

Abstract
In this article, we develop an evolutionary model for protein sequence evolution. Gene pleiotropy is characterized by K distinct but correlated components (molecular phenotypes) that affect the organismal fitness. These K molecular phenotypes are under stabilizing selection with microadaptation (SM) due to random optima shifts, the SM model. Random coding mutations generate a correlated distribution of K molecular phenotypes. Under this SM model, we further develop a statistical method to estimate the “effective” number of molecular phenotypes (Ke) of the gene. Therefore, for the first time we can empirically evaluate gene pleiotropy from the protein sequence analysis. Case studies of vertebrate proteins indicate that Ke is typically ∼6–9. We demonstrate that the newly developed SM model of protein evolution may provide a basis for exploring genomic evolution and correlations.
ALTHOUGH the wide availability of high-throughput data has greatly facilitated the study of evolutionary genomics, the foundation of molecular evolution has been relatively “static” (Kimura 1983). Basically, the evolutionary fate of a mutant is largely determined by the prespecified fitness value (the coefficient of selection) and the effective population size. Although this classical treatment has successfully removed the complexity of genotype–phenotype association, problems may appear when the association itself has been one of the research highlights. The recent debate about genomic correlations among evolutionary rate, interaction, expression, and dispensability has indicated the limitation of current evolutionary models (reviewed in Pal et al. 2006). Alternatively, researchers have to use the partial-correlation or principle-component methods to analyze major determinants governing protein evolution (e.g., Drummond et al. 2005; Wall et al. 2005; Wolf et al. 2006), because these approaches are not model dependent.
It is desirable to develop a novel framework that can provide an integrated view of protein evolution, but an unsolved key problem is how to deal with the sophisticated relationship between protein sequence and organismal fitness. We recognized that the substantial genotype–phenotype literature can be used to address this issue (e.g., Fisher 1930; Lande 1980; G. Wagner 1989; Waxman and Peck 1998; A. Wagner 2000; Poon and Otto 2000; Zhang and Hill 2003). As mutations are raw materials for evolution, the distribution of effects of mutations, f(s), has been a central issue in the genotype–phenotype study (Lynch et al. 1999; Bataillon 2000; Shaw et al. 2002). Under Fisher's geometric model (e.g., Fisher 1930; Hartl and Taubes 1998; Poon and Otto 2000) or multivariate model (e.g., Lande 1980; Welch and Waxman 2003; Martin and Lenormand 2006), f(s) can be predicted on the basis of selective and mutational assumptions. The dimensionality in each framework has been interpreted as the number of loci affecting a quantitative trait (reviewed in Kingsolver et al. 2001), or the number of affected phenotypic traits of a mutation, estimated from mutational-accumulation experiments (reviewed in Elena and Lenski 2003). On the other hand, an ad hoc gamma distribution for f(s) was introduced in the early days of the neutral theory of molecular evolution (Ohta 1973, 1977; Kimura 1983), which has been found useful to estimate the proportion of neutral or deleterious mutations (e.g., Nielsen and Yang 2003; Piganeau and Eyre-Walker 2003; Eyre-Walker et al. 2006).
Here we extend our recent work (Gu 2006). The principle behind our present work in modeling protein sequence evolution is as follows: First, multifunctionality of a gene (pleiotropy) is characterized by K distinct components in the fitness (called molecular phenotypes). Second, these K molecular phenotypes are under stabilizing selection (Fisher 1930; Waxman and Peck 1998) with microadaptations due to random shifts of the fitness optimum (Gillespie 1991). Third, random mutations in the coding region of the gene generate a mutational distribution for K molecular phenotypes. Correlated structures among K molecular phenotypes are taken into account at both selectional and mutational levels. More importantly, we develop a statistical method to estimate the “effective” number of molecular phenotypes (Ke) of the gene. Case studies of vertebrate proteins demonstrate that for the first time we can empirically evaluate gene pleiotropy only from phylogenetic sequence analysis.
THE MODEL
Molecular phenotypes and gene pleiotropy:
Pleiotropy of a gene (protein) function can be modeled by multiple (K) molecular phenotypes, denoted by (y1, …, yK). Each molecular phenotype yi represents a nontrivial component of genetic variation, contrasting with organismal fitness as a result of a specific (yet unknown) biological process (Figure 1). At one extreme, molecular phenotypes could correspond to subcomponents of protein function, regardless of the biological processes. At another extreme, molecular phenotypes could be determined mainly by distinct physiological processes. Since these underlying biological processes are usually intractable, the concept of molecular phenotypes based on fitness effects avoids this difficulty.
Figure 1.—

Schematic presentation for the concept of molecular phenotypes that affect the fitness of an organism.
Following the widely used procedure (e.g., Lande 1980; Turelli 1985; Waxman and Peck 1998), we assume that the fitness function of molecular phenotypes y = (y1, …, yK)′ is Gaussian-like; i.e.,
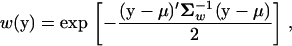 |
(1) |
where μ is the optimum of the fitness function, and Σw is a (positive definite) symmetric matrix characterizing correlated stabilizing selection on K molecular phenotypes. The ith diagonal element 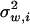 measures the strength of stabilizing selection on the ith molecular phenotype, while the ijth nondiagonal element σw,ij measures the correlated stabilizing selection on yi and yj.
measures the strength of stabilizing selection on the ith molecular phenotype, while the ijth nondiagonal element σw,ij measures the correlated stabilizing selection on yi and yj.
Let y0 be the population mean of molecular phenotypes of a gene, which may not necessarily be at the optimum (μ). By definition (Fisher 1930), the coefficient of selection for the molecular phenotype y is given by
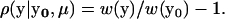 |
(2) |
Classical stabilizing selection (Turelli 1985; Waxman and Peck 1998) assumes that the population mean is always at the fitness optimum, i.e., y0 = μ, leading to purifying selection; that is, ρ(y | y0 = μ) ≤ 0 always holds.
Next, we consider the effects of mutations on molecular phenotypes. A random mutation in the coding region of a gene may affect molecular phenotypes in a correlated fashion (Figure 1). Such a pleiotropic mutational effect can be described by a multivariate normal distribution
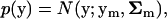 |
(3) |
where ym is the mean mutational effect on molecular phenotypes, and the covariance matrix Σm measures the correlated mutational effects. The coefficient of selection ρ(y | y0, μ) and the distribution of mutational effects p(y) provide a connection between organismal fitness and protein sequence evolution through molecular phenotypes y (Figure 1). Below we discuss the classical stabilizing selection model and the newly developed stabilizing selection with microadaptation (SM) model. See Figure 2 for illustration.
