

Abstract
Phenotypic discordance between monozygotic twins, such as a difference in disease susceptibility, implicates the role of the environment in determining phenotype. To assess genomewide environmental effects on “gene expression phenotype,” we employed a published microarray data set for twins. We found that variations in expression phenotypes between monozygotic twins have biases in their chromosomal locations. They also showed a strong inverse correlation with gene density. Genomic regions of low gene density were environmentally sensitive, containing genes involved in response to external signals, cell differentiation, and development, etc. Genetic factors were found to make no contribution to the observed regional biases, stressing the role of epigenetics. We propose that epigenetic modifications might occur more frequently in heterochromatic, gene-poor regions in response to environmental signals while gene-rich regions tend to remain in an active chromatin configuration for the constitutive expression of underlying genes.
GENETICALLY identical individuals display a remarkable variation in phenotype. Differing histories of environmental exposure produce the phenotypic discordance. Among examples are the different fingerprint patterns of identical twins and different coat patterns and personalities between cloned animals (Jain et al. 2002; Shin et al. 2002). The expression level of a gene, as a quantitative trait, can be considered a phenotype (Cheung and Spielman 2002). With the development of microarrays, genetic analysis of “expression phenotype” has recently attracted attention (Oleksiak et al. 2002; Cheung et al. 2003; Whitney et al. 2003). Although a fraction of expression phenotypes were successfully linked to particular genetic markers, genetic linkage or association was not found for many of them (Berm et al. 2002; Morley et al. 2004; Cheung et al. 2005). The observed expression variation, which has been accepted as genetic variation, may contain a substantial amount of environmental diversity. Therefore, there exists a great need to characterize the contribution of environmental factors to natural variation in gene expression.
Epigenetics serves as a link between the environment and gene expression. Epigenetics refers to a set of reversible heritable changes that occur without a change in DNA sequence. The best-known epigenetic signal is DNA methylation in CpG islands, which is generally associated with silencing of gene expression. Chromatin remodeling is another important epigenetic mechanism. Chemical modification of histone tails can alter chromatin structure, which in turn influences the activity of adjacent genes. For example, the transcription of genes with acetylated histones is usually switched on. These changes may be induced spontaneously or in response to environmental factors. Increasing evidence suggests that “epigenetic modifications can be a molecular substrate for the impact of the endogenous and exogenous environment” (Petronis 2006, p. 347). Indeed, Fraga et al. (2005) showed that monozygotic (MZ) twins have differential epigenetic patterns. Furthermore, the differences were greater in older twins, underscoring the responsiveness of epigenetics to environmental exposures. Of particular importance, epigenetics is also associated with MZ twin discordance for common diseases (Bertelsen et al. 1977; Poulsen et al. 1999; Bjornsson et al. 2004). These changes in epigenetic state lead to changes in the access of transcriptional machinery to the underlying genetic code. As such, epigenetic mechanisms can allow an organism to respond to the environment through gene expression changes.
We aimed to gain a genomewide insight into the environmental or epigenetic variation in expression phenotype. The global expression profiles of blood leukocytes from MZ twins were employed to this end (Sharma et al. 2005). We first characterized environmentally variable or invariable genes from a functional perspective and then attempted to associate the variations with epigenetic mechanisms. In doing so, we also assessed variations among genetically unrelated individuals to compare the effect of genetic factors with that of environmental factors.
MATERIALS AND METHODS
Measuring expression variations:
The microarray data produced by Sharma et al. (2005) were downloaded from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) with the following accession numbers: GSM14480, GSM14481, GSM29053, GSM29054, GSM29055, and GSM29056. These data correspond to three female MZ twin pairs (F1:F2, F5:F6, and F7:F8, following the notation by the original authors). They belonged to a similar age group (20–23 years), lived close to each other, and had similar nutrition habits and professions. Blood leukocytes were obtained at the same time of day. Expression variation was calculated for each gene by a log ratio between the expression values. Within-pair variation was given by averaging the three ratios for the three pairs: 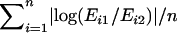 , where
, where 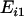 and
and 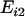 indicate the expression values for the two individuals of ith twin pair. To assess between-pair variation, we first calculated the mean expression of each pair as
indicate the expression values for the two individuals of ith twin pair. To assess between-pair variation, we first calculated the mean expression of each pair as 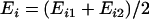 for
for 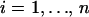 . All possible comparisons were made between the pairs and the average of the comparisons was calculated:
. All possible comparisons were made between the pairs and the average of the comparisons was calculated:  for
for 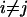 . To estimate experimental variation, we used the data for three replicates of the same individual (GSM14477, GSM14482, and GSM14485).
