

Summary
SIRPα and SIRPβ1, the two major isoforms of the signal regulatory protein (SIRP) family, are co-expressed in human leukocytes but mediate distinct extracellular binding interactions and divergent cell signaling responses. Previous studies have demonstrated that binding of SIRPα with CD47, another important cell surface molecule, through the extracellular IgV domain regulates important leukocyte functions including macrophage recognition, leukocyte adhesion and transmigration. Although SIRPβ1 shares highly homologous extracellular IgV structure with SIRPα, it does not bind to CD47. In this study, we defined key amino acid residues exclusively expressing in the IgV domain of SIRPα, but not SIRPβ1, which determine the extracellular binding interaction of SIRPα to CD47. These key residues include Gln67, a small hydrophobic amino acid (Ala or Val) at the 57th position and Met102. We found that Gln67 and Ala/Val57 are critical. Mutation of either of these residues abates SIRPα directly binding to CD47. Functional cell adhesion and leukocyte transmigration assays further demonstrated central roles of Gln67 and Ala/Val57 in SIRPα extracellular binding mediated cell interactions and cell migration. Another SIRPα-specific residue, Met102, appears to assist SIRPα IgV binding through Gln67 and Ala/Val57. An essential role of these amino acids in SIRPα binding to CD47 was further confirmed by introducing these residues into the SIRPβ1 IgV domain, which dramatically converts SIRPβ1 into a CD47-binding molecule. Our results thus revealed the molecular basis by which SIRPα selectively binds to CD47 and shed new light into the structural mechanisms of SIRP isoform mediated distinctive extracellular interactions and cellular responses.
Keywords: Ig superfamily protein, signal regulatory proteins, IgV domain, extracellular binding interaction, protein binding, mutagenesis, cell adhesion
Introduction
Signal regulatory proteins (SIRPs) are cell surface Ig superfamily proteins that mediate essential cell surface protein interactions and signal transduction. SIRPs are expressed predominantly in leukocytes under normal conditions 1 and play important roles in mediating immune cell functions, including macrophage recognition / interaction and leukocyte migration 2; 3; 4; 5. It is interesting that SIRPα is also found differentially expressing in certain cancer cells that are of non-hematopoietic origin 6; 7; 8.
SIRPs all contain an N-terminal extracellular domain, a single transmembrane domain and a C-terminal intracellular domain. Based on the structures of their transmembrane and/or intracellular domains, and their potential role in signal transduction, SIRP proteins can be separated into at least three subfamily isoforms, referred to as SIRPα, SIRPβ and SIRPγ 9. SIRPα has a long intracellular domain that comprises two putative immunoreceptor tyrosine-based inhibition motifs (ITIM). Studies have suggested that activation of SIRPα ITIMs delivers inhibitory signals that negatively regulate cell responses 10; 11. In contrast, both SIRPβ and γ have only minimal intracellular tails. Studies showed that SIRPβ1, the predominant form found in human leukocytes 1, can bind to the immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptor protein DAP12 and thus deliver positive regulatory signals 12; 13. It is unclear how SIRPγ mediates signal transduction.
Although the intracellular domains of SIRP isoforms and their mediated cell signals are strikingly different, the extracellular domains of all SIRPs share highly homologous amino acid sequences and similar structures that contain three Ig-like loops. However, despite these highly similarities, researches have revealed that SIRP isoforms mediate remarkably different extracellular ligand binding interactions. SIRPα was identified to be an extracellular receptor/ ligand for CD47, another important cell surface Ig superfamily protein 8; 14. SIRPα-CD47 binding interactions have been confirmed by direct protein binding assays using CD47 and SIRPα extracellular domain fusion proteins, as well as cell adhesion assays using CD47 or SIRPα expressing cells 5; 15. Studies further mapped that the CD47 binding site on SIRPα is located at the membrane distal extracellular IgV loop 15; 16; 17. Since being identified, SIRPα-CD47 binding interactions have been demonstrated to play a vital role in multiple important leukocyte functions, including neutrophil and monocyte migration 4; 5, macrophage recognition and apoptotic cell clearance 15; 18, monocyte / macrophage fusion 3, T cell and dendritic cell functions 19; 20, etc.
Although SIRPβ1 shares a highly homologous IgV domain with SIRPα, this SIRP isoform has no extracellular binding interaction with CD47. Given that SIRPα and β1 are co-expressed on the cell surfaces of granulocytes and mononuclear cells 1 while mediate opposite cellular responses, it is therefore highly important for these SIRPs to precisely differentiate their extracellular binding partner. In this research, we questioned how SIRPα compiles unique amino acid elements in the extracellular IgV domain to mediate specific binding interactions with CD47. As demonstrated in this study, we defined that three essential amino acids uniquely expressed in the IgV of SIRPα, but not SIRPβ1, determine SIRPα extracellular binding to CD47. Indeed, our data showed that constitution of these essential amino acids into the SIRPβ1 IgV domain can convert SIRPβ1 into a CD47-binding molecule. These findings, in combined with the structural information gained from protein modeling, provide new lights into the molecular mechanisms by which SIRP proteins mediate extracellular recognition and regulate leukocyte functions.
Results
Identification of the key residues in SIRPα IgV loop that mediate SIRPα binding to CD47
In previous studies, we and others have reported that, although SIRPα and SIRPβ1 share highly homologous extracellular domains, the extracellular domain of SIRPα, but not SIRPβ1, specifically binds to CD47 5; 16. Furthermore, studies also showed that the binding site on SIRPα is restricted at the N-terminus, the membrane distal-most IgV domain 15; 16; 17. Since the SIRPα subfamily contains multiple members, such as Bit, SIRP α1 and α2, which vary mainly in the membrane-distal IgV domains 10, we thus questioned whether these SIRPα members with different primary structures of IgVs assert differences in binding to CD47. By RT-PCR, we amplified the SIRPα isoforms, SIRPα1 and Bit, from their predominant expressing leukocytes, peripheral blood mononuclear cells (PBMC) and neutrophils, respectively 1. As shown in Figure 1A, 12 amino acid differences (underlined) were found between the primary structures of SIRPα1 and Bit IgV loops, including one missing residue from SIRPα1 that corresponds to Val132 of Bit. We thus constructed soluble SIRPα1.IgV-Fc and Bit.IgV-Fc fusion proteins, both comprising the entire IgV domains flanked by two Cysteine residues and the adjacent linking regions. After transfection into COS cells, the generated SIRPα1.IgV-Fc and Bit.IgV-Fc fusion proteins were purified and tested for binding to CD47 using CD47 extracellular domain fusion protein, CD47-AP, and our previously established in vitro SIRP-CD47 binding assays 5; 15. As shown in Figure 1B, despite the structural differences in their IgV domains, both Bit.IgV-Fc and SIRPα1.IgV-Fc directly bound to CD47-AP and exhibited equivalent binding capability. In the same experiments, we also generated SIRPβ1 IgV domain fusion protein and tested for binding to CD47. As shown in Figure 1B, SIRPβ1.IgV-Fc had no binding to CD47.
Figure 1.