Figure 2.—

(A) Stabilizing selection model with microadaptation in the case of a single molecular phenotype. (B) Distribution of mutational effects on the molecular phenotype. In both cases, we set K = 1 for simplicity.
Stabilizing selection model:
Under the classical stabilizing model, the population mean of molecular phenotypes (y0) is always fixed at the optimum (μ). Without loss of generality, we assume that y0 = μ = 0. From Equation 2 one can show that 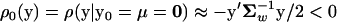 , reflecting the stabilizing (purifying) selection against deleterious mutations, which necessarily results in a deviation from the optimum. A consequence of this model is that sequence evolution is dominated by the fixation of very slightly deleterious mutations (Ohta 1973; Kimura 1979, 1983).
, reflecting the stabilizing (purifying) selection against deleterious mutations, which necessarily results in a deviation from the optimum. A consequence of this model is that sequence evolution is dominated by the fixation of very slightly deleterious mutations (Ohta 1973; Kimura 1979, 1983).
For mathematical convenience, we assume that the mutational distribution, p(y) in Equation 3, is centered at the population mean, namely, ym = y0 = 0. In the following we consider mainly the selection intensity S(y) = 4Neρ0(y) = −2Ne(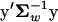 ), where Ne is the effective population size. Given Equation 3 of p(y), the distribution of S, f(S), can be uniquely determined by the probability theory (see the appendix). Although the analytical form of f(S) is available only for a few special cases, the moments of S can be obtained readily. For instance, the mean of S can be computed by
), where Ne is the effective population size. Given Equation 3 of p(y), the distribution of S, f(S), can be uniquely determined by the probability theory (see the appendix). Although the analytical form of f(S) is available only for a few special cases, the moments of S can be obtained readily. For instance, the mean of S can be computed by
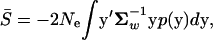 |
(4) |
which is always negative.
Stabilizing selection with microadaptation model:
Consider the case when the fitness optimum (μ) of a gene is no longer fixed during the evolution. Rather, μ can be shifted either by environmental changes or by internal physiological perturbations. Each shift of μ results in a microadaptation toward a new fitness optimum (Gillespie 1991). Since the direction and strength of the μ-shift are unobservable, we treat μ as a (K-dimensional) random variable. Similar to the above, we assume that the population mean and the mutation mean satisfy y0 = ym = 0. Then, under the SM model, the coefficient of correlation defined by Equation 2 can be specified as follows:
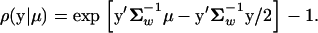 |
(5) |
After assuming that μ follows a multivariate normal distribution φ(μ) with mean 0 and covariance matrix Σμ, one can integrate out the unobservable μ by 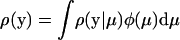 . Therefore, one can show that ρ(y), the coefficient of selection of y averaged over μ-shifts during the evolution, is given by
. Therefore, one can show that ρ(y), the coefficient of selection of y averaged over μ-shifts during the evolution, is given by
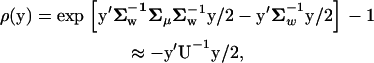 |
(6) |
where the matrix 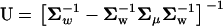 characterizes the correlated nature of molecular phenotypes in fitness after stabilizing selection and microadaptation. It follows that the selection intensity defined by S(y) = 4Neρ(y) is given by
characterizes the correlated nature of molecular phenotypes in fitness after stabilizing selection and microadaptation. It follows that the selection intensity defined by S(y) = 4Neρ(y) is given by
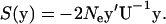 |
(7) |
Apparently, the stabilizing selection model is a special case when Σμ = 0 so that U = Σw. Moreover, unlike the stabilizing selection model that S(y) is always negative, the selection intensity S(y) under the SM model can be positive when the μ-shift becomes huge. See analytical results for more details.
Evolutionary rate of protein sequence:
Suppose that a protein carrying one (nonsynonymous) mutation has the molecular phenotype vector y. The selection intensity for this mutation, S(y), is given by Equation 7. Hence, the well-known formula for the evolutionary rate (Kimura 1983) depends on the molecular phenotypes y; that is,
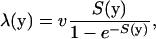 |
(8) |
where v is the mutation rate. At the gene level, molecular phenotypes y are random variables, resulting from random mutations at nucleotide sites (or amino acid residues). Therefore, the evolutionary rate is a random variable that varies among sites. Under the SM model, the mean evolutionary rate of a protein sequence is given by
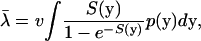 |
(9) |
given the distribution of mutational effects on y, p(y).
ANALYTICAL RESULTS
Here we present analytical results under the SM model, which are useful to study the underlying mechanisms governing protein sequence evolution and to develop a statistical method for estimating important parameters such as K, the number of molecular phenotypes of a gene.
Single molecular phenotype (K = 1):
When K = 1, the fitness function in Equation 1 can be reduced as 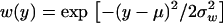 and the selection coefficient in Equation 6 as
and the selection coefficient in Equation 6 as 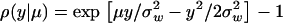 , where
, where 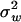 measures the strength of single stabilizing selection. Similarly, the shift of μ follows a single normal distribution φ(μ) with mean 0 and
measures the strength of single stabilizing selection. Similarly, the shift of μ follows a single normal distribution φ(μ) with mean 0 and 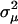 . Together, Equation 8 can be simplified as
. Together, Equation 8 can be simplified as
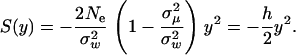 |
(10) |
Given the distribution of mutational effects 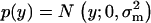 , one can show the mean of selection intensity,
, one can show the mean of selection intensity, 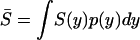 , as follows:
, as follows:
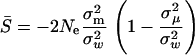 |
(11) |
Several interesting results need to be mentioned. First, 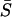 is determined by the mutational variance (
is determined by the mutational variance (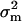 ) and the variance of microadaptation (
) and the variance of microadaptation (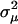 ) relative to the coefficient of stabilizing selection (
) relative to the coefficient of stabilizing selection (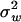 ). Hence, the scale of molecular phenotype is not important. Second,
). Hence, the scale of molecular phenotype is not important. Second, 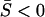 if
if 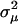 <
< 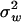 , and vice versa. In particular, the microadaptation model is reduced to the stabilizing selection model when
, and vice versa. In particular, the microadaptation model is reduced to the stabilizing selection model when 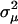 = 0 (Gu 2007). Third, microadaptation reduces the sequence conservation by decreasing the absolute values of
= 0 (Gu 2007). Third, microadaptation reduces the sequence conservation by decreasing the absolute values of 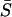 , although
, although 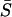 may remain negative. In other words, microadaptation may increase the evolutionary rate of protein sequence. And fourth, from Equation 10, one can show that S follows a negative gamma distribution with the shape parameter ½. For instance, when
may remain negative. In other words, microadaptation may increase the evolutionary rate of protein sequence. And fourth, from Equation 10, one can show that S follows a negative gamma distribution with the shape parameter ½. For instance, when 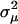 <
< 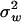 , S follows a negative gamma distribution
, S follows a negative gamma distribution
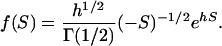 |
(12) |
K molecular phenotypes:
For K ≥ 2, the distribution of selection intensity S, f(S), is generally not analytically tractable. Nevertheless, one can derive the analytical forms of moments on the basis of the theory of multiple normal distribution. These results are crucial for estimating K from the sequence data.