. To estimate experimental variation, we used the data for three replicates of the same individual (GSM14477, GSM14482, and GSM14485).
Identification of housekeeping genes:
We measured expression breadth of a gene as the number of tissues where the gene is expressed. Whether or not a gene is expressed in a tissue was determined as previously described (Su et al. 2004). A gene was identified as a housekeeping gene if expressed in all 79 tissues analyzed. Of 9233 genes, 1605 were identified as housekeeping genes.
Functional category analysis:
For each gene ontology (GO) category with >50 genes, we computed a normalized expression variation (z-score) from the variations of all members of that category. The z-score is defined as 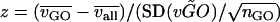 , where
, where 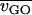 is the average variation for the genes in the GO category,
is the average variation for the genes in the GO category, 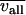 is the average variation for all genes in any GO category,
is the average variation for all genes in any GO category, 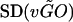 is the standard deviation for the genes in the category, and
is the standard deviation for the genes in the category, and 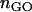 is the number of the genes in the category. The z-scores for within-pair variation and for between-pair variation are shown in supplemental Table 1 at http://www.genetics.org/supplemental/.
is the number of the genes in the category. The z-scores for within-pair variation and for between-pair variation are shown in supplemental Table 1 at http://www.genetics.org/supplemental/.
Positional effects on gene expression:
Each transcript was mapped to the human genome. Expression variation correlation between neighboring genes was calculated. The significance of the correlation was estimated by permuting gene order on the genome. The permutation was repeated 10,000 times. Gene density was defined as the inverse of the average distance between neighboring genes in a window of 40 adjacent transcripts (Caron et al. 2001). The correlation between gene density and the average expression variation of 40 adjacent transcripts was computed and statistically assessed by the permutation test as described above.
RESULTS
Housekeeping genes are impervious to environmental changes:
Environmental variation was measured by expression differences within MZ twin pairs (within-pair variation). Expression differences between MZ twin pairs (between-pair variation) were deemed to contain both genetic and environmental variation. We refer to them as total variation. T-tests demonstrate significant differences between environmental and total variation (P < 2 × 10−6). Figure 1 shows the scatterplot of environmental vs. total variation. Most of the genes show a substantially higher level of between-pair variation, indicating large effects of genetic factors on gene expression. However, environmental variation is not negligible. To assess pure genetic variation, environmental components should be taken into account and distinguished from genetic components.
Figure 1.—

Comparison of within-pair variation and between-pair variation. Within-pair variation is expected to represent environmental variation while between-pair variation may contain genetic factors as well as environmental influences. The higher degree of between-pair variation indicates the contribution of genetic factors to creating expression variation.
Sharma et al. (2005) showed that housekeeping genes had a low expression variation within twin pairs. We confirmed their result: Spearman's rank correlation between expression breadth and environmental variation was significant (R = −0.242). We identified 1605 housekeeping genes of 9233 genes whose tissue profile was available, in a different way from that of Sharma et al. (2005). However, we reached the same conclusion that housekeeping genes exhibit markedly low environmental variations. It may be that the different histories of environmental exposure between individuals play only a minor role in expression polymorphisms of housekeeping genes. These results are not surprising considering their essential functions for cellular maintenance. Random changes in expression level as well as in amino acid sequence should be minimized. On the other hand, between-pair variation did not negatively correlate with expression breadth (R = 0.088). Thus, it is likely that housekeeping genes may be subject to genetic effects while remaining resistant to environmental stimuli.
