD mapping methods. The results of this study will aid in the proper design and analysis of LD mapping studies in populations of limited (historical) effective size, in which most LD is generated by drift. Such populations are common in domestic animals but also include some closed human populations and plant and wildlife populations.
METHODS
Simulated populations:
A QTL region of 11 cM containing 1000 SNPs with frequencies 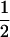 and in equilibrium and a central QTL with uniquely numbered alleles was simulated. LD was created by g (g = 50, 100, 200) generations of random mating in a population of effective size Ne (Ne = 50, 100, 200). In generation g, N (N = 200, 500, 1000) individuals were generated by randomly sampling N mating pairs with replacement and the QTL was converted to be biallelic by making the allele with frequency closest to 0.5 and between 0.3 and 0.7 the favorable allele, with an additive effect that was standardized to explain x% (x = 2, 5) of the phenotypic variance. The remainder of an individual's phenotypic value was generated as a random standard normal deviate. The k-medoids clustering (Speed 2003) method was used to identify k (k = 6, 10, 20, 38) well-spaced SNPs over the QTL region that were still segregating in generation g (minor allele frequency ≥0.2). Phase-known genotypes of SNPs at the median of each cluster were used for analysis. For each scenario, a total of 10,000 viable replicates, i.e., for which at least one QTL allele had a frequency between 0.3 and 0.7 in generation g, were used for analysis. Significance thresholds at a 1% regionwide level were determined empirically on the basis of 10,000 replicates where the QTL had no effect on phenotype (x = 0).
and in equilibrium and a central QTL with uniquely numbered alleles was simulated. LD was created by g (g = 50, 100, 200) generations of random mating in a population of effective size Ne (Ne = 50, 100, 200). In generation g, N (N = 200, 500, 1000) individuals were generated by randomly sampling N mating pairs with replacement and the QTL was converted to be biallelic by making the allele with frequency closest to 0.5 and between 0.3 and 0.7 the favorable allele, with an additive effect that was standardized to explain x% (x = 2, 5) of the phenotypic variance. The remainder of an individual's phenotypic value was generated as a random standard normal deviate. The k-medoids clustering (Speed 2003) method was used to identify k (k = 6, 10, 20, 38) well-spaced SNPs over the QTL region that were still segregating in generation g (minor allele frequency ≥0.2). Phase-known genotypes of SNPs at the median of each cluster were used for analysis. For each scenario, a total of 10,000 viable replicates, i.e., for which at least one QTL allele had a frequency between 0.3 and 0.7 in generation g, were used for analysis. Significance thresholds at a 1% regionwide level were determined empirically on the basis of 10,000 replicates where the QTL had no effect on phenotype (x = 0).
Regression-based LD mapping:
Following Grapes et al. (2004), QTL detection and fine mapping was by regression of phenotypes in the final generation on genotypes or haplotypes of m neighboring SNPs (m = 1, 2, 4) for each window of m SNPs within the k-SNP interval. For regression on genotypes, the model for the window starting with SNP j (j = 1 to k − m + 1) was 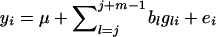 , where yi is the phenotype of individual i,
, where yi is the phenotype of individual i, 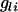 is the number of copies of allele 1 (vs. allele 0) at SNP l for individual i, bl is the allele substitution effect, and ei is the residual. The model of regression on haplotypes for each window of m SNPs was
is the number of copies of allele 1 (vs. allele 0) at SNP l for individual i, bl is the allele substitution effect, and ei is the residual. The model of regression on haplotypes for each window of m SNPs was 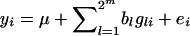 , where
, where 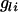 is the number of copies of haplotype l carried by individual i (assumed known) and bl is the haplotype effect. The window with the most significant F-value was chosen as the best model and the center of that window as the estimate of QTL position.
is the number of copies of haplotype l carried by individual i (assumed known) and bl is the haplotype effect. The window with the most significant F-value was chosen as the best model and the center of that window as the estimate of QTL position.
IBD-based LD mapping:
Following Meuwissen and Goddard (2000), phenotypic records in the last generation were analyzed by y = Xβ + Zh + e, where y is the vector of N phenotypic records, β is a vector of fixed effects, which reduces to the overall mean here, X is an incidence matrix for β, which reduces to a vector of N ones, h is a (q × 1) vector of random effects of q unique marker haplotypes present in the final generation, Z is the known incidence matrix for h, and e is the vector of residuals. The variance of the residuals is 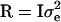 , where
, where 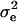 is the residual variance and I is an identity matrix. The variance of the haplotype effects is
is the residual variance and I is an identity matrix. The variance of the haplotype effects is 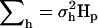 , where
, where 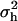 is the variance contributed by the QTL, and Hp is a (q × q) matrix with probabilities that QTL alleles at the assumed position p are IBD given a pair of marker haplotypes. Thus, covariances between QTL alleles carried by alternate marker haplotypes were modeled using IBD. The probability of IBD of a pair of alleles at the putative QTL position increased as the number of markers surrounding the QTL that are consecutively identical in state increased (Meuwissen and Goddard 2000). It was derived with assumptions about historical population structure, such as Ne and mutation age, using the method of Meuwissen and Goddard (2001).
is the variance contributed by the QTL, and Hp is a (q × q) matrix with probabilities that QTL alleles at the assumed position p are IBD given a pair of marker haplotypes. Thus, covariances between QTL alleles carried by alternate marker haplotypes were modeled using IBD. The probability of IBD of a pair of alleles at the putative QTL position increased as the number of markers surrounding the QTL that are consecutively identical in state increased (Meuwissen and Goddard 2000). It was derived with assumptions about historical population structure, such as Ne and mutation age, using the method of Meuwissen and Goddard (2001).