Mean and variance of S—general results:
Denote matrix A = U−1Σm = Z − Ω, where 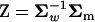 and
and 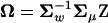 . Briefly, under stabilizing selection, matrix Z describes correlated mutational effects, while Ω describes correlated microadaptations. Together, matrix A characterizes the net effects of correlated mutations on fitness under the SM model. In the appendix (also see Martin and Lenormand 2006 for phenotypic data), we show that the joint effects of all selective, microadaptive, and mutational covariances can be reduced to K eigenvalues of matrix A, α1, …, αK. Consequently, the mean and variance of S are given by
. Briefly, under stabilizing selection, matrix Z describes correlated mutational effects, while Ω describes correlated microadaptations. Together, matrix A characterizes the net effects of correlated mutations on fitness under the SM model. In the appendix (also see Martin and Lenormand 2006 for phenotypic data), we show that the joint effects of all selective, microadaptive, and mutational covariances can be reduced to K eigenvalues of matrix A, α1, …, αK. Consequently, the mean and variance of S are given by
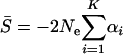 |
(13) |
and
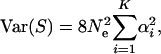 |
(14) |
respectively. In short, each αi corresponds to an independent molecular phenotypic direction, on which mutation, microadaptation, and stabilizing selection act with an average effect −2Neαi on the mean selection intensity (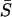 ) of the gene.
) of the gene.
Canonical representation of S̄:
Although Equation 13 is mathematically elegant, we wish to find an equivalent representation of 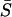 that is biologically more interpretable. Due to the arbitrary nature of the original K molecular phenotypes, without loss of generality one may choose a convenient coordinate system. Here we adopt a canonical form of K molecular phenotypes, to define independent phenotypes experiencing stabilizing selection. This leads to a diagonal matrix Σw; each diagonal element
that is biologically more interpretable. Due to the arbitrary nature of the original K molecular phenotypes, without loss of generality one may choose a convenient coordinate system. Here we adopt a canonical form of K molecular phenotypes, to define independent phenotypes experiencing stabilizing selection. This leads to a diagonal matrix Σw; each diagonal element 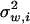 measures the independent stabilizing selection on the ith (canonical) molecular phenotype. Let
measures the independent stabilizing selection on the ith (canonical) molecular phenotype. Let 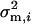 be the ith diagonal element of matrix Σw or the mutational variance for the ith (canonical) molecular phenotype. We have shown that the canonical presentation for the mean of selection intensity is given by
be the ith diagonal element of matrix Σw or the mutational variance for the ith (canonical) molecular phenotype. We have shown that the canonical presentation for the mean of selection intensity is given by
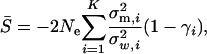 |
(15) |
where the parameter γi measures the net effect of microadaptation on the ith (canonical) molecular phenotype. In particular, when the mutational effect or microadaptation is independent among the canonical molecular phenotypes, we have 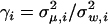 . It should be noted that the pleiotropic nature of mutations affects the variance of S, Var(S) (see the appendix), while
. It should be noted that the pleiotropic nature of mutations affects the variance of S, Var(S) (see the appendix), while 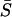 is not affected, as shown in Equation 15.
is not affected, as shown in Equation 15.
Model classification:
As the stabilizing selection acts against nucleotide substitutions (negative selection), microadaptation may provide an opposite force (positive selection) to increase the evolutionary rate. The mean selection intensity given by Equations 13 and 15 can be used to classify the pattern of sequence conservation under the SM model:
Strong stabilizing selection with weak microadaptation (SMw): In this case, the magnitude of μ-shift of molecular phenotypes during the evolution is small, relative to the strength of stabilizing selection. It indicates a dominant purifying selection in the protein sequence evolution. The SMw model can be defined as having matrix A positive definite, such that each eigenvalue αi > 0 and
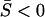 . In the canonical form of Equation 15, it means that γi < 1 for any i = 1, … , K. The pure stabilizing selection is a special case of γi = 0 (no microadaptation). Moreover,
. In the canonical form of Equation 15, it means that γi < 1 for any i = 1, … , K. The pure stabilizing selection is a special case of γi = 0 (no microadaptation). Moreover, 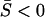 implies that the ratio of nonsynonymous to synonymous substitutions (dN/dS) is <1. Therefore, the SMw model may describe the general pattern in protein sequence evolution, evidenced by genomewide analyses that have shown dN/dS < 1 for most genes.
implies that the ratio of nonsynonymous to synonymous substitutions (dN/dS) is <1. Therefore, the SMw model may describe the general pattern in protein sequence evolution, evidenced by genomewide analyses that have shown dN/dS < 1 for most genes.Episodic microadaptation under strong stabilizing selection (SME): Some genes may have experienced episodic adaptive processes in a few molecular phenotypes, resulting in negative eigenvalues of matrix A (i.e., αi < 0 for some i's, but the mean selection intensity remains negative,
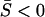 ). That is, matrix A is no longer positive definite but a positive trace
). That is, matrix A is no longer positive definite but a positive trace 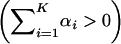 remains. In the canonical form, some of γi are >1, although
remains. In the canonical form, some of γi are >1, although 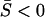 . Apparently, the dN/dS ratio under the SME model may be increased by episodic microadaptations, but dN/dS < 1 remains. A special case is the neutral-like behavior of protein sequence evolution when
. Apparently, the dN/dS ratio under the SME model may be increased by episodic microadaptations, but dN/dS < 1 remains. A special case is the neutral-like behavior of protein sequence evolution when 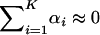 , resulting in
, resulting in 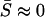 . That is, the observed neutral-like evolution may reflect the zero net effect on fitness, canceling each other between episodic microadaptation and stabilizing selection.