Functional classification of the genes sensitive to environmental stimuli:
To gain insights into functional aspects of environmentally susceptible expression phenotypes, we grouped genes into GO categories. The average environmental variation for each category was calculated and normalized as a z-score. The z-score indicates the deviation of the average variation of the genes in that category from the average variation of all genes in any category. Therefore, a high z-score for a category means that the genes in that category rank high in expression variations. The z-scores for total variation and for environmental variation are shown in supplemental Table 1 at http://www.genetics.org/supplemental/. The list of the GO terms with the environmental z-score >2.0 having >100 genes is given in Table 1. The list is remarkably enriched for genes involved in response to external signals (shown in boldface type), cell differentiation and development (in italics), cell division and proliferation (underlined), and cellular transport (double underlined). A common feature of them is responsiveness to environmental signals or conditions. Therefore, their expression phenotype may reflect the history or current state of the individual's adaptations to the environment.
TABLE 1.
Functional classification of genes with high environmental variation
| GO category | No. of genes | Between-pair variation | Within-pair variation |
|---|---|---|---|
| Extracellular region | 440 | 1.896 | 5.940 |
| Extracellular space | 463 | 5.131 | 5.419 |
| Muscle development | 110 | 3.027 | 4.659 |
| Ion transport | 319 | 0.562 | 3.871 |
| Hormone activity | 101 | 4.541 | 3.730 |
| Transport | 408 | 1.383 | 3.522 |
| Structural molecule activity | 292 | 1.507 | 3.447 |
| Muscle contraction | 116 | 2.180 | 3.349 |
| Lipid metabolism | 245 | 0.119 | 3.213 |
| Cell division | 141 | −0.945 | 3.108 |
| Cell differentiation | 300 | 1.966 | 3.094 |
| Ion channel activity | 112 | 0.449 | 3.091 |
| Development | 461 | 1.489 | 2.832 |
| Proteolysis | 463 | 2.960 | 2.691 |
| Extracellular matrix (sensu Metazoa) | 170 | 2.432 | 2.549 |
| DNA replication | 127 | −1.991 | 2.539 |
| Cell cycle | 454 | −3.949 | 2.476 |
| Nervous system development | 289 | 1.024 | 2.455 |
| Potassium ion transport | 154 | 1.611 | 2.453 |
| Sensory perception | 181 | 3.149 | 2.432 |
| Inflammatory response | 180 | −0.791 | 2.424 |
| Cytoskeleton | 390 | 2.551 | 2.383 |
| RNA polymerase II transcription factor activity | 152 | −1.026 | 2.307 |
| Iron ion binding | 215 | 0.815 | 2.296 |
| Sensory perception of sound | 120 | 0.697 | 2.295 |
| Cell–cell signaling | 317 | 3.636 | 2.260 |
| Visual perception | 161 | 4.177 | 2.163 |
| Positive regulation of cell proliferation | 139 | 0.177 | 2.154 |
| Regulation of transcription from RNA polymerase II promoter | 205 | −3.425 | 2.013 |
Listed are the GO categories with z > 2.0 (for environmental variation) having >100 genes, sorted by the environmental z-scores. The full list can be found in supplemental Table 1 at http://www.genetics.org/supplemental/. The GO categories were further grouped into four higher categories as follows: response to external signals (boldface type), cell differentiation and development (italics), cell division and proliferation (underline), and cellular transport (double underline).
Of great importance are genes involved in cell differentiation and development. Phenotypic discordance between MZ twins can be best explained in terms of differences in developmental processes. Here we show that the expression polymorphisms of developmental genes, which can contribute most to phenotypic discordance, are highly affected by environmental differences. Functions associated with response to external signals are also enriched in the list. Examples include genes located in the extracellular region and those involved in hormone activity, sensory perception, etc. The regulation of cell proliferation may also be affected by environmental conditions, growth signals, etc. In a dramatic contrast, apoptosis seems to rely on the intracellular state independently of the extracellular environment (z = −6.471 for apoptosis, z = −5.998 for anti-apoptosis, and z = −4.729 for regulation of apoptosis). We also found that RNA polymerase II transcription factors show significantly high environmental variation (Table 1). On the contrary, translation initiation factors have markedly low environmental variation (z = −3.298, see supplemental Table 1 at http://www.genetics.org/supplemental/). These findings imply that transcription, rather than translation, may be responsive to environmental signals. Some categories, especially cell cycle- or polymerase-related ones, show a low between-pair variation (Table 1). Expression polymorphisms of these genes seem attributable only to environmental factors, independently of genetic factors.