For each window of m neighboring SNPs (m = 1, 2, 4, 6, 8), the full model was fitted sequentially in the m − 1 marker intervals by assuming that the QTL was at the center of each interval. The parameters were estimated using the following mixed-model equations (MME):
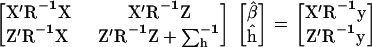 |
The residual log likelihood under multivariate normality was
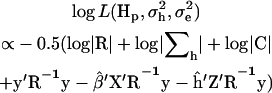 |
(Meyer and Smith 1996), where C is the coefficient matrix of the MME.
Given a QTL position p, i.e., given Hp, the log L was maximized using the Newton–Raphson algorithm to obtain estimates of the variance components 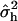 and
and 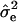 . The
. The 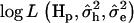 was calculated for every putative QTL position and the position with the highest
was calculated for every putative QTL position and the position with the highest 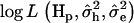 was identified as the most likely QTL position. To test for significance of the QTL, a reduced model y = Xβ + e was also fitted. Under the reduced model
was identified as the most likely QTL position. To test for significance of the QTL, a reduced model y = Xβ + e was also fitted. Under the reduced model
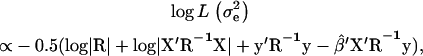 |
which was maximized with respect to 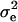 using the Newton–Raphson algorithm.
using the Newton–Raphson algorithm.
For the Newton–Raphson algorithm, object-oriented programming was used to implement automatic differentiation (Tsukanov and Hall 2003) to calculate the first and second derivatives of log L. We constrained 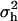 and
and 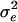 to their parameter spaces by setting
to their parameter spaces by setting 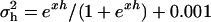 and
and 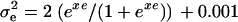 and maximizing log L with regard to xh and xe. A grid of starting values was used to improve convergence of the Newton–Raphson algorithm to the global maximum. The difference between the maximum of
and maximizing log L with regard to xh and xe. A grid of starting values was used to improve convergence of the Newton–Raphson algorithm to the global maximum. The difference between the maximum of 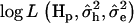 under the full model and the maximum of
under the full model and the maximum of 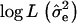 under the reduced model was used to test for significance at the most likely QTL position.
under the reduced model was used to test for significance at the most likely QTL position.
RESULTS
Power of regression methods:
The impacts on power to detect a QTL of sample size (N), SNP density, QTL effect, Ne, number of generations since mutation, and model of regression analysis are presented in Figure 1. Regardless of the model of analysis, power increased with sample size, SNP density, and QTL effect (Figure 1, A and B), but decreased with increasing Ne (Figure 1C), and increased a little with the number of generations since mutation (Figure 1D).
Figure 1.—

Effects of sample size, marker density (“# SNPs”), QTL effect, effective population size (Ne), number of generations since mutation (“# generations”), and model of analysis [regression on genotype (G) for 1, 2, or 4 SNPs or on haplotype (H) for 2 or 4 SNPs] on power to detect QTL. Based on 10,000 replicates.
The reason behind the increase in power with mutation age was explored further. The difference was not caused by a difference in average LD in the population because average LD at a given distance of c morgans was close to its expectation of 1/(1 + 4Nec) (Sved 1971) regardless of mutation age. However, segregating SNPs that were used for analysis did tend to be closer to the QTL after 200 generations than after 100 generations. This was caused by the fact that we kept only replicates that were segregating for the QTL for analysis, and those replicates were more likely to have a cluster of segregating SNPs around the QTL. For example, with Ne = 100 and N = 500, 17 and 50% of replicates were removed after 100 and 200 generations of random mating, respectively. The averages of the shortest SNP–QTL distances across 10,000 replicates were 0.27 and 0.21 cM for 100 and 200 generations, respectively.
In general, sample size had a greater effect on power than marker density. For example, doubling the number of genotypes by increasing density from 10 to 20 for N = 500 resulted in a smaller increase in power than doubling N from 500 to 1000 (Figure 1, A and B). The significance thresholds became more stringent as marker density increased. So the increase in the F-value with increasing numbers of markers was to some degree offset by an increase in the threshold. This did, however, depend on the extent of LD; with Ne = 200, an increase in density had a substantial impact on power (Figure 1C).
To test the effect of QTL position on power to detect a QTL, a noncentral QTL was also simulated, at 3.25 cM from the left end of the 11-cM chromosome region. Power was, however, very similar for central and noncentral QTL (results not shown). The effect of allele frequencies of SNPs in the base population was tested by simulating SNPs with allele frequencies randomly chosen from 0.2 to 0.8 in generation zero. Again, power was affected little (results not shown).
Differences between models of analysis were generally small; 1-SNP regression had very similar power as regression on 2 or 4 SNPs (Figure 1). Regression on haplotypes of 2 SNPs had similar power as genotype regression, but 4-SNP haplotype regression generally had lower power (Figure 1).