. That is, the observed neutral-like evolution may reflect the zero net effect on fitness, canceling each other between episodic microadaptation and stabilizing selection.Strong microadaptation under stabilizing selection (SMs): In a few genes, positive selection and adaptation may dominate the evolution in many molecular phenotypes. In these cases,
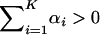 results in
results in 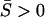 and the ratio dN/dS > 1. An extreme case occurs if αi < 0 or γi > 1 (the canonical form) for all i = 1, …, K. In this case, matrix A is negative definite, indicating the overwhelming adaptation forces for virtually all of the substitutions.
and the ratio dN/dS > 1. An extreme case occurs if αi < 0 or γi > 1 (the canonical form) for all i = 1, …, K. In this case, matrix A is negative definite, indicating the overwhelming adaptation forces for virtually all of the substitutions.
The W-model for S-gamma distribution:
Under some strong assumptions, Gu (2007) showed that S is distributed as a (negative) gamma. A more general condition for S-gamma distribution exists when stabilizing selection, mutations, and microadaptation have very similar correlation structures among molecular phenotypes. Indeed, when the three corresponding matrices can be written as 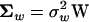 ,
, 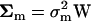 , and
, and 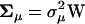 , respectively, where W is a (positive-definite) symmetric matrix, the eigenvalues of matrix A become identical; i.e., α1, …, αK = α, as given by
, respectively, where W is a (positive-definite) symmetric matrix, the eigenvalues of matrix A become identical; i.e., α1, …, αK = α, as given by
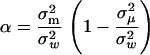 |
(16) |
Then, one can easily show that when α > 0 (the SMw model), f(S) follows a negative gamma distribution
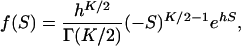 |
(17) |
where the parameter h = 1/(4Neα). Although it is rare, when α < 0 (the SMs model), f(S) follows a (positive) gamma distribution. In both situations, the gamma shape parameter of S is the (half) number of molecular phenotypes (K/2) (Gu 2007).
ESTIMATION AND EXAMPLES
The newly developed SM model is useful in practice only if the underlying parameters can be estimated. But this is difficult to accomplish using conventional statistical approaches, since the number of unknown parameters may be huge. Instead, we attempt to focus on two important parameters, K, critical for understanding gene pleiotropy, and 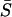 , the measure for overall sequence conservation. Our purpose here is to estimate K and
, the measure for overall sequence conservation. Our purpose here is to estimate K and 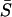 , without specifying other more detailed parameters.
, without specifying other more detailed parameters.
Moments of evolutionary rate:
Under the SM model, the mean evolutionary rate of a protein sequence (λ) is given by Equation 9. Therefore, any kth moment of the evolutionary rate is given by
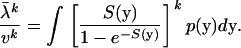 |
(18) |
To apply Equation 18 to estimate unknown parameters, we use the approximation S/(1 − e−S) ≈ e−|S|(1 + c|S|), where c ≈ 0.5772 is Euler's constant. As shown by Figure 3, this approximation is satisfactory for S ≤ 0. Thus, under the SMw model (stabilizing selection with weak microadaptation), which assures S(y) = −2Ney′U−1y < 0, the kth moment of the evolutionary rate λ can be approximated as follows:
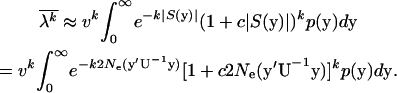 |
(19) |
Figure 3.—

(A) The λ/v − S (the rate ratio of evolution to mutation vs. the selection intensity) relationship, for the accurate formula and the approximation. (B) The gK ∼ K plotting for estimating the effective number of molecular phenotypes.
As shown in the appendix, the mean evolutionary rate (k = 1) is given by
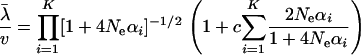 |
(20) |
and the second moment of λ (k = 2) by
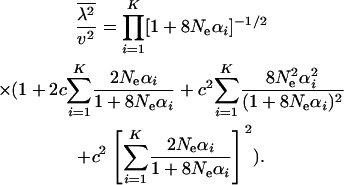 |
(21) |
Effective number (Ke) of molecular phenotypes:
In general, the mean evolutionary rate depends on K distinct eigenvalue parameters (Neαi's), while the second moment depends on Neαi's and 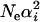 , and so forth. To develop a simple method for estimating K on the basis of Equations 20 and 21, we invoke the assumption of Neαi ≫ 1, leading to
, and so forth. To develop a simple method for estimating K on the basis of Equations 20 and 21, we invoke the assumption of Neαi ≫ 1, leading to
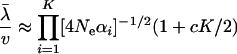 |
(22) |
and
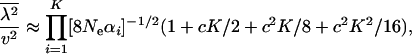 |
(23) |
respectively. Notably, the first and second moments of evolutionary rate depend only on two parameters, K and the product 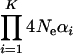 . Further, we found that the ratio of the second moment
. Further, we found that the ratio of the second moment 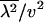 to the first moment (
to the first moment (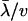 ), denoted by
), denoted by 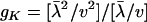 , is given by
, is given by
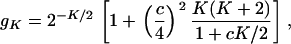 |
(24) |
which is only K dependent. As shown in Figure 3, gK decreases when K increases; gK = 1 when K = 0, and 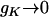 when
when 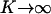 .
.
Equations 22–24 provide the theoretical foundation for estimating K when Neαi ≫ 1. In this case, the parameter K is interpreted as the number of molecular phenotypes that have experienced strong stabilizing selection. Thereafter we refer to Ke as the effective number of molecular phenotypes, which is less than the true number of molecular phenotypes. If the ratio gK is known, Ke can be estimated according to Equation 24.