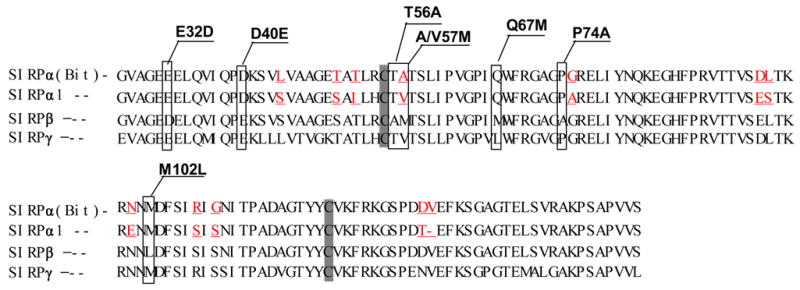
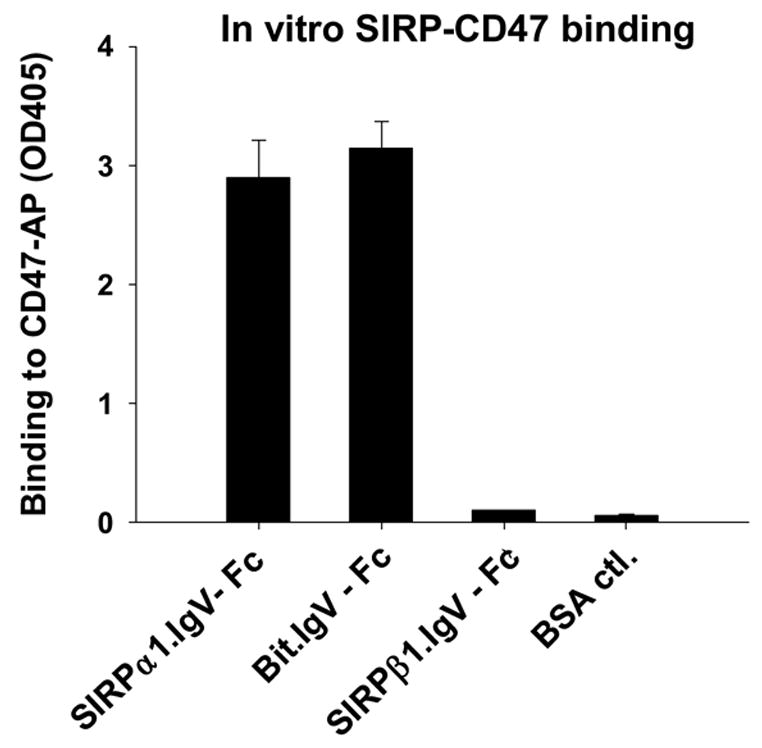
Extracellular binding interactions of SIRP family proteins mediated by the membrane distal IgV domains. A) Primary structure alignments of the extracellular most distal IgV loops of SIRPα members, Bit and SIRPα1, reveal 12 amino acid differences (labeled in red) between these two SIRPα members. The sequence alignments also reveal 7 amino acid (selectively labeled) differences between SIRPα isoforms and SIRPβ1. The primary structure of SIRPγ IgV is also aligned. The shaded Cys residues form the putative disulfide bond in the IgV structures. B) In vitro SIRP-CD47 binding assays. In these experiments, SIRP.IgV-Fc fusion proteins were generated by transfection of COS cells followed by protein purification using protein-A sepharose. The purified fusion proteins (5μg/ml) were used to coat the microtiter wells. After blocking, the wells were incubated with CD47-AP (5μg /ml). After washing, CD47-AP binding to immobilized SIRP.IgV-Fc was detected by assaying AP activity. The data shows that the IgV domains of SIRPα (Bit and SIRPα1), but not SIRPβ1, bind to CD47 extracellular domain fusion protein (CD47-AP).
To define the critical amino acid residues in SIRPα that mediate CD47 binding, we further compared the IgV structures of SIRPα1 and Bit to that of SIRPβ1. Within the 128 amino acids located in IgV and the adjacent area, only 7 varied amino acids were found between the CD47-binding SIRPα (SIRPα1 or Bit) and the non-binding SIRPβ1. As marked in the sequences shown in Figure 1A, these unique residues in SIRPα include Glu32, Asp40, Thr56, Ala57 (in Bit) or Val57 (in SIRPα1), Gln67, Pro74 and Met102. To test if these were the essential residues that determined SIRPα binding to CD47, we performed site-directed mutagenesis and changed these residues in Bit IgV loop to the corresponding counter amino acids in SIRPβ1 (Figure 1A). After affinity purification, we performed ELISA to confirm the equal expression of wild-type and all mutant Bit.IgV-Fc fusion proteins using an anti-Fc antibody (supplemental Fig. 2S-a). In addition, we also tested the chimeric SIRPα portion in these fusions using multiple antibodies against SIRPα extracellular domain. Our results indicated that these fusion proteins were all equally reactive to anti-SIRPα mAbs SE5A5 and SE7C2 4, and a polyclonal anti-SIRPα antibody (anti-SIRPα.ex 1) (data not shown), suggesting that mutations at these residues may not globally affect the Bit IgV structure.
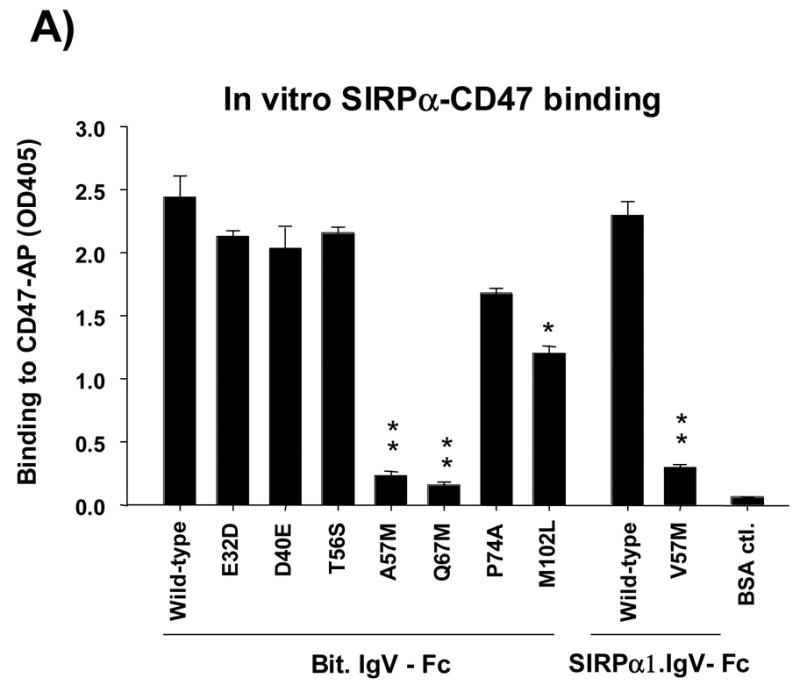
We then tested the direct binding of these Bit.IgV-Fc mutants to CD47-AP. In these assays, microtiter plate wells were coated with purified Bit.IgV-Fc followed by incubation with CD47-AP. CD47 directly binding to Bit.IgV was detected by assaying AP activity. As shown in Figure 2A, compared to CD47 binding to wild-type Bit.IgV, mutations at Glu32 (E32D), Asp40 (D40E) and Thr56 (T56A) did not affect Bit-CD47 binding interaction. Mutations at Ala57 (A57M) and Gln67 (Q67M), however, significantly inhibited Bit IgV binding to CD47 (>80%) (Fig. 2A). Since SIRPα1 expresses Val at the 57th position, which differs from that of Bit (Ala57), we further constructed a V57M mutant in SIRPα1.IgV. As can be seen in Figure 2A, similar to that of A57M mutation, changing of Val57 in SIRPα1 to Met (V57M) resulted in markedly diminished CD47-AP binding to SIRPα1.IgV-Fc. As also shown in Figure 2A, mutation of Met102 to Leu (M102L) partially inhibited (30–50%) Bit.IgV-Fc binding to CD47-AP (Fig. 2A).
Figure 2.
Site-directed mutagenesis of the unique amino acids expressed in the IgV of SIRPα but not SIRPβ1.