Precision of regression methods:
Mapping precision was quantified as the mean absolute error of position estimates and is summarized in Figure 2 for significant replicates. Similar to power, precision increased with sample size, SNP density, and QTL effect (Figure 2, A and B), but was affected little by Ne (Figure 2C). Precision increased with the number of generations since mutation (Figure 2D) because, as described previously, after many generations, replicates that were segregating for the QTL were also likely to be segregating for SNPs close to the QTL. Sample size and density had similar effects on precision (Figure 2, A and B). The magnitude of the QTL effect had less impact on precision (Figure 2, A and B) than on power (Figure 1, A and B).
Figure 2.—

Effects of sample size, marker density (“# SNPs”), QTL effect, effective population size (Ne), number of generations since mutation (“# generations”), and model of analysis [regression on genotype (G) for 1, 2, or 4 SNPs or on haplotype (H) for 2 or 4 SNPs] on precision of estimates of position for significant QTL. Based on 10,000 replicates.
Similar to power, precision was very similar for central and noncentral QTL (results not shown), except for a SNP density of 6. In that case, the precision of 4-SNP genotype or haplotype regression was poorer for noncentral QTL compared to central QTL (results not shown). With a window of 4 of 6 SNPs, it is impossible for the estimate of QTL position, which is the center of the best-fitting window, to be at the edges of the 11-cM region. Precision was affected little by allele frequencies of SNPs in the base population (results not shown).
Figure 3 shows mapping precision of regression methods for all replicates. Average precision was poorer when considering all (Figure 3, A and B) vs. only significant replicates (Figure 2, A and B). In contrast to considering significant replicates only (Figure 2), precision across all replicates decreased with increasing Ne (Figure 3C) and the QTL effect had much greater impact on precision (Figure 3, A and B); other results were the same as those for significant replicates only.
Figure 3.—

Effects of sample size, marker density (“# SNPs”), QTL effect, effective population size (Ne), number of generations since mutation (“# generations”), and model of analysis [regression on genotype (G) for 1, 2, or 4 SNPs or on haplotype (H) for 2 or 4 SNPs] on precision of estimates of position for all QTL. Based on 10,000 replicates.
When comparing alternate methods of analysis, single-marker regression resulted in similar or better precision than all other methods, which was the same when considering all and only significant replicates (Figures 2 and 3). When considering multiple markers, regression on genotypes resulted in similar precision to regression on haplotypes (Figures 2 and 3).
Power of the IBD method:
The power of the IBD method is presented in Tables 1 and 2 for QTL effects of 5 and 2% of the phenotypic variance, respectively, in comparison with regression methods. Because of high computing requirements, the IBD method was evaluated only for a limited number of scenarios. Power of the IBD method generally increased with SNP density and QTL effect, although this increase was not consistent when SNP density increased from 10 to 20 (Tables 1 and 2). In general, power increased with the number of SNPs used to compute IBD probabilities (window size) but appeared to reach an asymptote for window sizes of 6 and 8 markers (Tables 1 and 2). Window sizes >4 were not evaluated for regression methods because it would require a large numbers of parameters to be estimated, resulting in lower power. Single-marker regression had higher power than the 1-SNP IBD method (Tables 1 and 2). When considering windows of 2 or 4 SNPs, the IBD method resulted in similar or higher power than regression (Tables 1 and 2), except for a SNP density of 20 and a QTL effect of 5% (Table 1). In that case, regression on genotypes had better power than the IBD method (Table 1). For SNP densities of 6 and 10, power of the IBD method increased with the number of SNPs included in the model up to 4 or 6, and IBD using >1 SNP had similar or higher power than single-marker regression (Tables 1 and 2); these same patterns were, however, not clear for a SNP density of 20 (Tables 1 and 2). IBD using 6 and 8 SNPs had similar power for a SNP density of 10 (Tables 1 and 2).
TABLE 1.
Comparison of regression-based LD mapping methods with identity-by-descent (IBD) methods when the QTL explains 5% of the phenotypic variance
| No. SNPs included in model | Marker density (no. SNPs in 11-cM region)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 6
|
10
|
20
|
|||||||
| Geno | Haplo | IBD | Geno | Haplo | IBD | Geno | Haplo | IBD | |
| Power to detect QTL (%) | |||||||||
| 1 | 67 | — | 48 | 77 | — | 52 | 85 | — | 53 |
| 2 | 69 | 69 | 69 | 78 | 79 | 78 | 85 | 84 | 81 |
| 4 | 68 | 59 | 76 | 79 | 70 | 82 | 84 | 74 | 77 |
| 6 | — | — | 75 | — | — | 85 | — | — | 83 |
| 8 | — | — | — | — | — | 84 | — | — | — |
| Mean absolute error of position (cM) for significant QTL | |||||||||
| 1 | 1.05 | — | 1.14 | 0.79 | — | 0.90 | 0.64 | — | 0.81 |
| 2 | 1.20 | 1.21 | 1.17 | 0.92 | 0.92 | 0.87 | 0.67 | 0.64 | 0.64 |
| 4 | 1.38 | 1.34 | 1.11 | 1.31 | 1.30 | 0.85 | 0.88 | 0.91 | 0.62 |
| 6 | — | — | 1.21 | — | — | 0.86 | — | — | 0.62 |
| 8 | — | — | — | — | — | 0.93 | — | — | — |
| Mean absolute error of position (cM) for all QTL | |||||||||
| 1 | 1.34 | — | 1.33 | 0.96 | — | 1.00 | 0.74 | — | 0.82 |
| 2 | 1.40 | 1.36 | 1.32 | 1.05 | 1.05 | 0.98 | 0.76 | 0.74 | 0.69 |
| 4 | 1.38 | 1.36 | 1.22 | 1.37 | 1.37 | 0.93 | 0.94 | 1.01 | 0.66 |
| 6 | — | — | 1.31 | — | — | 0.93 | — | — | 0.66 |
| 8 | — | — | — | — | — | 1.01 | — | — | — |
Power (detection at 1% regionwise level) and precision are shown for each LD mapping method: (1) Geno, regression on genotypes at 1, 2, or 4 adjacent SNPs; (2) Haplo, regression on assumed known haplotypes of 2 or 4 adjacent SNPs; and (3) IBD, identity-by-descent methods using single SNP genotype or assumed known haplotypes of 2, 4, 6, or 8 adjacent SNPs. In the base population, SNPs were simulated with allele frequency of 0.5 and in linkage equilibrium, and a QTL was simulated with unique alleles at the center of the 11-cM region. The other parameters are Ne = 100, number of generations since mutation = 100, and sample size in generation 100 = 500. Results are based on 10,000 replicates.