A widely used measure for 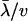 is dN/dS, the ratio of nonsynonymous to synonymous substitutions, under the assumption that synonymous substitutions are almost neutral. We have realized that the second moment of rate
is dN/dS, the ratio of nonsynonymous to synonymous substitutions, under the assumption that synonymous substitutions are almost neutral. We have realized that the second moment of rate 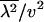 is related to the H-measure (Gu et al. 1995) for the rate variation among sites (Uzzel and Corbin 1971), defined by
is related to the H-measure (Gu et al. 1995) for the rate variation among sites (Uzzel and Corbin 1971), defined by 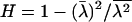 . Ranging from 0 to 1, a higher value of H means a greater rate variation among sites and vice versa. One can easily verify
. Ranging from 0 to 1, a higher value of H means a greater rate variation among sites and vice versa. One can easily verify
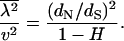 |
(25) |
Therefore, the effective number of molecular phenotypes (Ke) is the solution of the following equation:
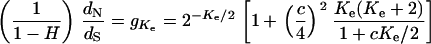 |
(26) |
Effective selection intensity (Se):
Equation 13 indicates that the mean selection intensity 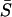 can be written as
can be written as 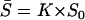 , where
, where 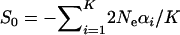 is the (arithmetic) average of selection intensity over molecular phenotypes. However, without knowing each Neαi, it is difficult to estimate
is the (arithmetic) average of selection intensity over molecular phenotypes. However, without knowing each Neαi, it is difficult to estimate 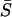 . After realizing that the product
. After realizing that the product 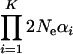 is actually estimable, as implied by Equation 22, we define the effective selection intensity as
is actually estimable, as implied by Equation 22, we define the effective selection intensity as 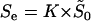 , where
, where 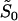 is the geometric average of selection intensity per molecular phenotype; that is,
is the geometric average of selection intensity per molecular phenotype; that is,
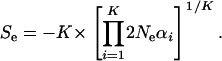 |
(27) |
In general, 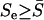 , and
, and 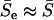 when all αi's are roughly the same. At any rate, both geometric and arithmetric means of selection intensity have virtually the same effect on the rate of protein evolution. Replacing
when all αi's are roughly the same. At any rate, both geometric and arithmetric means of selection intensity have virtually the same effect on the rate of protein evolution. Replacing 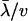 by dN/dS in Equation 22 and K by Ke, the effective selection intensity can be estimated by
by dN/dS in Equation 22 and K by Ke, the effective selection intensity can be estimated by
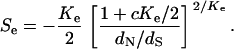 |
(28) |
Estimation procedure:
We propose an analytical pipeline to estimate the effective number of molecular phenotypes (Ke) and the effective selection intensity (Se). Although it needs to be refined both statistically and computationally, our procedure consists of the following steps:
Infer the phylogenetic tree from a multiple alignment of homologous protein sequences. Although there is no methodological preference, we require that the inferred tree topology should be roughly the same over methods.
Estimate the nonsynonymous to synonymous ratio (dN/dS) from closely related coding sequences that satisfy dN/dS < 1.
Estimate the H-measure for rate variation among sites: Use the method of Gu and Zhang (1997) to infer the (corrected) number of changes at each site, under the inferred phylogeny.
Let 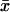 and V(x) be the mean and variance of number of changes over sites, respectively. Assuming a Poisson process at each site, we obtain the mean evolutionary
and V(x) be the mean and variance of number of changes over sites, respectively. Assuming a Poisson process at each site, we obtain the mean evolutionary 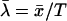 , where T is the total evolutionary time along the tree. Similarly, the variance of evolutionary rate among sites is given by
, where T is the total evolutionary time along the tree. Similarly, the variance of evolutionary rate among sites is given by 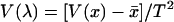 . Then, H can be estimated by
. Then, H can be estimated by
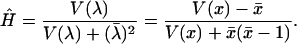 |
(29) |
And finally, one can estimate Ke and Se according to Equations 26 and 28, respectively.
Examples:
For case studies, we compiled 20 vertebrate protein families. Each family includes eight complete sequences from human, mouse, dog, cow, chicken, Xenopus, fugu, and zebrafish, respectively, which were downloaded from Ensembl EnsMar (http://www.ensembl.org/Multi/martview) (Kasprzyk et al. 2004). The synonymous distance (dS) and the nonsynonymous distance (dN) between mammalian orthologs were estimated by codeml of the PAML package (Yang 1997). We used the same phylogeny (Figure 4) to estimate the H-index of rate variation among sites; using an alternative tree gave a very similar result (not shown).
Figure 4.—

Phylogenetic tree of vertebrates used in our data analysis.
Table 1 shows the results. The dN/dS ratio varies substantially among genes (from 0.001 to 0.274), and the H-index ranges from 0.286 to 0.886. Consequently, the estimated effective number of molecular phenotypes Ke ranges from 2.3 to 20.4, with an average of 8.77 (the median is 8.23). Our analysis suggests that most genes are highly pleiotropic. That is, there are on average eight distinct components in the fitness that may be related to a gene. Roughly, ∼73% of the among-gene variation in the dN/dS ratio can be explained by the variation in Ke among genes. In other words, the level of gene pleiotropy associated with a gene may be the dominant factor for predicting sequence conservation, as predicted by Fisher (1930). The effective mean selection intensity (Se) ranges from 5.39 to 23.63, with a median of 12.6. Note that dN/dS ratios in Table 1 were estimated from the human and mouse orthologous genes; using other mammals or taking an average would lead to ∼5% difference in the Ke estimation.
TABLE 1.
Estimation of Ke and Se for 20 vertebrate proteins
| Genes | dN/dS (H–M) | H | Ke | Se |
|---|---|---|---|---|
| Adiponectin receptor protein 2 | 0.109 | 0.785 | 2.30 | −12.32 |
| GTP cyclohydrolase I | 0.188 | 0.586 | 2.68 | −7.16 |
| F-box protein 34 | 0.274 | 0.355 | 2.90 | −5.39 |
| RNA methyltransferase-like 1 | 0.221 | 0.422 | 3.26 | −6.18 |
| Galectin-3 | 0.148 | 0.393 | 4.79 | −7.63 |
| Glia maturation factor (GMF)β | 0.047 | 0.729 | 5.97 | −11.65 |
| Metalloprotease 1 | 0.076 | 0.514 | 6.31 | −9.94 |
| Nucleolar GTP-binding protein 1 | 0.049 | 0.650 | 6.64 | −11.34 |
| Sperm tail protein SHIPPO1 | 0.063 | 0.286 | 8.18 | −10.80 |
| Transaldolase | 0.042 | 0.524 | 8.23 | −11.96 |
| CGTHBA protein | 0.030 | 0.654 | 8.23 | −12.93 |
| Rhomboid family 1 | 0.024 | 0.582 | 9.64 | −13.77 |
| Nucleoredoxin | 0.020 | 0.641 | 9.80 | −14.38 |
| Myosin lc (Myosin Iβ) | 0.023 | 0.573 | 9.85 | −13.93 |
| Exocyst complex component 5 | 0.023 | 0.566 | 9.94 | −13.97 |
| Plakophilin-3 | 0.034 | 0.343 | 9.97 | −12.89 |
| Homeobox protein OTX2 | 0.014 | 0.360 | 12.74 | −15.83 |
| Cornichon homolog (TGAM77) | 0.001 | 0.886 | 15.83 | −23.63 |
| 14-3-3 protein epsilon (14-3-3E) | 0.001 | 0.777 | 17.77 | −23.61 |
| F-box/LRR-repeat protein 14 | 0.001 | 0.507 | 20.40 | −24.27 |
The dN/dS ratio was calculated from the human (H) and mouse (M) orthologous genes.