Environmental effects have regional biases across the genome:
There is increasing evidence that neighboring genes show similar expression patterns in species as diverse as humans (Lercher et al. 2002), flies (Spellman and Rubin 2002), worms (Roy et al. 2002), and yeast (Cohen et al. 2000). The role of higher-order chromatin structure has been suggested in respect to these patterns. Here we first asked whether neighboring genes maintain similar levels of environmental changes in expression. Spearman's rank correlation of environmental variations between neighboring genes on the real genome was highly significant as compared to that on randomly permuted genomes (Figure 2A). These findings imply that environmental effects have regional biases in the genome, creating hot and cold regions with housekeeping genes probably located in the cold ones. More interestingly, the hot domains tend to be rich in genes while the cold domains are poor in genes: we observed a strikingly high correlation between gene density and environmental variation (Figure 2B). This suggests that the expression of dispersed genes may be prone to environmental stimuli while that of clustered genes may be resistant.
Figure 2.—

Chromosomal location effects on expression variation. The arrows indicate the correlation computed on the real chromosome and the histogram shows the distribution of the correlations computed on permuted chromosomes. The long arrows indicate environmental and total variation. The short arrow indicates experimental variation that was obtained from three replicates of the same individual. The permuted chromosomes have randomized gene order. (A) Correlation of expression variation between neighboring genes on the chromosome. The left long arrow is for environmental variation (R = 0.106) and the right one is for total variation (R = 0.125). (B) Correlation between gene density and expression variation. Gene density was defined as the inverse average distance between neighboring genes in a window of 40 adjacent transcripts. The average expression variation was calculated in the same window. The left long arrow is for total variation (R = −0.506) and the right one is for environmental variation (R = −0.498).
Experimental variation was calculated from three replicates of the same individual. We observed marginal correlations as indicated by the short arrows in Figure 2. Considering that high expression levels tend to produce low variations, the correlations are possibly due to the clustering of highly expressed genes (Caron et al. 2001). However, as shown in Figure 3, this effect seems only marginal. Moreover, Lercher et al. (2002) showed that the apparent clustering of highly expressed genes is an artifact of the clustering of housekeeping genes.
Figure 3.—

Whole-chromosome view of gene density, inverse environmental variation, inverse total variation, and open chromatin signature. Gene density is shown on a log scale. Open chromatin signature was obtained by the hybridization of the open chromatin fraction from sucrose gradient sedimentation onto a genomic DNA microarray (Gilbert et al. 2004). The array was assembled from clones spaced at ∼1-Mb intervals. Mean log 2 ratios of open to control hybridization signal for four replicate experiments were mapped to corresponding chromosomal positions. The similar pattern between environmental and total variation indicates that chromosomal distribution of expression variation should be due to environmental, not genetic, effects. Shown is a view for chromosome 7. The other chromosomes are shown in supplemental Figure 1 at http://www.genetics.org/supplemental/.
Housekeeping genes exhibit low environmental variation. Does the clustering of housekeeping genes explain the biased distribution of environmental variation? Lercher et al. (2002) were not able to find chromosomal dispersion of tissue-specific genes. A significant correlation was not found between expression breadth (b) and gene density (d), Rbd = 0.042. From our data, the correlation of expression breadth (b) and environmental variation (v) was found to be Rbv = −0.242. Therefore, we cannot expect Rbd and Rbv to produce the correlation of gene density (d) and environmental variation (v), Rdv = −0.498. Taken together, the clustering and dispersion of environmentally invariable and variable genes cannot be explained by expression level effects or expression breadth effects.