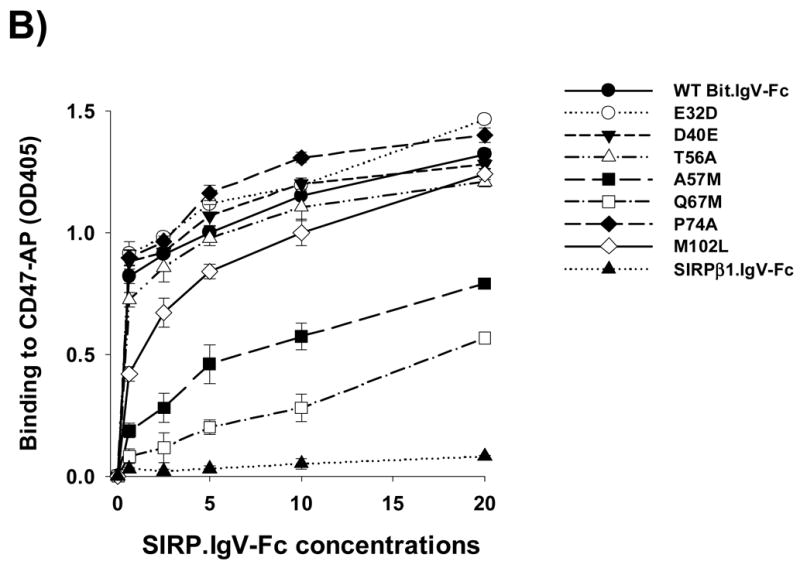
Seven amino acids uniquely expressed in the SIRPα IgV loop (Fig. 1A) were mutated to their counter residues in SIRPβ1. These mutations, including E32D, D40E, T56A, A57M (Bit) or V57M (SIRPα1), Q67M, P74A and M102L, were performed using wild-type Bit.IgV-Fc or SIRPα1.IgV-Fc fusion in pCDNA3.1 as the templates. A) The purified wild-type and mutant Bit and SIRPα1 IgV-Fc fusion proteins (5μg/ml) were coated in microtiter plate wells and assayed for directly binding to CD47-AP as described in Fig. 1. (**, p≤0.01; *, p≤0.05) B) Wild-type and mutant Bit.IgV-Fc fusion proteins were assayed for binding to CD47-AP in a does-dependent manner. In these assays, affinity purified CD47-AP (5μg/ml) was coated in the wells followed by blocking and incubation with varied concentrations of purified Bit.IgV-Fc in HBSS. Binding of Bit.IgV-Fc was detected by a peroxidase-conjugated anti-rabbit Fc antibody.
Further analysis of each SIRPα mutants binding to CD47-AP in a does-dependent setting confirmed our results. In these experiments, microtiter wells were coated with purified CD47-AP followed by incubation with varied concentrations of wild-type and mutant Bit.IgV-Fc proteins. As shown in Figure 2B, significant decreased binding to CD47-AP was observed for mutants Q67M and V57M. In particular, binding of Q67M to CD47-AP required even higher concentrations of the fusion protein than that of V57M, suggesting a key role of Gln67 in SIRPα IgV for mediating its binding with CD47. Mutant M102L also displayed decreased binding to CD47-AP at lower range of protein concentrations (Fig. 2B).
Function of Gln67 in SIRPα IgV
Given the important role of Gln67 in SIRPα IgV binding to CD47 suggested by Figure 2, we further analyzed the function of this residue by mutating Gln67 to various other amino acids. Changing Gln to the similar amino acid Asn (Q67N), as shown in Figure 3, did not affect Bit.IgV-Fc binding to CD47. Changing Gln to another similar but charged residue Glu (Q67E) maintained 70% of Bit IgV binding. Changing Gln to residues with other polar or charged side-chains, including Ser (Q67S), Cys (Q67C), Arg (Q67R) and Lys (Q67K), partially or completely inhibited Bit IgV binding to CD47 (Figure 3). Changing Gln to Gly (Q67G) or Pro (Q67P), however, completely blocked Bit IgV binding (Fig. 3). Since Gly and Pro are the amino acids that promote kinks or turns in peptide chain, we surmised that changing to these amino acids likely disrupted the β-sheet structure predicted at this region (Fig. 9). It was surprising to observe that changing Gln67 to Val (Q67V) completely preserved Bit.IgV-Fc binding to CD47 (Fig. 3). Further mutagenesis showed that changing Gln to Ile, but not Leu, also largely maintained Bit IgV binding (Fig. 3). These results strongly argue that the importance of Gln67 resides on the shape and length of the side-chain instead of the reactive polar group (NH2). Our results also shows that replacing this residue with Met (Q67M), the counter residue expressed in SIRPβ1, completely abrogated the SIRP IgV domain binding capability.
Figure 3.
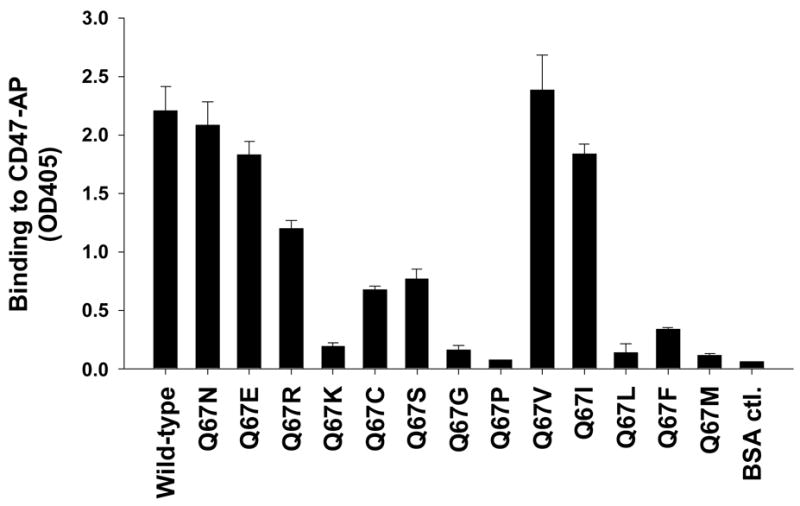
Mutagenesis study of Gln67. Gln67 in Bit IgV was changed to various amino acids using a degenerate primer as described in the Methods. The obtained mutant plasmids were DNA-sequenced to confirm the amino acid changes. After being transiently expressed in COS cells, mutant Bit.IgV-Fc fusion proteins were purified and tested for binding to CD47-AP by in vitro SIRPα-CD47 binding assays.
Figure 9.