DISCUSSION
Estimation of gene pleiotropy:
Although the theory of gene pleiotropy, the capacity of a gene to affect multiple phenotypic characters, has been used to explain many biological phenomena (Fisher 1930; Wright 1968; Barton 1990; Waxman and Peck 1998; Otto 2004; MacLean et al. 2004; Dudley et al. 2005), the characteristic level of pleiotropy remains largely unknown. Many phenotype–genotype models (e.g., Fisher 1930; Wright 1968; Hartl and Taubes 1996, 1998; Waxman and Peck 1998; Poon and Otto 2000) have linked gene pleiotropy to the dimensionality of the fitness function of the organism. The major contribution of this article is to formulate a statistical framework for estimating the capacity of a gene to significantly affect distinct components in the fitness, the effective number of molecular phenotypes (Ke). Thus, for the first time we have an empirical measure of gene pleiotropy. Examples in Table 1 show that Ke is typically ∼6–9, the number of distinct fitness components a gene may affect, which has been further confirmed by a large-scale data analysis (not shown).
Our SM model provides an approach to understanding the role of gene pleiotropy in a gene network. Although a detailed analysis will be published elsewhere, we test the hypothesis that gene pleiotropy underlies genomic correlations related to the evolutionary rate of protein sequence (He and Zhang 2006; Pal et al. 2006; Wolf et al. 2006). That is, (i) evolutionary rate is inversely related to gene pleiotropy (K), (ii) gene pleiotropy determines gene dispensability, and (iii) gene pleiotropy underlies “hub” genes. In short, estimation of gene pleiotropy (Ke) provides a model-based data-analysis approach that avoids the interpretative difficulty in principle-component or partial-correlation analysis.
In the future, we shall refine the analytical pipeline for estimating Ke. We shall examine how statistical properties of dN/dS and H would affect the estimation efficiency of Ke. In addition to the delta method, another method to estimate the sampling variance of the estimated Ke is nonparametric bootstrapping. Under the W-model when the selection intensity S follows a (negative) gamma distribution, a likelihood approach can be developed. Since the method of Nielsen and Yang (2003) is applicable only for closely related sequences, we shall develop a protein sequence-based likelihood approach.
Gene pleiotropy and allelic pleiotropy:
It should be noted that the effective number of molecular phenotypes (Ke) measures the functional pleiotropy of a gene, rather than a single mutation. For instance, Ke = 8.23 for trans-aldolase is the estimated number of distinct fitness dimensions implied by all random mutations in the coding sequence. Although the estimated Ke indicates that most genes may be highly pleiotropic, what it exactly means is very much gene specific.
The number of fitness dimensions affected by a single mutation (Ω) could be much less than Ke, say, ∼2–4 (Dudley et al. 2005). Waxman and Peck (1998) argued that a single optimal gene sequence may become common when three or more characters (as molecular phenotypes here) are affected by each mutation, (i.e., Ω ≥ 3). The problem of estimating Ω effectively from sequence data remains a question that requires further study.
Several studies (Nielsen and Yang 2003; Piganeau and Eyre-Walker 2003; Eyre-Walker et al. 2006) estimated the shape parameter of gamma distribution for S. Nielsen and Yang (2003) obtained the shape parameter of 3.22 from the primate mitochondrial genome sequences. Under the SM framework, it can be interpreted as Ke/2; see the above section for the W-model. That is, Ke = 2 × 3.22 = 6.44 for primate mitochondria proteins, consistent with our results (Table 1). In contrast, Eyre-Walker et al. (2006) obtained a much smaller value (0.23) of the shape parameter from the human SNP data. We speculate that the estimate from population genetics data perhaps reflects the allelic pleiotropy (Ω). Hence, Eyre-Walker et al. (2006) may provide a rough estimate for Ω = 2 × 0.23 = 0.46.
Assumptions and alternative models:
The SM model involves several simplifying assumptions, since an exhaustive consideration of all possible mutational effects on molecular phenotypes is intractable. Many are shown with previous phenotype–genotype models. One may refer to Martin and Lenormand (2006) for a recent summary about rationales and criticisms. The first assumption is the single fitness optimum. Under this condition we assume that disruptive selection with alternative optima following an adaptive process (reviewed in Kingsolver et al. 2001 and Elena and Lenski 2003) occurs rarely at the molecular level. Moreover, we used the Gaussian fitness function (see Equation 1) to measure the fitness consequences of a deviation from the optimum. Lande (1980) showed that when the population mean is close to the optimum, a Gaussian-like function can be a local kernel approximation for many arbitrary fitness functions.
Second, the effect of mutations on molecular phenotypes is assumed to be continuous and symmetric. The continuous property is consistent with some empirical evidence from random mutations of genes (Keightley 1994; Keightley and Caballero 1997; Imhof and Schlotterer 2001). The symmetric assumption implies that even if mutations bias toward higher or lower values of molecular phenotypes, the trend should be small compared to the mutational variance. In practice we used a multiple Gaussian distribution for mutational effects. In the non-Gaussian case, it is possible to reduce kurtosis after an appropriate transformation such that mutational effects on a “transformed” molecular phenotype are roughly Gaussian-like. In short, we conclude that in the absence of practically useful alternatives, single optimum, Gaussian fitness function, and continuous symmetric effect of mutations remain useful working assumptions. Further work will examine the effects of these assumptions using computer simulations.
A more important issue is whether the estimation procedure for the effective number of molecular phenotypes (Ke) is robust under various alternative models. The first alternative model is Fisher's geometric model, in which adaptive change is represented by stepwise movement of a point toward the center of the hypersphere. In this case, the number of molecular phenotypes (K) is Fisher's geometric dimension. Several studies (Hartl and Taubes 1996, 1998; Sella and Hirsh 2005) showed that at equilibrium, Fisher's geometric model can be approximated by allowing the current population to vary randomly around the fixed optimum. A second alternative model is to define the fitness to be a function of flux through a metabolic pathway (Kacser and Burns 1979; Hartl and Taubes 1996), rather than a Gaussian-like function. Preliminary results have indicated that in both cases, estimation of Ke varies only slightly. It seems that Ke is a robust measure of gene pleiotropy, which may not be very sensitive to the specific evolutionary model. We shall address this issue more extensively in future studies.
Acknowledgments
The author is grateful to Adam Eyre-Walker and the reviewing editor Antony Long for constructive comments on the manuscript. Assistance in data analysis from Dongping Xu and Zhixi Su is appreciated.