TABLE 2.
Comparison of regression-based LD mapping methods with identity-by-descent (IBD) methods when the QTL explains 2% of the phenotypic variance
| No. SNPs included in model | Marker density (no. SNPs in 11-cM region)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 6
|
10
|
20
|
|||||||
| Geno | Haplo | IBD | Geno | Haplo | IBD | Geno | Haplo | IBD | |
| Power to detect QTL (%) | |||||||||
| 1 | 26 | — | 18 | 31 | — | 21 | 34 | — | 22 |
| 2 | 25 | 23 | 25 | 28 | 27 | 30 | 31 | 28 | 34 |
| 4 | 24 | 15 | 28 | 28 | 18 | 32 | 30 | 19 | 31 |
| 6 | — | — | 27 | — | — | 34 | — | — | 32 |
| 8 | — | — | — | — | — | 32 | — | — | — |
| Mean absolute error of position (cM) for significant QTL | |||||||||
| 1 | 1.13 | — | 1.26 | 0.93 | — | 1.16 | 0.85 | — | 1.03 |
| 2 | 1.33 | 1.31 | 1.27 | 1.10 | 1.13 | 1.06 | 0.96 | 0.94 | 0.95 |
| 4 | 1.39 | 1.36 | 1.23 | 1.42 | 1.48 | 1.06 | 1.15 | 1.25 | 0.99 |
| 6 | — | — | 1.36 | — | — | 1.10 | — | — | 0.96 |
| 8 | — | — | — | — | — | 1.20 | — | — | — |
| Mean absolute error of position (cM) for all QTL | |||||||||
| 1 | 1.71 | — | 1.67 | 1.41 | — | 1.45 | 1.28 | — | 1.28 |
| 2 | 1.71 | 1.69 | 1.64 | 1.51 | 1.55 | 1.44 | 1.37 | 1.41 | 1.25 |
| 4 | 1.41 | 1.38 | 1.54 | 1.64 | 1.66 | 1.38 | 1.50 | 1.64 | 1.27 |
| 6 | — | — | 1.69 | — | — | 1.41 | — | — | 1.25 |
| 8 | — | — | — | — | — | 1.50 | — | — | — |
Power (detection at 1% regionwise level) and precision are shown for each LD mapping method: (1) Geno, regression on genotypes at 1, 2, or 4 adjacent SNPs; (2) Haplo, regression on assumed known haplotypes of 2 or 4 adjacent SNPs; and (3) IBD, identity-by-descent methods using single SNP genotype or assumed known haplotypes of 2, 4, 6, or 8 adjacent SNPs. In the base population, SNPs were simulated with allele frequency of 0.5 and in linkage equilibrium, and a QTL was simulated with unique alleles at the center of the 11-cM region. The other parameters are Ne = 100, number of generations since mutation = 100, and sample size in generation 100 = 500. Results are based on 10,000 replicates.
Precision of the IBD method:
Tables 1 and 2 also show the precision of the IBD method compared with regression methods. Precision of the IBD method increased with SNP density and QTL effect (Tables 1 and 2). Single-marker regression, in general, had similar or higher precision than the 1-SNP IBD method, which was more obvious when considering only significant replicates than all replicates (Tables 1 and 2). When IBD probabilities were determined using 2 or 4 SNPs, the IBD method had better precision than regression using 2 or 4 SNPs, which was true when considering all and only significant replicates (Tables 1 and 2), except for a SNP density of 6 and a QTL effect of 2%, where regression on 4-SNP haplotypes gave the best precision for all replicates (Table 2). Comparing mapping precision of the IBD method using different window sizes both for significant and for all replicates, 4-SNP IBD, in general, resulted in the best precision (Tables 1 and 2); the IBD method using 8 SNPs resulted in as poor precision as IBD using 1 SNP (Tables 1 and 2). Comparing the 4-SNP IBD method with single-marker regression, which gave the best precision among regression methods, single-marker regression was in general better than 4-SNP IBD when considering only significant replicates (Tables 1 and 2); however, 4-SNP IBD had better precision when considering all replicates (Tables 1 and 2).