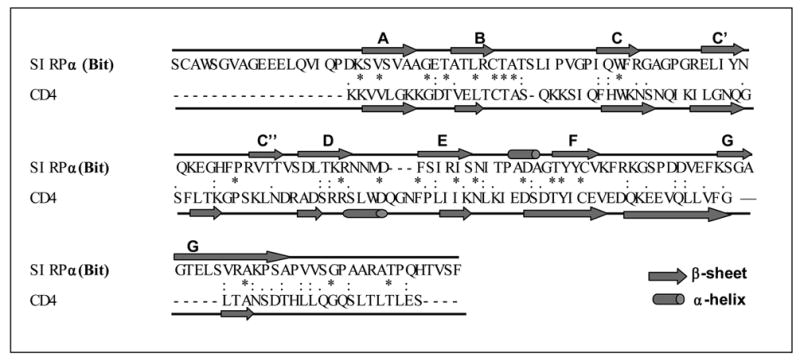
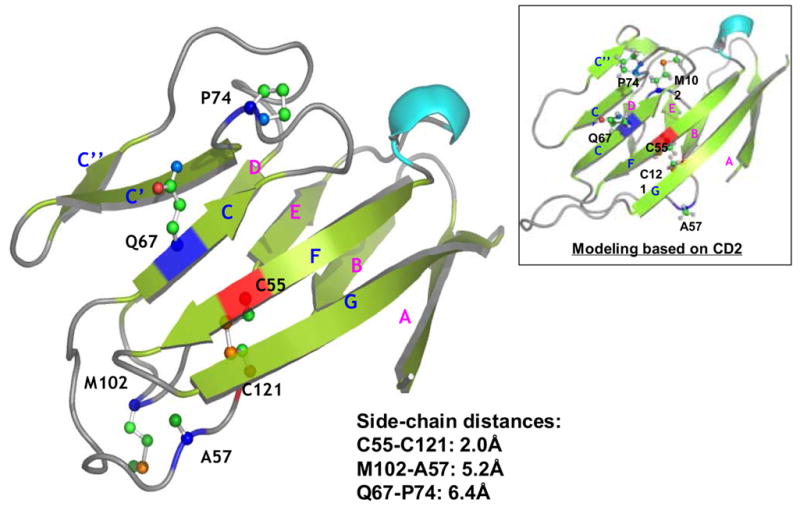
Computational homology modeling of SIRPα (Bit) extracellular IgV domain. A) Sequence alignment of SIRPα (Bit) with CD4. The predicted secondary structural elements, α-helices and βsheets, are shown as rods and arrows, respectively (“*”, identical residues; “:”, highly homologous residues; “.”, homologous residues). B) The predicted three-dimensional model of the Bit IgV domain based on the structures of CD4. The putative disulfide bond in the IgV structure is linked by the corresponding cysteine residues C55 and C121 (red). Q67 is located in the C β-sheet strand and A57 in the loop between the B and C strands. Met102 is predicted in the flexible loop between strands D and E. Another potential residue, Pro74, is predicted in the loop between strands of C and C’. The distances between the side-chains of individual residues are listed. Figure inset shows the result of protein modeling based on the structure of CD2 revealing another possible position of Met102 within the flexible D–E loop.
Function of Ala/Val57 in SIRPα IgV
We also further analyzed the function of Ala/Val57 in SIRPα IgV binding to CD47. In addition to changing this residue to Met, we substituted this residue with various other amino acids. As shown in Figure 4, changing Ala57 to structural similar hydrophobic residues, such as Leu (A57L), completely retained Bit.IgV binding to CD47. Changing Ala57 to other hydrophobic residues, including Phe (A57F), Pro (A57P), Typ (W), but not Met (A57M), partially maintained Bit IgV binding ability (Fig. 4). However, replacing Ala57 with charged or polar residues, such as Glu (A57E), Arg (A57R), Thr (A57T), Ser (A57S) and Tyr (A57Y) diminished Bit.IgV binding to CD47 (Fig. 4). As observed in the same assays, no binding was detected after changing of Ala57 to Gly (A57G), suggesting a side chain at this position is required for SIRPα IgV domain binding interaction with CD47.
Figure 4.
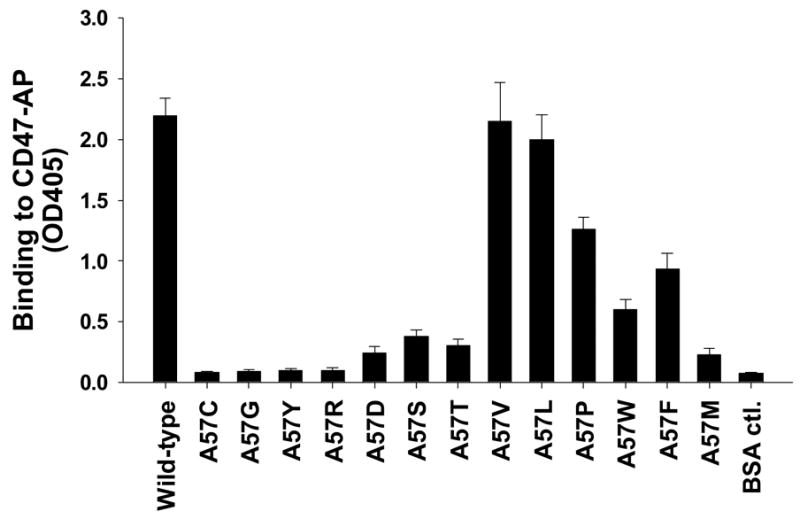
Mutagenesis study of Ala57 in Bit IgV. Residue Ala57 in Bit IgV was changed to various amino acids as described in the Methods. After DNA sequencing confirmed the amino acid changes, the mutant Bit.IgV-Fc fusion proteins were expressed in COS cells, purified and tested for binding to CD47-AP.
Ala/Val57 and Gln67 are critical in SIRPα-CD47 interaction-mediated cell adhesion and migration
Given that SIRPα-CD47 binding mediated cell surface interactions play an essential role in leukocyte recognition, phagocytosis and other functions 2; 3; 4; 18; 21, we examined the role of identified key residues in the SIRPα extracellular IgV domain in mediating cell adhesion via SIRPα extracellular binding interactions with CD47. In these experiments, wild-type and mutant SIRP IgV fusion proteins were immobilized on microtiter plate well surfaces. Cells with CD47 expressing on the cell surfaces, including peripheral blood mononuclear leukocytes (PBMC), neutrophil-like differentiated HL-60 cells, red blood cells (RBC), intestinal epithelial cells (HT-29) and CD47-transfected CHO (CHO-CD47) cells, were tested for the ability of adhesion. As shown in the microimages in Figure 5A, PBMC, differentiated HL-60 cells and RBC all demonstrated remarkable binding / adhesion to wild-type Bit.IgV and SIRPα1.IgV fusion proteins. Significant adhesion to wild-type SIRPα fusion proteins was also observed for HT29 and CHO-47 cells. On the contrast, no cell adhesion to SIRPβ1.IgV was observed (Fig. 5). In addition, cells without CD47 expressing (CHO) also displayed no adhesion (Fig. 5). Compared to differentiated HL-60 cells that readily adhered to Bit IgV coated surfaces, un-differentiated HL60 cells, which expressed less amount of CD47, exhibited decreased adhesion (data not shown). Thus, these results demonstrated that SIRPα IgV directly interacts with cell surface CD47 and mediates cell adhesion. As shown in Figure 5B, specificity of cell adhesion via SIRPα-CD47 interactions was also confirmed by including inhibitory anti-SIRPα (SE5A5) or anti-CD47 (C5D5) antibodies in the assays.
Figure 5.
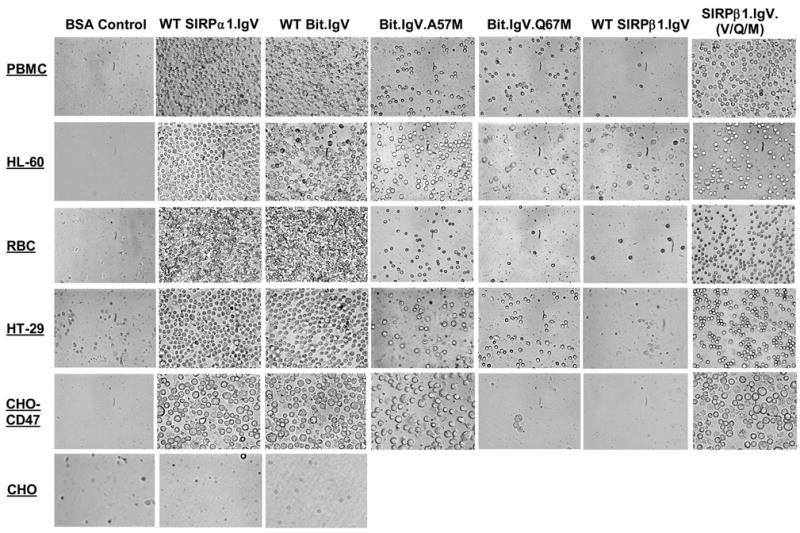
SIRPα-CD47 interactions mediate cell adhesion. In these experiments, microtiter plate wells were coated with wild-type and mutant SIRPα/β1.IgV-Fc fusion proteins (5μg/ml of each in HBSS), or control BSA (1% in HBSS). Suspensions of PBMC, neutrophil-like differentiated HL-60 cells, RBC, HT29 epithelial cells, CD47-transfected CHO cells (CD47-CHO) and control CHO cells were pre-loaded with the fluorescence dye BCECF (Molecular Probe) and allowed to adhere to the protein-coated wells (at 1–5x105 cells / per well), in the presence or absence of mAbs (20μg/ml of each) against CD47 (C5D5) or SIRPα (SE5A5), for 30min at 4ºC. After gentle washing to remove non-adherent cells, the adherent cells were imaged by a CCD camera under a microscope with a 40x projector (A). Cell adhesion to the wells was also analyzed by recording the fluorescence intensity using a fluorescence plate reader before and after washing. This figure (B) selectively presents the data of PBMC adhesion while similar cell adhesion results were obtained using differentiated HL-60 cells, RBC, HT29 cells, CD47-transfected CHO cells.