Aside from environmental effects, gene expression is greatly affected by genetic factors (as shown in Figure 1). Surprisingly, however, we observed that the correlations for total variation were almost the same as or only marginally higher than those for environmental variation (R = 0.125 and 0.106 for the correlations of neighboring genes; R = −0.506 and −0.498 for the correlations with gene density, shown in Figure 2, A and B). This suggests that environmental factors alone should create the biased distribution of expression variations on the chromosomes. Genetic polymorphisms seem to make little or no contribution to the regional biases. On the assumption that the distribution of genetic polymorphisms over the genome has a similar pattern to that of somatic mutations, we can also rule out the effect of somatic mutations. If this is the case, environmental factors may act mainly through epigenetic mechanisms. Aside from this speculation, the accumulation of genetic mutations is expected to occur at a much slower rate than that of epigenetic mutations (Fraga et al. 2005). First, the consequences of genetic mutations are probably more dramatic in the survival of cells. Second, cells have correction mechanisms for genetic mutations, but not for epigenetic defects. Indeed, epigenetic mutations in aging mouse tissues were shown to be two orders of magnitude greater than genetic mutations (Bennett-Baker et al. 2003). Therefore, we can safely rule out genetic effects (genetic polymorphisms) and environment–gene interactions (genetic mutations), leaving environmental effects (epigenetic mutations) alone.
Potential role of epigenetics in regional biases of environmental variation:
How is epigenetics associated with the regional biases in expression variation? A study by Gilbert et al. (2004) provides a clue for this issue. They identified open chromatin fiber structures by sucrose sedimentation and hybridization to genomic microarrays. It was shown that open chromatin structures correlated with regions of high gene density: regions that were rich in genes tended to be in open chromatin structures, whereas regions that were poor in genes tended to be in close or compact chromatin. In addition, their results show that it is not simply expression level that determines or is determined by chromatin structure. They suggest that open chromatin indicates transcriptional constancy rather than transcriptional activity, supporting the clustering of environmentally robust (or housekeeping) genes but not that of highly expressed genes (Lercher et al. 2002).
We also made use of the data of Kim et al. (2005). They detected active promoters using antibodies that recognize the RNA polymerase II (RNAP), transcription factor IID (TFIID), dimethylated lysine 4 on histone H3 (MeH3K4), and acetylated histone H3 (AcH3). Interestingly, we found that despite tight correlations between these four signals, chromatin modification signals (MeH3K4 and AcH3) showed significant correlations (R = 0.295 and 0.149, respectively) with gene density while transcriptional activity signals (RNAP and TFIID) showed relatively low correlations with gene density (R = 0.083 and 0.075, respectively). Again, gene distribution seems to correlate better with chromatin modifications than with expression level.
Gene density, expression variations, and open chromatin signature are plotted together for each chromosome (supplemental Figure 1 at http://www.genetics.org/supplemental/). As an example, chromosome 7 is shown in Figure 3. Again, the similar pattern between environmental and total variation indicates that the chromosomal distribution of expression variation should result from epigenetic, not genetic, factors. These findings hint that the effect of gene distribution on expression variation might be attributable to epigenetics such as chromatin structure. Chromatin structure of high-density regions may constitutively remain in an active configuration to ensure the constitutive activation of housekeeping genes. By contrast, epigenetic states of low-density regions may undergo sporadic changes in response to internal or external signals. The consequent alteration in the expression of the underlying genes may lead to variations in phenotype, which are subject to selection under environmental pressure. Selected epigenetic phenotypes of progenitor cells can be inherited through many cell divisions to give rise to expression variations between cell populations from individuals.
DISCUSSION
Our study demonstrated chromosomal location effects on variations in gene expression. Surprisingly, environmental components were able to account for most of the positional variation in gene expression changes. Genetic variation, although responsible for much expression variation, does not seem to contribute to the regional biases in it. These findings support the key role of epigenetics, adding to a growing body of evidence that it mediates the translation of environmental signals into biochemical changes. Importantly, we proposed that epigenetic changes tend to occur in gene-poor regions, which are mostly maintained in an inactive chromatin state.