DISCUSSION
Power of QTL detection:
This study compared several methods for LD-based QTL fine mapping: regression on SNP genotypes, regression on SNP haplotypes, and the IBD method of Meuwissen and Goddard (2000). Regression on SNP genotypes does not require knowledge of SNP haplotypes and is therefore easier to implement. One of the main findings of our study is that single-marker regression provided similar or higher power than other regression-based methods for SNP densities ranging from 6 to 38/11 cM, while fitting haplotypes of 4 markers generally had low power (Figure 1). Part of the limited extra or lower power of multimarker and, in particular, haplotype methods over single-marker regression may be caused by the additional parameters fitted, which would be avoided when using the IBD method. And indeed, power of the IBD method did tend to increase with the number of SNPs used to compute IBD probabilities but only up to a point, and extra power over 1-SNP regression was limited (Tables 1 and 2). Several other studies also found that single-marker tests provide as much or greater power than haplotype-based tests (Long and Langley 1999; Nielsen et al. 2004).
Using haplotypes of more SNPs in the IBD method is expected to improve power because it improves the accuracy of IBD probabilities (Grapes et al. 2006) without fitting additional parameters. The IBD method with 1 SNP had much lower power than using more SNPs (Tables 1 and 2) because of the poor accuracy of IBD probabilities (Grapes et al. 2006). The power of the IBD method kept increasing until the window size reached 4 or 6, although this trend was not clear for a SNP density of 20 (Tables 1 and 2). The IBD method with 8 SNPs was evaluated for a SNP density of 10 and found to be similar in power as using 6 SNPs (Tables 1 and 2). It appears that the power of the IBD method can be improved only up to a certain point by using more SNPs to derive IBD probabilities because more distant SNPs provide little additional information to determine if QTL alleles are IBD. Grapes et al. (2006) quantified the accuracy of IBD probabilities by calculating the correlation between the true IBD state of two QTL alleles and the IBD probabilities obtained given the individuals' marker haplotypes. The correlations were 0.34, 0.5, and 0.52 for window sizes of 1, 4, and 10, respectively (Grapes et al. 2006). Therefore, the accuracy of IBD probabilities was only slightly increased by using >4 SNPs, which is consistent with the trend of power observed in our study.
Precision of QTL detection:
Precision was evaluated for all and only significant replicates. Significant replicate results should be considered if there is no prior information on QTL position (i.e., if QTL detection is part of the experiment). Results for all replicates would apply when the objective is to fine map a QTL in an already identified region, as was assumed in Grapes et al. (2004, 2006).
Our greater mapping precision for 1- vs. multi-SNP regression is in contrast to Grapes et al. (2004), who found that 2-SNP haplotype regression performed better at estimating the position of the QTL than single-marker regression for the same SNP density (Grapes et al. 2004). They, however, simulated the QTL at the center of a SNP interval, which advantaged 2-SNP regression. Here, SNP positions varied, which resulted in an average distance of the QTL to the closest SNP of 0.52 cM for 6 SNPs and 0.27 cM for 10 SNPs, compared to an average distance to the center of the flanking SNP interval of 0.41 cM for 6 SNPs and 0.28 cM for 10 SNPs, resulting in no inherent bias of using 1 vs. 2 SNPs with 10 (or more) SNPs and a slight disadvantage to the 1-SNP method with 6 SNPs. Grapes et al. (2004) also found greater precision for 2-SNP haplotype than for 2-SNP genotype regression, while we showed no benefit to using haplotypes. This may be due to their much larger QTL effects (x > 15%).
Grapes et al. (2004, 2006) also compared regression to the IBD method and found that single-marker regression was not significantly different in precision from IBD with a single SNP (Grapes et al. 2006), while we found that single-marker regression had similar or higher mapping precision than 1-SNP IBD (Tables 1 and 2). On the basis of Grapes et al. (2004, 2006), 2-SNP haplotype regression gave similar precision to the IBD method that used all 10 SNPs in the region to determine IBD. We observed similar precision for all replicates between 2-SNP haplotype regression and 8-SNP IBD using a SNP density of 10 (Tables 1 and 2). Grapes et al. (2004, 2006) also showed that doubling the number of SNPs genotyped for single-marker regression gave similar precision to the IBD method using 4 or 6 SNPs, except when marker spacing was small (0.125 cM for 1-SNP regression and 0.25 cM for IBD); however, the IBD method using 4 or 6 SNPs had better precision than single-marker regression if the same number of SNPs was genotyped in the two approaches (Grapes et al. 2004, 2006), which was also observed in our study for all replicates (Tables 1 and 2). On the basis of our study, even with the same number of SNPs genotyped, single-marker regression, in general, had better precision than the 4-SNP IBD method when considering only significant replicates (Tables 1 and 2).
The most interesting finding in Grapes et al. (2006) was that for the IBD approach, derivation of IBD based on haplotypes of 4–6 markers around the postulated QTL position resulted in greater mapping precision than IBD derived using all 10 markers. This is in good agreement with our study. Although the QTL effect in our study (x = 5%) was much smaller than that in Grapes et al. (2006) (x > 15%), we found that fitting a 4-SNP haplotype in the IBD method, in general, resulted in the best precision compared to other haplotype sizes (Tables 1 and 2) and IBD using 8 SNPs gave precision as poor as IBD using 1 SNP (Tables 1 and 2). As explained by Grapes et al. (2006), the use of 4 SNPs provides enough information to accurately derive IBD probabilities while allowing for discrimination between alternate QTL positions. Fitting 8 SNPs in our case may reduce the sensitivity of IBD probabilities to the QTL position and therefore reduce mapping precision.