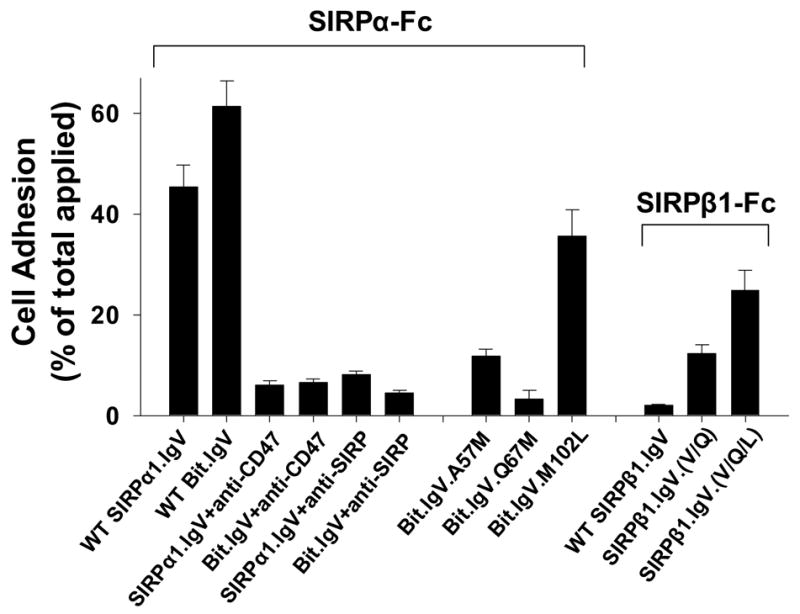
As also shown in Fig. 5A, in the same cell adhesion experiments, all tested cells demonstrated significantly decreased adhesion to A57M or Q67M mutant SIRPα IgV fusion proteins. As shown in Figure 5B, compared to that 45–60% of applied PBMC directly bound to immobilized wild-type SIRPα IgV fusions (46.4±4.2% and 61.1±6.1% of PBMC binding to SIRPα1.IgV-Fc and Bit.IgV-Fc, respectively), only less than 15% and 5% cells adhered to A57M and Q67M, respectively (Fig. 5B). These results thus confirmed our finding that Ala/Val57 and Gln67 are the key residues in the SIRPα extracellular IgV domain in mediating SIRPα binding interactions with CD47. Slightly decreased cell adhesion to mutant M102L (35.6±4.4%) was also observed (Fig. 5B), while no cell adhesion decrease when using other SIRPα IgV mutants comparing to wild-type (not shown).
Since T84 intestinal epithelial cells form monolayers with characteristic apical and basolateral polarization when grown on the transfilter supports 22; 23; 24 and previous studies showed that CD47 distributes along the basolateral surfaces of the monolayers 24, we thus tested wild-type and mutant SIRPα IgV fusion proteins for the ability of directly binding to cell surface CD47 on functional epithelial monolayers. In these experiments, SIRP IgV fusion proteins were incubated with intact T84 monolayers. After washing, the bound proteins to the monolayers were detected with a fluorescence-conjugated antibody against the Fc. As shown in Figure 6, wild-type SIRPα1.IgV and Bit.IgV fusion proteins directly labeled T84 monolayers. In particular, the fluorescence labeling patterns by these proteins were specific to the basolateral surfaces and indistinguishable from the labeling by anti-CD47 mAb C5D5 (Fig. 6). As shown, including of soluble CD47 (CD47-AP) or anti-CD47 mAb (not shown) in the labeling inhibited wild-type SIRPα IgV binding to the monolayers, confirming these proteins binding via interactions with basolateral surface CD47. As to our prediction, mutations of SIRPα IgV residues essential for CD47 binding interactions, Gln67 (Q67M) and Ala57 (A67M), resulted in diminishing of labeling to the monolayers (Fig. 6, lower panel). In addition to label epithelial monolayers, we also performed experiments to label leukocyte cell surfaces using these fusion proteins without cell permeablization. Figure 6, lower panel, shows wild-type and mutant SIRPα IgV labeling on differentiated HL-60 cells indicating that mutations at Gln67 (Q67M) and Ala57 (A67M) inhibited SIRPα IgV binding.
Figure 6.
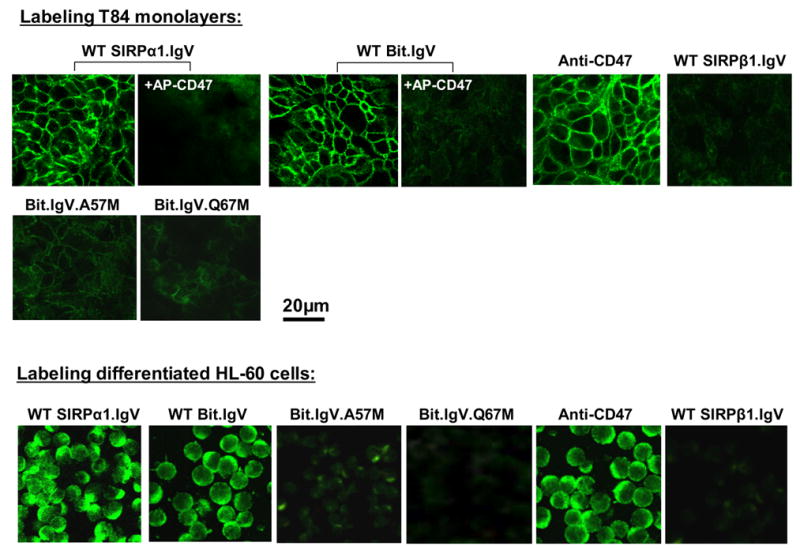
Cell surface labeling epithelial monolayers and HL-60 leukocytes by SIPRα.IgV-Fc fusion proteins. Upper panel: T84 epithelial monolayers were incubated with wild-type or mutant SIRP IgV fusion proteins as labeled in the figure, in the presence or absence of CD47-AP (5μg/ml), followed by detection using fluorescence-conjugated secondary antibody. For control staining, the monolayers were also labeled with anti-CD47 mAb C5D5 (10μg/ml), SIRPβ1.IgV-Fc or Fc only (not shown). Lower panel: similar labeling procedures were also performed to analyze wild-type and mutant SIRPα.IgV-Fc binding to differentiated HL-60 cell surfaces. Images shown represent one of at least three individual experiments, with multiple images taken per slide.