The extension of primary sequence organization to a three-dimensional (3D) chromosomal structure may provide another clue for understanding the effect of gene distribution on environmental expression variation. At the chromosomal level, most gene-dense regions of the human genome are preferentially located in the nuclear interior with gene-poor regions located progressively toward the nuclear periphery (Boyle et al. 2001). This is in accordance with the most widely known feature of genomic 3D organization, namely the differential enrichment of euchromatin and heterochromatin in the nuclear interior and periphery, respectively. These patterns tend to be recapitulated by local chromosomal regions, where gene deserts are located more peripherally while gene clusters aggregate and locate themselves more centrally (Shopland et al. 2006). Gene expression was also shown to correspond with these patterns (Lukasova et al. 2002). The effects of chemical, electrical, and mechanical signals from the outside of the nucleus may be reduced progressively toward the nuclear interior. In this regard, it is tempting to postulate that chromatin modifications and subsequent gene expression changes may frequently occur in exposed gene-poor regions rather than in interior gene-rich regions.
Epigenetic variation can be also generated by stochastic events during development and aging (Barbot et al. 2002; Bennett-Baker et al. 2003). Stochastic variations can possibly occur without any specific environmental effect (Petronis 2006). Since our study focused on the MZ twins who usually lived in the same macroenvironment, some of the observed expression changes may be attributable to stochastic alterations in epigenetic patterns (Richards 2006). Developmental noise is an example of the stochastic variations in epigenetics. We found high levels of expression variation in genes involved in cellular differentiation (Table 1). In embryonic stem cells, chromatin is globally in an open, decondensed state. As differentiation progresses, cells accumulate condensed heterochromatin structures with an increase in silencing histone marks (Arney and Fisher 2004; Meshorer and Misteli 2006). We speculate that stochasticity in this process may create variations in heterochromatin distribution between individuals. As such, developmental noise in epigenetics may also explain expression variation mostly observed in gene-poor regions.
Although we are convinced of the role of epigenetics, further experimental evidence should be pursued. First of all, the correlation of gene distribution pattern and epigenetic variation should be confirmed by experiments. There is a need for a global high-density measure of epigenetic differences between MZ twins. Also, it should be determined if they are coupled with gene expression divergences. Further, it would be of great interest to see if epigenetic variations are higher in chromatin regions located toward the nuclear periphery rather than in the interior. Interestingly, Fraga et al. (2005) showed that most epigenetic changes between monozygotic twins are found at repeated and heterochromatic DNA regions, which tend to be located near the nuclear periphery. High environmental variations observed in gene-poor regions may be linked to the frequent epigenetic changes of heterochromatic regions that may be close to environmental signals from the outside of the nucleus. Another issue to be dealt with is the characterization of pure stochastic variation that is independent of environmental stimuli.
Acknowledgments
We thank Jong Bhak (Korean BioInformation Center) and Sangsoo Kim (Soongsil University) for their valuable comments and suggestions. We are grateful to the original authors of the microarray data used in this analysis. This work was supported by the Korean Ministry of Science and Technology under grant no. M10407010001-04N0701-00110.