It should be noted that Grapes et al. (2004, 2006) simulated 10 or 20 evenly spaced SNPs in the base population and used them for QTL fine mapping in the final generation although some of them became fixed after 100 generations of random mating. In practice, SNPs that are not informative will not be used for analysis. By simulating 1000 SNPs initially and identifying 6–38 SNPs that were still segregating in the last generation (minor allele frequency ≥0.2) and that were well spaced over the QTL region, our study reflects the real situation better.
Uncertainty of haplotype information:
The results for haplotype regression and the IBD method assumed that haplotypes are known, which would not be true in practice. This will obviously reduce power and precision of these methods. Genotype regression methods do not require knowledge of haplotypes. The extent to which power or mapping precision is compromised by approximations in haplotype reconstruction will depend on the degree of uncertainty in haplotype estimation, which in turn depends on family structure and the extent of LD between markers. For example, Lee and Van Der Werf (2005) found haplotype uncertainty to have little impact on mapping precision and power, which they suggested was because their design of two to three progeny per family with a 1-cM marker distance provided sufficient information to estimate haplotypes accurately. Morris et al. (2004), however, found that assuming the most likely haplotype configuration to be true resulted in substantial loss of mapping precision compared to integrating over all haplotype configurations, which had similar precision to using the true haplotypes.
Impact of the nature of LD on QTL detection:
In our study, LD was generated by drift and mutation, starting from a base population that was in complete LD. To evaluate the impact of LD generated by mutation vs. drift on power to detect QTL, populations described in the Table 1 legend were simulated. Allele Q was either simulated to be unique in the base population, representing complete LD, or biallelic with frequency 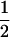 and in linkage equilibrium (LE). QTL detection by single-SNP regression showed limited difference in power between the LE and LD scenarios (0.7 for LE vs. 0.77 for LD with 10 SNPs and 0.82 vs. 0.85 with 20 SNPs). The average absolute error of QTL position was also only slightly increased for LE (0.88 cM for LE vs. 0.79 cM for LD with 10 SNPs and 0.73 vs. 0.64 with 20 SNPs). With LE, all Q alleles traced back to a single ancestral allele, which makes it equivalent to the LD case, for only 21% of all and 24% of significant replicates. The numbers of common ancestors of the Q allele were two, three, and four or greater for 56, 33, and 11% of the other replicates, for which the most frequent common ancestor accounted for only 67% of all Q alleles. These results demonstrate that mutation is not essential for sufficient LD to detect QTL and that QTL can be detected even if substantial heterogeneity exists with regard to ancestral origin of the Q alleles. Abdallah et al. (2003) found that power to detect QTL by single-marker regression was even greater with LE than with LD when using multiallelic markers and similar when using SNPs.
and in linkage equilibrium (LE). QTL detection by single-SNP regression showed limited difference in power between the LE and LD scenarios (0.7 for LE vs. 0.77 for LD with 10 SNPs and 0.82 vs. 0.85 with 20 SNPs). The average absolute error of QTL position was also only slightly increased for LE (0.88 cM for LE vs. 0.79 cM for LD with 10 SNPs and 0.73 vs. 0.64 with 20 SNPs). With LE, all Q alleles traced back to a single ancestral allele, which makes it equivalent to the LD case, for only 21% of all and 24% of significant replicates. The numbers of common ancestors of the Q allele were two, three, and four or greater for 56, 33, and 11% of the other replicates, for which the most frequent common ancestor accounted for only 67% of all Q alleles. These results demonstrate that mutation is not essential for sufficient LD to detect QTL and that QTL can be detected even if substantial heterogeneity exists with regard to ancestral origin of the Q alleles. Abdallah et al. (2003) found that power to detect QTL by single-marker regression was even greater with LE than with LD when using multiallelic markers and similar when using SNPs.
Random drift plays an important role in generating LD in livestock breeding populations, which are typically of limited size (Terwilliger et al. 1998). Because of sampling, drift creates a random pattern of LD around the QTL, without distinct haplotype signatures (Terwilliger et al. 1998). To illustrate the random nature of LD generated by drift, Zhao et al. (2005) evaluated the correlation of LD between markers 2 cM apart with LD of those markers with a central QTL flanked by these markers. After 100 generations of random mating, resulting correlations ranged from 0.06 for Ne = 100 to 0.21 for Ne = 25, indicating nearly complete randomness of LD within a region.
It appears, at least for SNP maps of medium density evaluated here, that haplotype information may not be essential for QTL detection, which is consistent with the more random pattern of LD expected from drift, and that rather simple regression methods can provide sufficient power to detect QTL in data of reasonable size (Long and Langley 1999).
Our study used only consecutive SNPs when multiple SNPs were included in a model. Because of the rather random nature of LD generated by drift, it seems reasonable to fit all possible combinations of SNPs within the chromosome region, which was used by Bonnen et al. (2006). This strategy, however, did not improve power of QTL detection because of more stringent significance thresholds (results not shown).
Impact of population structure on QTL detection:
Although Ne may be ∼100 in current livestock populations, Ne was likely much greater in history (Hayes et al. 2003). To investigate the impact of this on power and precision of QTL detection, a historical population was simulated with Ne = 500 for the first 85 generations and Ne = 100 for the last 15 generations. This population resulted in much lower power and precision of QTL mapping than the population with Ne = 100 for 100 generations (Table 3). Compared to Ne = 100 for 100 generations, the level of LD within 2 cM was much lower when Ne was 500 for the first 85 generations (results not shown), because LD at short distances is more affected by Ne in remote generations than in recent ones (Hayes et al. 2003). However, in the population with Ne = 500 for the first 85 generations, 4-SNP haplotype regression provided higher power and precision than single-marker regression (Table 3).