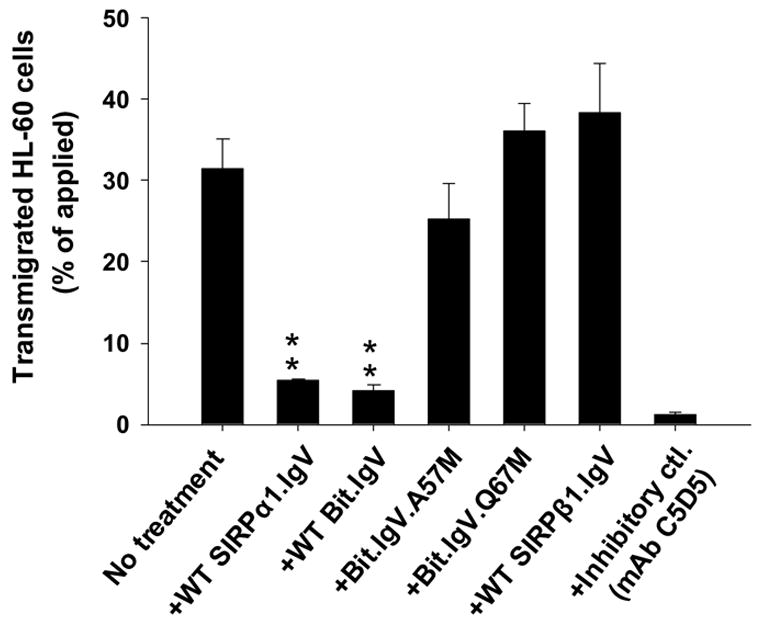
Given the previous studies demonstrating that cell surface CD47-SIRPα interaction plays an important role in modulating leukocyte transmigration, we further examined the critical role of SIRPα IgV domain Ala/Val57 and Gln67 by leukocyte transmigration assays. In these experiments, neutrophil-like differentiated HL-60 cells were induced to transmigrate across collagen-coated permeable filters towards chemoattractant fMLP (1μM) using a well-established transwell system 5; 22; 25. As shown in Figure 7, compared to the control in the absence of fusion protein (no treatment) or with wild-type SIRPβ1.IgV-Fc (40μg/ml) that significant amount of HL-60 cells (30–40% of applied) transmigrated after 1hr, the presence of wild-type SIRPα IgV fusion proteins, SIRPα1.IgV and Bit.IgV, resulted in decreased HL-60 cell transmigration, with only minimal cells (<6%) migrating across after the same time period. This result is not surprising given that wild-type SIRPα IgV is capable of binding to cell surface CD47 (Fig. 5) and previous studies indicating that ligation of cell surface CD47 inhibited leukocyte migration 22. As also can been seen, mutating SIRPα (Bit) residues Ala57 (A57M) and Gln67 (Q67M), which abolishes the extracellular IgV domain binding to CD47, diminished the inhibitory effects by soluble SIRPα IgV on HL-60 cell migration. This result further confirmed the key role of SIRPα residues Ala/Val57 and Gln67 in SIRPα–CD47 cell surface binding mediated leukocyte function.
Figure 7.
Effects of soluble SIRPα IgV fusion proteins in leukocyte transmigration.
Leukocyte transmigration across matrix (collagen)-coated filters (transwell) using neutrophil-like differentiated HL-60 cells were performed as described in the methods. In these experiments, HL-60 cells (2.5x106) in the upper chambers of the transwells in 150μl HBSS with or without SIRP fusion proteins (40μg/ml), or mAbs (20μg/ml) against CD47 (C5D5) or SIRPα (SE5A5), were induced to migrate into fMLP (1μM) containing lower chambers at 37ºC. Cells that migrated into the lower chamber after 1hr were quantified by MPO assay. Data represents one of three experiments with three individual transwells in each condition + SD.
SIRPβ1 can be converted to be a CD47-binding protein after introducing Ala57, Gln67 and M102
As shown in Figure 2, SIRPβ1.IgV had no binding to CD47 even at high concentrations (>50μg/ml, not shown). In addition, SIRPβ1.IgV also failed to mediate cell adhesion (Fig. 5) and had no inhibition in leukocyte migration (Fig. 7). However, it is somewhat surprising that only two residues (Ala/Val57 and Gln67) on the IgV domain determine SIRP extracellular specific binding to CD47. Given the overall high similarities of the IgV structures between SIRPα and SIRPβ1, we wondered whether SIRPβ1 could be converted to a CD47-binding protein by introducing the identified functional essential amino acids in SIRPα. Site-directed mutagenesis was performed and Met57 and Met67 in SIRPβ1.IgV were changed to the corresponding SIRPα residues Val and Gln at these positions, respectively (see Figure 1A). As shown in Figure 8, changing these residues individually did not convert SIRPβ1.IgV into a CD47-binding protein. Double mutations of Met57 and Met67 to Val and Gln (mutant M57V/M67Q) resulted in slight SIRPβ1.IgV binding to CD47 (Fig. 8). Since our data also suggested a role of Met102 in SIRPα binding (Fig. 2), we further mutated Leu102 in SIRPβ1 to Met and created a M57V/M67Q/L102M triple mutant. As shown in Figure 8, M57V/M67Q/L102M triple mutant dramatically converted SIRPβ1 into a CD47-binding protein that directly bound to CD47-AP (Fig. 8). The capability of M57V/M67Q/L102M triple mutant binding to CD47 was also demonstrated by cell adhesion assays shown in Figure 5. As can be seen, compared to hardly any cell adhering to SIRPβ1.IgV-coated surfaces, increased cell adhesion were found when SIRPβ1.IgV with M57V/M67Q/L102M (SIRPβ1.IgV.(V/Q/L), Fig. 5A&B) were used. Figure 8 inset shows the quantities of HT-29 cell adhesion to SIRPβ1.IgV mutants comparing to wild-type SIRPβ1.IgV protein. However, as can be seen in Figure 8 and Figure 5, although M57V/M67Q/L102M triple mutant of SIRPβ1.IgV directly bound to CD47, it had lower affinity compared to wild-type Bit.IgV-Fc. Thus, additional structural elements are required for SIRPβ1 IgV fully mimicking SIRPα-specific conformation and function. Another triple mutant, M57V/M67Q/A74P, had only trivial effect in SIRPβ1 binding to CD47 (Fig. 8).
Figure 8.
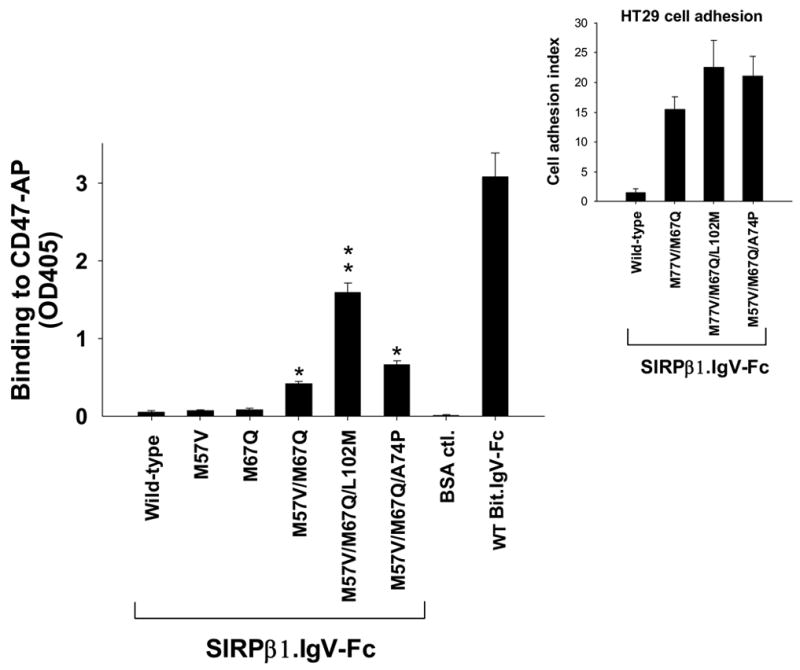
Engineering SIRPβ1 to be a CD47-binding protein. Mutagenesis reactions were performed in the SIRPβ1 IgV loop to replace SIRPβ1-specific residues with the counter residues in SIRPα (also see Fig. 1). These changes in SIRPβ1.IgV included Met57 to Val, Met67 to Gln, Leu102 to Met and Ala74 to Pro with single, double and triple mutations as indicated in the figure. The binding of these mutant SIRPβ1.IgV-Fc fusion proteins to CD47-AP were then assayed and compared to minimal binding of wild-type SIRPβ1.IgV-Fc (**, p≤0.01; *, p≤0.05). Figure inset shows the results of HT-29 cell adhesion (% of applied) to wild-type and mutant SIRPβ1.IgV-Fc fusion proteins. Adhesion results of other cells were also shown in Figure 5A & B.