References
- Arney, K. L., and A. G. Fisher, 2004. Epigenetic aspects of differentiation. J. Cell Sci. 117: 4355–4363. [DOI] [PubMed] [Google Scholar]
- Barbot, W., A. Dupressoir, V. Lazar and T. Heidmann, 2002. Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res. 30: 2365–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Baker, P. E., J. Wilkowski and D. T. Burke, 2003. Age-associated activation of epigenetically repressed genes in the mouse. Genetics 165: 2055–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berm, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Bertelsen, A., B. Harvald and M. Hauge, 1977. A Danish twin study of manic-depressive disorders. Br. J. Psychiatry 130: 330–351. [DOI] [PubMed] [Google Scholar]
- Bjornsson, H. T., M. D. Fallin and A. P. Feinberg, 2004. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 20: 350–358. [DOI] [PubMed] [Google Scholar]
- Boyle, S., S. Gilchrist, J. M. Bridger, N. L. Mahy, J. A. Ellis et al., 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10: 211–219. [DOI] [PubMed] [Google Scholar]
- Caron, H., B. van Schaik, M. van der Mee, F. Baas, G. Riggins et al., 2001. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science 291: 1289–1292. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., and R. S. Spielman, 2002. The genetics of variation in gene expression. Nat. Genet. 32: 522–525. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., L. K. Conlin, T. M. Weber, M. Arcaro, K. Y. Jen et al., 2003. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat. Genet. 33: 422–425. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., R. S. Spielman, K. G. Ewens, T. M. Weber, M. Morley et al., 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437: 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, B. A., R. D. Mitra, J. D. Hughes and G. M. Church, 2000. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 26: 183–186. [DOI] [PubMed] [Google Scholar]
- Fraga, M. F., E. Ballestar, M. F. Paz, S. Ropero, F. Setien et al., 2005. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102: 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, N., S. Boyles, H. Fiegler, K. Woodfine, N. P. Carter et al., 2004. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118: 555–566. [DOI] [PubMed] [Google Scholar]
- Jain, A. K., S. Prabhakar and S. Pankanti, 2002. On the similarity of identical twin fingerprints. Pattern Recogn. 35: 2653–2663. [Google Scholar]
- Kim, T. H., L. O. Barrera, M. Zheng, C. Qu, M. A. Singer et al., 2005. A high-resolution map of active promoters in the human genome. Nature 436: 876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher, M. J., A. O. Urrutia and L. D. Hurst, 2002. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 31: 180–183. [DOI] [PubMed] [Google Scholar]
- Lukasova, E., S. Kozubek, M. Kozubek, M. Falk and J. Amrichova, 2002. The 3D structure of human chromosomes in cell nuclei. Chromosome Res. 10: 535–548. [DOI] [PubMed] [Google Scholar]
- Meshorer, E., and T. Misteli, 2006. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 7: 540–546. [DOI] [PubMed] [Google Scholar]
- Morley, M., C. M. Molony, T. M. Weber, J. L. Devlin, K. G. Ewens et al., 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430: 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak, M. F., G. A. Churchill and D. L. Crawford, 2002. Variation in gene expression within and among natural populations. Nat. Genet. 32: 261–266. [DOI] [PubMed] [Google Scholar]
- Petronis, A., 2006. Epigenetics and twins: three variations on the theme. Trends Genet. 22: 347–350. [DOI] [PubMed] [Google Scholar]
- Poulsen, P., K. O. Kyvik, A. Vaag and H. Beck-Nielsen, 1999. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—a population-based twin study. Diabetologia 42: 139–145. [DOI] [PubMed] [Google Scholar]
- Richards, E. J., 2006. Inherited epigenetic variation- revisiting soft inheritance. Nat. Rev. Genet. 7: 395–401. [DOI] [PubMed] [Google Scholar]
- Roy, P. J., J. M. Stuart, J. Lund and S. K. Kim, 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975–979. [DOI] [PubMed] [Google Scholar]
- Sharma, A., V. K. Sharma, S. Horn-Saban, D. Lancet, S. Ramachandran et al., 2005. Assessing natural variations in gene expression in humans by comparing with monozygotic twins using microarrays. Physiol. Genomics 21: 117–123. [DOI] [PubMed] [Google Scholar]
- Shin, T., D. Kraemer, J. Pryor, L. Liu, J. Rugila et al., 2002. Cell biology: a cat cloned by nuclear transplantation. Nature 415: 859. [DOI] [PubMed] [Google Scholar]
- Shopland, L. S., C. R. Lynch, K. A. Peterson, K. Thornton, N. Kepper et al., 2006. Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J. Cell Biol. 174: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman, P. T., and G. M. Rubin, 2002. Evidence for large domains of similarly expressed genes in the Drosophila genome. J. Biol. 1: 5.1–5.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, A. I., M. P. Cooke, K. A. Ching, Y. Hakak, J. R. Walker et al., 2004. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA 99: 4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney, A. R., M. Diehn, S. J. Popper, A. A. Alizadeh, J. C. Boldrick et al., 2003. Individuality and variation in gene expression patterns in human blood. Proc. Natl. Acad. Sci. USA 100: 1896–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]