APPENDIX
Mean and variances of S—derivation of Equations 13 and 14:
For the distribution of y that follows a multi(K)-variate normal distribution,
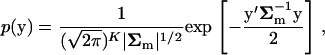 |
(A1) |
we note that for any quadratic function y′U−1y, the first and second moments can be expressed as
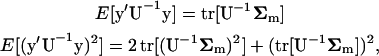 |
(A2) |
respectively, where the operator tr[.] means the trace of a square matrix. From Equation 7 it is straightforward that 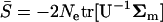 and
and 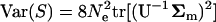 . Denote the matrix A = U−1Σm. After noting that the trace of the matrix is the sum of the eigenvalues, i.e.,
. Denote the matrix A = U−1Σm. After noting that the trace of the matrix is the sum of the eigenvalues, i.e., 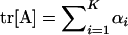 and
and 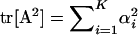 , we obtain Equations 13 and 14.
, we obtain Equations 13 and 14.
Canonical representation of S—derivation of Equation 15:
The canonical representation will choose molecular phenotypes such that Σw = diag( ,1, …,
,1, …, 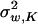 ). Recall that A = U−1Σm = Z − Ω, where
). Recall that A = U−1Σm = Z − Ω, where 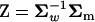 and
and 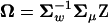 . Then, one can verify
. Then, one can verify 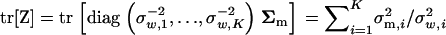 , since the trace of a matrix is the sum of its diagonal elements. Next, matrix Ω can be written as
, since the trace of a matrix is the sum of its diagonal elements. Next, matrix Ω can be written as 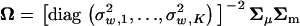 . It follows that
. It follows that  , where
, where 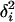 is the ith eigenvalue of matrix ΣμΣm. From the relation tr[A] = tr[Z] − tr[Ω], together we have
is the ith eigenvalue of matrix ΣμΣm. From the relation tr[A] = tr[Z] − tr[Ω], together we have
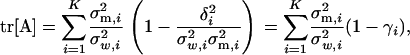 |
(A3) |
leading to Equation 15, where 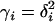 /[
/[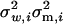 ].
].
The canonical form for the variance of S can be obtained similarly, but the algebra is somewhat tedious, largely due to the detail of A2 = Z2 + Ω2 − 2ZΩ. Let 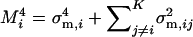 . One thus can show the trace
. One thus can show the trace 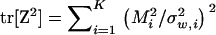 and
and 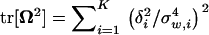 . Noting that ZΩ = diag(
. Noting that ZΩ = diag(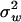 ,1, …,
,1, …, 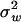 ,K)ΣmΣμΣm, we obtain
,K)ΣmΣμΣm, we obtain 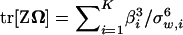 , where
, where 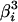 is the ith eigenvalue of matrix ΣmΣμΣm. Therefore, from the fact tr[A2] = tr[Z2] + tr[Ω2] − 2 tr[ZΩ], we have
is the ith eigenvalue of matrix ΣmΣμΣm. Therefore, from the fact tr[A2] = tr[Z2] + tr[Ω2] − 2 tr[ZΩ], we have
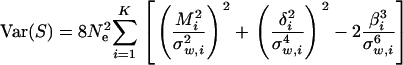 |
(A4) |
Condition of S-gamma distribution:
Given p(y) characterized by the covariance Σw, the distribution of selection intensity, f(S), can be uniquely determined by the probability theory that for any S* and y* that satisfy S* = −2Ne(y*′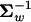 y*) in Equation 7, the relationship
y*) in Equation 7, the relationship 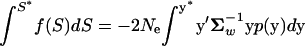 holds. However, the analytical form of f(S) is not available, except for the case when the matrix U in the quadratic form and the covariance in p(y), Σm, satisfies A = ΣmU−1 = αI. In other words, the matrix A is diagonal, having the same eigenvalues α.
holds. However, the analytical form of f(S) is not available, except for the case when the matrix U in the quadratic form and the covariance in p(y), Σm, satisfies A = ΣmU−1 = αI. In other words, the matrix A is diagonal, having the same eigenvalues α.
Moments of evolutionary rate—derivation of Equations 20 and 21:
Here we show several analytical integral results. First we consider 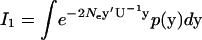 , which is given by
, which is given by
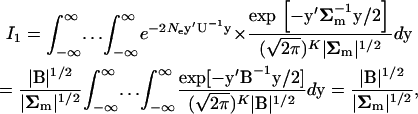 |
(A5) |
where 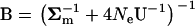 . It follows that
. It follows that
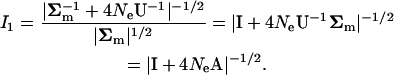 |
(A6) |
In the same manner, we have 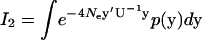 given by
given by
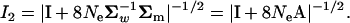 |
(A7) |
Next we consider 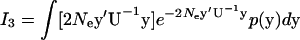 . From the derivation of I1, one can verify
. From the derivation of I1, one can verify
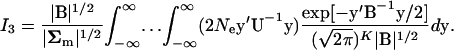 |
(A8) |
From Equation A2, we obtain
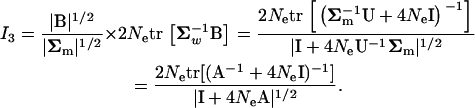 |
(A9) |
In the same manner, we show that 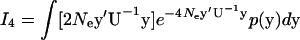 as follows:
as follows:
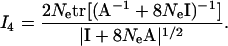 |
(A10) |
Finally, we consider the integral 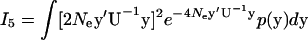 . Similar to the derivations of I1–I5, one can write
. Similar to the derivations of I1–I5, one can write
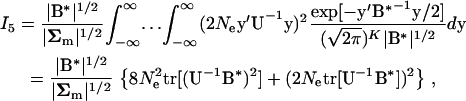 |
(A11) |
where matrix B* = (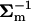 + 8NeU−1)−1. Since U−1B* = (
+ 8NeU−1)−1. Since U−1B* = (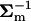 U + 8NeI)−1 = (A−1 + 8NeI)−1, and |B*/Σm| = |I + 8NeA|, I5 can be written as follows:
U + 8NeI)−1 = (A−1 + 8NeI)−1, and |B*/Σm| = |I + 8NeA|, I5 can be written as follows:
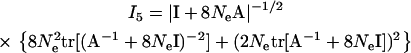 |
(A12) |
From Equation 19, one can show that 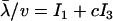 and
and 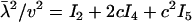 , both of which are determined a single matrix A. Hence, we have derived Equations 20 and 21 on the basis of the matrix theory that, for a square matrix A, the trace is the sum of the eigenvalues, the determinant is the production of the eigenvalues, and any eigenvalue of the matrix that is a function of A is the function of the corresponding eigenvalue of A.
, both of which are determined a single matrix A. Hence, we have derived Equations 20 and 21 on the basis of the matrix theory that, for a square matrix A, the trace is the sum of the eigenvalues, the determinant is the production of the eigenvalues, and any eigenvalue of the matrix that is a function of A is the function of the corresponding eigenvalue of A.