Discussion
SIRPs are a family of transmembrane proteins comprising multiple subfamilies that are predominantly expressed on leukocytes, including granulocytes and mononuclear cells 21; 26; 27. Expression of SIRPs in cancer cells and neuronal cells has also been reported 7; 14; 28; 29. Among SIRP proteins, SIRPα was identified to be a counter extracellular receptor for CD47 and growing evidence has demonstrated the vital role of SIRPα-CD47 binding interactions in leukocyte-mediated cell functions. For example, studies showed that macrophages recognize phagocytic targets, such as apoptotic cells during embryonic development or opsonized RBCs in autoimmune anemia, via macrophage surface SIRPα trans interactions with CD47 on the target cells 18; 30. SIRPα-CD47 interactions were also shown to regulate neutrophil and monocyte transmigration across tissues 4; 5. Although there may be other SIRPβ isoforms, SIRPβ1 is the major isoform found in human leukocytes 1 and it shares a high extracellular structural homology with SIRPα. However, unlike SIRPα, SIRPβ1 does not bind to CD47 5. Furthermore, cell surface interactions mediated by SIRPα and SIRPβ1 extracellular domains elicit divergent signal transduction pathways in cells. SIRPα binding to CD47 activates the intracellular immunotyrosine inhibitory motifs (ITIMs) and mediates inhibitory cellular responses 10. Conversely, ligation of SIRPβ1 delivers activating cell signals through binding to the immunotyrosine activating motif (ITAM)-containing adaptor proteins 12; 13.
As evident by recent research, SIRPα and β1 are commonly co-expressed on the same cell surfaces of neutrophils and mononuclear cells 1. Given that these proteins share strikingly similar extracellular domain structures with highly homologous amino acid sequences 10, it is essential for these co-expressed SIRP isoforms to exploit their limited extracellular structural differences to discrete individual extracellular ligands. As shown in Figure 1, comparing the primary structures of CD47-binding IgV domains 5; 15 of multiple SIRPα members with that of the IgV of SIRPβ1 only found seven amino acid differences. This suggests that these few unique amino acids likely form a functional motif on SIRPα protein surface that mediates specific binding interaction with CD47. Studies of these unique amino acid residues in SIRP isoforms would also explain, in part, the striking differences of the extracellular binding between SIRPα and β subfamilies.
In this study, we performed site-directed mutagenesis and studied in detail the seven unique residues found in the CD47-binding SIRPα IgV domain. To our surprise, our results revealed that only 3 amino acids, including Ala/Val57, Gln67 and Met102, are essential for SIRPα extracellular binding interaction with CD47. In particular, we found that Gln67 is the key and mutation of this residue to the counter amino acid in SIRPβ1 completely abrogated SIRPα IgV binding to CD47 or CD47-expressing cells. Further replacing Gln67 with any other amino acids revealed interesting feature of this residue. It appears that changing this to the similar side-chain residues Asn and Glu, and (to our surprise) non-polar amino acids Val and Ile, completely or partially maintained SIRPα IgV binding to CD47 (Fig. 3). It is also surprising to observe that, although Val and Ile at this position supported SIRPα IgV interactions with CD47, Leu, as well as SIRPβ1-specific Met completely inhibited SIRPα IgV binding. We thus surmise that a short and “tight” side-chain structure as that in Val and Ile may be essential for SIRPα IgV binding interaction.
The second important residue that we identified in this study is a small hydrophobic amino acid located at the 57th position of SIRPα IgV. This residue is either Ala or Val in all the SIRPα members10. Replacement of this residue with the SIRPβ1 counter residue also diminished SIRPα IgV binding to CD47. Further studies by replacing the residue with other amino acids found that, at this position (the 57th), the hydrophobic property and the side-chain length / shape are important for SIRPα IgV binding function. As shown in Fig. 4, changing this residue to any polar or charged residues blocked SIRPα IgV binding to CD47 while replacing this residue (Ala or Val) to amino acids with similar short or small, “branched”, hydrophobic side-chains, such as Leu, Phe and Pro, completely or partially maintained SIRPα IgV binding to CD47. Conversely, changing to the hydrophobic residues with long and “bulky” side-chains such as Trp resulted in inhibition of the binding. Along this line, it is interesting to note that both of these key residues in SIRPα, Gln67 and Ala/Val57, are Met in SIRPβ1 IgV. Since Met has a relatively long, “narrow” and “protrusion” side-chain, it may impose a specific feature into the SIRPβ1 IgV domain that impedes the binding interactions with CD47. The unique thiol (S) group in Met side-chain may also play a role in this regard.
In addition to Gln67 and Ala/Val57, SIRPα IgV-specific Met102 also significantly contributes to its binding interaction. Although changing Met102 to the SIRPβ1-specific Leu (M102L) only moderately affected SIRPα binding to CD47 (Fig. 2A), constitution of this amino acid into the IgV domain of SIRPβ1 in addition to Gln67 and Ala57 dramatically converted this non-CD47 binding SIRP to bind to CD47 (Fig. 8). Thus, although Met102 may not directly bridge the key interactions between SIRPα and CD47, it contributes to the essential structural motif on SIRPα IgV for binding interaction to occur.
Since the sequence alignment of the SIRPα IgV with IgV domains from several other Ig superfamily proteins revealed that SIRPα IgV shares characteristic sequence criteria similar to other IgV members (result nort shown) 5; 31, it is thus possible to predict the three-dimensional SIRPα IgV model based on other known IgV structures. Figure 9A shows the predicted secondary structural elements including α helices and β sheets in Bit IgV and the homology modeling of the Bit IgV based on the structure of CD4 IgV domain 32; 33; 34. As can be seen in Figure 9B, similar to other IgVs, Bit IgV comprises two layers of antiparallel β-strand sheets with strands C, C’, C” (part), F and G located on one layer and strands A, B, C” (part), D and E on the other layer. The two Cys residues, Cys55 and Cys121, which form a disulfide bond in the IgV structure, are located in close proximity. As predicted by this model, Gln67 is in the C strand β sheet with the side-chain facing to the protein surface. Since previous studies of other IgV proteins suggest that the homophilic and heterophilic interactions of IgV are localized to the layer containing G, F and C strands 32; 33; 35; 36, it is thus highly possible that Gln67 in the C strand mediates direct interaction through the protruding side-chain. The Ala57 is predicted to be in the loop connecting the B and C strands, which is, however, distant from Gln67 but is adjacent to the disulfide bond. Thus, the side-chain of Ala57 may influence the SIRPα-specific IgV conformation and stability. Met102 is localized in the loop between strands D and E. From the structure, this loop appears highly dynamic and may situate Met102 close to Ala57 (5.2Å). However, this loop may also lay inside as a “closed” position that situates Met102 near by Gln67 (as predicted based on CD2 IgV (Fig. 9, inset)). We speculate that Met102 can be even closer to Gln67 or Ala/Val57 during SIRPα binding to CD47, given the flexible nature of the loop.