TABLE 3.
Effect of population structure on power and precision of regression-based LD mapping of QTL
| No. SNPs included in regression methods
|
|||||
|---|---|---|---|---|---|
| Genotype regression
|
Haplotype regression
|
||||
| Population structure | 1 | 2 | 4 | 2 | 4 |
| Power to detect QTL (%) | |||||
| Ne = 100 for 100 generations | 70 | 73 | 74 | 74 | 68 |
| Ne = 500 for 85 and then 100 for 15 generations | 39 | 43 | 45 | 49 | 50 |
| Ns = 30 and Nd = 150 for 100 generations | 64 | 65 | 67 | 67 | 63 |
| Mean absolute error of position (cM) for significant QTL | |||||
| Ne = 100 for 100 generations | 0.88 | 1.00 | 1.32 | 0.97 | 1.30 |
| Ne = 500 for 85 and then 100 for 15 generations | 1.78 | 1.68 | 1.62 | 1.62 | 1.54 |
| Ns = 30 and Nd = 150 for 100 generations | 0.92 | 1.03 | 1.34 | 1.01 | 1.32 |
| Mean absolute error of position (cM) for all QTL | |||||
| Ne =100 for 100 generations | 1.13 | 1.18 | 1.41 | 1.12 | 1.38 |
| Ne = 500 for 85 and then 100 for 15 generations | 2.12 | 1.99 | 1.81 | 1.86 | 1.66 |
| Ns = 30 and Nd = 150 for 100 generations | 1.23 | 1.26 | 1.45 | 1.20 | 1.40 |
Power (detection at 1% regionwise level) and precision for regression-based LD mapping methods are shown under three population structures: (1) Ne =100 for 100 generations, unrelated individuals; (2) Ne = 500 for first 85 generations and Ne = 100 for last 15 generations, unrelated individuals; and (3) 30 sires, each mated to 5 dams (Ns = 30 and Nd = 150) for 100 generations, which provides Ne =100 but related individuals. In the base population, SNPs and a central biallelic QTL were simulated with allele frequency of 0.5 and in linkage equilibrium. The other parameters are sample size in generation 100 = 500, QTL effect = 5% of the phenotypic variance, and marker density = 10 SNPs/11 cM. Results are based on 10,000 replicates.
To evaluate whether power and precision are affected by population structure beyond Ne, a population with related individuals was generated using 30 sires, each mated to 5 dams (i.e., Ns = 30 and Nd = 150) for 100 generations. On the basis of Ne = 4NsNd/(Ns + Nd) (Falconer and Mackay 1996), this provides the same Ne = 100 as the idealized population simulated throughout this article. In the analysis of the population with related individuals, we did not account for genetic relationships because a polygenic effect was not simulated. This population showed slightly lower power and precision than the idealized population where Ne = 100 for 100 generations with unrelated individuals (Table 3). To try to explain this difference, we evaluated the extent of LD in each population by r2 (Hill and Robertson 1968) for a SNP density of 10. The average LD between loci at a given distance was somewhat lower when individuals are related compared to a random sample of unrelated individuals (e.g., 0.35 vs. 0.41 for loci within 0.5 cM and 0.13 vs. 0.15 for loci at 1.5–2 cM). In addition, the LD of the QTL with the best-fitting SNP, which is what drives power to detect the QTL, was on average lower with than without relationships (0.51 vs. 0.57). These differences explain the reduced power with relationships and show that population structure beyond Ne may be important for LD mapping.
Our simulation parameters reflect populations of limited (historical) effective size, in which most LD is generated by drift. This is a situation commonly encountered in domestic animal populations, as well as in some closed human populations and in several closed populations in plants, wildlife, and model organism populations. In human populations that have undergone exponential growth, the effective size is of the order of 10,000 (Eller et al. 2004) and most LD is created by mutation and selective sweeps (Reed and Tishkoff 2006). Thus, results presented herein cannot necessarily be extrapolated to human populations. Human populations that have undergone rapid growth will require much greater marker density and are characterized by different patterns of LD than those generated by drift. Further work is needed to compare alternate methods of LD mapping in such populations.
Conclusions:
With adequate sample size, and levels of LD expected on the basis of limited Ne, most livestock populations lend themselves to QTL detection by LD with SNPs at medium density (1–2/cM). Because of the rather random nature of LD generated by drift and when using marker maps of limited density, use of haplotype information may not increase power to detect QTL. For rapid initial screening, QTL can be detected and mapped by regression on SNP genotypes without recovering haplotypes. In addition to computational speed, regression offers flexibility to include dominance and epistatic effects. To account for relationships, a random polygenic effect should be added.
Acknowledgments
We thank Laura Grapes for her previous work, as well as Long Qu and Dorian Garrick for their valuable advice and discussion. This work was funded by Monsanto and Genus Plc.