References
- Barton, N., 1990. Pleiotropic models of quantitative variation. Genetics 124: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillon, T., 2000. Estimation of spontaneous genome-wide mutation rate parameters: Whither beneficial mutations? Heredity 84: 497–501. [DOI] [PubMed] [Google Scholar]
- Drummond, D. A., J. D. Bloom, C. Adami, C. O. Wilke and F. H. Arnold, 2005. Why highly expressed proteins evolve slowly. Proc. Natl. Acad. Sci. USA 102: 14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, A., D. Janse, A. Tanay, R. Shamir and G. Church, 2005. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol. Syst. Biol. 1: 2005.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena, S. F., and R. E. Lenski, 2003. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4: 457–469. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., M. Woolfit and T. Phelps, 2006. The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173: 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The genetical theory of natural selection. Oxford University Press, Oxford.
- Gillespie, J. H., 1991. The Causes of Molecular Evolution. Oxford University Press, Oxford.
- Gu, X., and J. Zhang, 1997. A simple method for estimating the parameter of substitution rate variation among sites. Mol. Biol. Evol. 14: 1106–1113. [DOI] [PubMed] [Google Scholar]
- Gu, X., 2007. Stabilizing selection of protein function and distribution of selection coefficient among sites. Genetica (in press). [DOI] [PubMed]
- Gu, X., Y. X. Fu and W. H. Li, 1995. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 12: 546–557. [DOI] [PubMed] [Google Scholar]
- Hartl, D. L., and C. H. Taubes, 1996. Compensatory nearly neutral mutations: selection without adaptation. J. Theor. Biol. 182: 303–309. [DOI] [PubMed] [Google Scholar]
- Hartl, D. L., and C. H. Taubes, 1998. Towards a theory of evolutionary adaptation. Genetica 102/103: 525–533. [PubMed] [Google Scholar]
- He, X., and J. Zhang, 2006. Toward a molecular understanding of pleiotropy. Genetics 173: 1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof, M, and C. Schlotterer, 2001. Fitness effects of advantageous mutations in evolving Escherichia coli populations. Proc. Natl. Acad. Sci. USA 98: 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser, H., and J. A. Burns, 1979. Molecular democracy: Who shares the controls? Biochem. Soc. Trans. 7: 1149–1161. [DOI] [PubMed] [Google Scholar]
- Kasprzyk, A., D. Keefe, D. Smedley, D. London, W. Spooner et al., 2004. EnsMart: a generic system for fast and flexible access to biological data. Genome Res. 14: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1994. The distribution of mutation effects of viability in Drosophila melanogaster. Genetics 138: 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and A. Caballero, 1997. Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 94: 3823–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1979. Model of effectively neutral mutations in which selective constraint is incorporated. Proc. Natl. Acad. Sci. USA 75: 1934–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kingsolver, J. G., H. E. Hoekstra, J. M. Hoekstra, D. Berrigan, S. N. Vignieri et al., 2001. The strength of phenotypic selection in natural populations. Am. Nat. 157: 245–261. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1980. The genetic covariance between characters maintained by pleiotropic mutations. Genetics 94: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., J. Blanchard, D. Houle, T. Kibota, S. Schultz et al., 1999. Perspective: spontaneous deleterious mutation. Evolution 53: 645–663. [DOI] [PubMed] [Google Scholar]
- MacLean, R. C., G. Bell and P. B. Rainey, 2004. The evolution of a pleiotropic fitness tradeoff in Pseudomonas fluorescens. Proc. Natl. Acad. Sci. USA 101: 8072–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, G., and T. Lenormand, 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 2003. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol. Biol. Evol. 20: 1231–1239. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1973. Slightly deleterious mutant substitutions in evolution. Nature 246: 96–98. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1977. Extension to the neutral mutation random drift hypothesis, pp. 148–176 in Molecular Evolution and Polymorphism, edited by M. Kimura. National Institute of Genetics, Mishima, Japan.
- Otto, S. P., 2004. Two steps forward, one step back: the pleiotropic effects of favoured alleles. Proc. Biol. Sci. 271: 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, C., B. Papp and M. J. Lercher, 2006. An integrated view of protein evolution. Nat. Rev. Genet. 7: 337–348. [DOI] [PubMed] [Google Scholar]
- Piganeau, G., and A. Eyre-Walker, 2003. Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl. Acad. Sci. USA 100: 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, A., and S. P. Otto, 2000. Compensating for our load of mutants: freezing the meltdown of small populations. Evolution 54: 1467–1479. [DOI] [PubMed] [Google Scholar]
- Shaw, F. H., C. J. Geyer and R. G. Shaw, 2002. A comprehensive model of mutations affecting fitness and inferences for Arabidopsis thaliana. Evolution 56: 453–463. [DOI] [PubMed] [Google Scholar]
- Sella, G., and A. E. Hirsh, 2005. The application of statistical physics to evolutionary biology. Proc. Natl. Acad. Sci. USA 102: 9541–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli, M., 1985. Effects of pleiotropy on predictions concerning mutation-selection balance for polygenic traits. Genetics 111: 165–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzzel, T., and K. W. Corbin, 1971. Fitting discrete probability distribution to evolutionary events. Science 172: 1089–1096. [DOI] [PubMed] [Google Scholar]
- Wagner, A, 2000. The role of population size, pleiotropy and fitness effects of mutations in the evolution of overlapping gene functions. Genetics 154: 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, G. P., 1989. Multivariate mutation-selection balance with constrained pleiotropic effects. Genetics 122: 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, D. P., A. E. Hirsh, H. B. Fraser, J. Kumm, G. Giaever et al., 2005. Functional genomic analysis of the rates of protein evolution. Proc. Natl. Acad. Sci. USA 102: 5483–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman, D., and J. R. Peck, 1998. Pleiotropy and the preservation of perfection. Science 279: 1210–1213. [PubMed] [Google Scholar]
- Welch, J. J., and D. Waxman, 2003. Modularity and the cost of complexity. Evolution 57: 1723–1734. [DOI] [PubMed] [Google Scholar]
- Wolf, Y. I., L. Carmel and E. V. Koonon, 2006. Unifying measures of gene function and evolution. Proc. Biol. Sci. 273: 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1968. Evolution and the Genetics of Populations. University of Chicago Press, Chicago.
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Zhang, X. S., and W. G. Hill, 2003. Multivariate stabilizing selection and pleiotropy in the maintenance of quantitative genetic variation. Evolution 57: 1761–1775. [DOI] [PubMed] [Google Scholar]