References
- Abdallah, J. M., B. Goffinet, C. Cierco-Ayrolles and M. Pérez-Enciso, 2003. Linkage disequilibrium fine mapping of quantitative trait loci: a simulation study. Genet. Sel. Evol. 35: 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnen, P. E., I. Pe'er, R. M. Plenge, J. Salit, J. K. Lowe et al., 2006. Evaluating potential for whole-genome studies in Kosrae, an isolated population in Micronesia. Nat. Genet. 38: 214–217. [DOI] [PubMed] [Google Scholar]
- Dekkers, J. C. M., and F. Hospital, 2002. The use of molecular genetics in the improvement of agricultural populations. Nat. Rev. Genet. 3: 22–32. [DOI] [PubMed] [Google Scholar]
- Eller, E., J. Hawks and J. H. Relethford, 2004. Local extinction and recolonization, species effective population size, and modern human origins. Hum. Biol. 76: 689–709. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Addison-Wesley Longman, Harlow, UK.
- Farnir, F., W. Coppieters, J.-J. Arranz, P. Berzi, N. Cambisano et al., 2000. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 10: 220–227. [DOI] [PubMed] [Google Scholar]
- Farnir, F., B. Grisart, W. Coppieters, J. Riquet, P. Berzi et al., 2002. Simultaneous mining of linkage and linkage disequilibrium to fine map quantitative trait loci in outbred half-sib pedigrees: revisiting the location of a quantitative trait locus with major effect on milk production on bovine chromosome 14. Genetics 161: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapes, L., J. C. M. Dekkers, M. F. Rothschild and R. L. Fernando, 2004. Comparing linkage disequilibrium-based methods for fine mapping quantitative trait loci. Genetics 166: 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapes, L., M. Z. Firat, J. C. M. Dekkers, M. F. Rothschild and R. L. Fernando, 2006. Optimal haplotype structure for linkage disequilibrium-based fine mapping of quantitative trait loci using identity by descent. Genetics 172: 1955–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmegnies, N., F. Farnir, F. Davin, N. Buys, M. Georges et al., 2006. Measuring the extent of linkage disequilibrium in commercial pig populations. Anim. Genet. 37: 225–231. [DOI] [PubMed] [Google Scholar]
- Hayes, B. J., P. M. Visscher, H. C. Mcpartlan and M. E. Goddard, 2003. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 13: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Lee, S. H., and J. H. J. Van Der Werf, 2005. The role of pedigree information in combined linkage disequilibrium and linkage mapping of quantitative trait loci in a general complex pedigree. Genetics 169: 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., and C. H. Langley, 1999. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 9: 720–731. [PMC free article] [PubMed] [Google Scholar]
- Mcrae, A. F., J. C. Mcewan, K. G. Dodds, T. Wilson, A. M. Crawford et al., 2002. Linkage disequilibrium in domestic sheep. Genetics 160: 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2000. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 155: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2001. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 33: 605–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., A. Karlsen, S. Lien, I. Olsaker and M. E. Goddard, 2002. Fine mapping of a quantitative trait locus for twinning rate using combined linkage and linkage disequilibrium mapping. Genetics 161: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, K., and S. P. Smith, 1996. Restricted maximum likelihood estimation for animal models using derivatives of the likelihood. Genet. Sel. Evol. 28: 23–49. [Google Scholar]
- Morris, A. P., J. C. Whittaker and D. J. Balding, 2004. Little loss of information due to unknown phase for fine-scale linkage-disequilibrium mapping with single-nucleotide-polymorphism genotype data. Am. J. Hum. Genet. 74: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, D. M., M. G. Ehm, D. V. Zaykin and B. S. Weir, 2004. Effect of two- and three-locus linkage disequilibrium on the power to detect marker/phenotype associations. Genetics 168: 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nsengimana, J., P. Baret, C. S. Haley and P. M. Visscher, 2004. Linkage disequilibrium in the domesticated pig. Genetics 166: 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen, L., 2000. Positional cloning of disease genes: advantages of genetic isolates. Hum. Hered. 50: 66–75. [DOI] [PubMed] [Google Scholar]
- Reed, F. A., and S. A. Tishkoff, 2006. Positive selection can create false hotspots of recombination. Genetics 172: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed, T., 2003. Statistical Analysis of Gene Expression Microarray Data. Chapman & Hall/CRC Press, London/New York/Cleveland/Boca Raton, FL.
- Sved, J. A., 1971. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2: 125–141. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., S. A. Knott, D. Ward, D. Smith, J. L. Williams et al., 2003. Estimation of linkage disequilibrium in a sample of the United Kingdom dairy cattle population using unphased genotypes. J. Anim. Sci. 81: 617–623. [DOI] [PubMed] [Google Scholar]
- Terwilliger, J. D., S. Zöllner, M. Laan and S. Pääbo, 1998. Mapping genes through the use of linkage disequilibrium generated by genetic drift: ‘drift mapping’ in small populations with no demographic expansion. Hum. Hered. 48: 138–154. [DOI] [PubMed] [Google Scholar]
- Tsukanov, I., and M. Hall, 2003. Data structure and algorithms for fast automatic differentiation. Int. J. Numer. Methods Eng. 56: 1949–1972. [Google Scholar]
- Zhao, H., D. Nettleton, M. Soller and J. C. M. Dekkers, 2005. Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet. Res. 86: 77–87. [DOI] [PubMed] [Google Scholar]
- Zöllner, S., and J. K. Pritchard, 2005. Coalescent-based association mapping and fine mapping of complex trait loci. Genetics 169: 1071–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]