In summary, this study has identified for the first time the key amino acid elements in the SIRPα extracellular IgV that are essential for SIRPα extracellular binding interaction with CD47. In addition, as shown by our study, these key residues play a fundamental role in SIRPα-CD47 interaction mediated cell adhesion and leukocyte transmigration. Thus, these results shed new light on a molecular basis of how structurally similar SIRP isoforms discern their individual extracellular ligands through their unique IgV domains. Given the vital role of SIRPα-CD47 interactions in innate immune responses, future studies combined with crystal structure information are required for further understanding these protein interactions on the leukocyte surfaces.
Materials and Methods
Antibodies, chemicals and cells
Monoclonal anti-SIRPα antibodies SE5A5 and SE7C2, anti-SIRPβ1 mAb B4B6 and inhibitory anti-CD47 mAb C5D5 were used previously 1; 5; 16. Mouse polyclonal antibody against the extracellular domain of SIRPα (anti-SIRPα.ex) was generated previously 1. Goat anti-rabbit Fc specific antibody, mouse anti-alkaline phosphatase (AP) antibody and antibody-conjugated agarose and the AP substrate p-nitrophenyl phosphate were all purchased from Sigma. The human intestinal epithelial cells T84 were grown in DMEM/F-12 medium supplemented with 5% fetal bovine serum. Another human intestinal epithelial cell line HT29 was maintained in DMEM medium supplemented with 10% fetal bovine serum. These cells were also grown on collagen-coated permeable filters (3μm pore size; Fisher Scientific) to generate epithelial monolayers as described previously 37. Human peripheral blood mononuclear cells (PBMC) and red blood cells were obtained from healthy donors following a protocol described previously 25. Promyelocytic leukemic cell line HL-60 were obtained from American Tissue Culture Collection (ATCC) and maintained in IMDM medium containing 20% fetal bovine serum. To differentiate HL-60 cells into granulocytic lineage, HL-60 cells were cultured for 6 days in the same medium supplemented with 1.25% DMSO 38; 39. After differentiation, these cells were capable of chemotaxis in a similar pattern as that for neutrophils.
Recombinant SIRPα and SIRPβ1
SIRPα has several subfamily isoforms including Bit, SIRPα1 and others. The extracellular domains of the SIRPα isoform Bit and SIRPβ1 were RT-PCR amplified from human neutrophils as described previously 40. Plasmid consisting the DNA sequences encoding the membrane distal-most IgV loop of SIRPα1 extracellular domain and rabbit Fc (pCDNA3.1. SIRPα1.IgV-Fc) was generated previously 15. Similar strategies were used to generate Fc fusion constructs containing the IgV domains of Bit and SIRPβ1 (pCDNA3.1.Bit.IgV-Fc and pCDNA3.1.SIRPβ1.IgV-Fc, respectively). To express Fc fusion proteins containing Bit.IgV, SIRPα1.IgV and SIRPβ1.IgV, plasmids were transiently transfected into COS cells using a standard DEAE-dextran method 41. Cell medium was harvested at day 3, 5, 7, 9 and 11 post-transfection and SIRP-Fc chimeras were purified using protein-A Sepharose. Purified fusion proteins were then examined by anti-rabbit Fc antibody and multiple monoclonal antibodies against the SIRPα and β1 extracellular domains.
Recombinant CD47 extracellular domain fusion protein (CD47-AP)
Recombinant CD47 extracellular domain fused to alkaline phosphatase (AP) was produced as described previously 5. The CD47-AP in the medium was affinity purified using anti-AP agarose and eluted at pH10.5 for use in CD47-SIRP binding assays.
Site-directed mutagenesis of SIRPα-specific amino acid residues
Site-directed mutagenesis reactions were performed using the GeneTailor Site-Directed Mutagenesis System (Invitrogen). Wild-type Bit.IgV-Fc or SIRRPα1.IgV-Fc in pCDNA3.1 were used as the templates for mutagenesis reactions. Glu32 was mutated to Asp using the forward primer 5’tcaggagtggcgggtgaggatgagctgcagg and reverse primer 5’tcctcacccgccactcctgaccaggcgcag. Asp40 was mutated to Glu using the forward primer 5’ctgcaggtgattcagcctgaaaagtccgtgt and the reverse primer 5’tcaggctgaatcacctgcagctcctcctca. Leu44 was mutated to Ser using the forward primer 5’cagcctgacaagtccgtgtcggttgcagct and the reverse primer 5’acacggacttgtcaggctgaatcacctgca. Thr56 was mutated to Ala using the forward primer 5’gagtcggccattctgcactgcgctgtgacctc and the reverse primer 5’gcagtgcagaatggccgactctccagctgca. Ala57 in Bit was mutated to Met using the forward primer 5’acagccactctgcgctgcactatgacctctctg and the reverse primer 5’agtgcagcgcagagtggctgtctctccagct. Ala57 was also mutated to other amino acids using a degenerate forward primer 5’acagccactctgcgctgcactnnnacctctctgatccct. Val57 in SIRPα1 was mutated to Met using the forward primer 5’ acagccactctgcgctgcactatgacctctctg and the same reverse primer as was used for Ala57. Gln67 was mutated to Met using the forward primer 5’tgatccctgtggggcccatcatgtggttcaga and the reverse primer 5’gatgggccccacagggatcagagaggtcg. Pro74 was mutated to Ala using the forward primer 5’agtggttcagaggagctggagcagcccgggaa and the reverse primer 5’tccagctcctctgaaccactggatgggccc. Met102 was mutated to Leu using the forward primer 5’gacctcacaaagagaaacaacctggacttttcc and the reverse primer 5’gttgtttctctttgtgaggtctgaaacagtt. To further elucidate the role of Gln67, this residue was further mutated to other amino acids using a degenerate forward primer 5’tgatccctgtggggcccatcnnntggttcagaggagct with the reverse primer 5’gatgggccccacagggatcagagaggtcg. Mutated plasmids were confirmed by DNA sequencing and at least two clones of each mutant were used to transfect into COS cells and the resulting SIRPα-Fc fusion proteins were purified and analyzed for binding to antibodies and CD47-AP.
Site-directed mutagenesis of SIRPβ1-specific residues
To change the unique residues in the SIRPβ1 IgV domain to their corresponding amino acids in SIRPα, mutagenesis reactions were performed using the wild-type SIRPβ1.IgV-Fc in pCDNA3.1 as the template. Met57 was changed to Val using the forward primer 5’cggccactctgcgctgtgctgtgacgtcc and the reverse primer 5’agcacagcgcagagtggccgactctccag. Met67 was changed to Glu using the forward primer 5’tgatccctgtggggcccatccagtggtttaga and the reverse primer 5’gatgggccccacagggatcagggacgtca. Sequential changes of Met57 to Val and Met67 to Glu on the same template were also performed to create M57V/M67Q (VQ) double mutations. Additional mutation of Leu102 to Met was performed using forward primers 5’ctcacaaagagaaacaacatggacttttcc and the reverse primer 5’gttgtttctctttgtgagttctgaaac to generate M57V/M67Q/L102M (VQL) triple mutations. Ala74 was also mutated to Pro using the forward primers 5’tggtttagaggagctggaccaggccgggaa and the reverse primer 5’tccagctcctctaaaccacatgatggg to construct M57V/M67Q/A74P (VQP) triple mutations.
In vitro CD47-SIRP binding assays
The assays were performed as described previously 5. Briefly, 96-well microtiter plate wells were coated with SIRP-Fc fusion proteins (2–5μg/ml each) for 2hr at 25ºC. After blocking with 1% BSA (30min, 25ºC), the wells were incubated with CD47-AP (2–5μg/ml) for 30min. After washing, CD47 binding to immobilized SIRP protein was detected by assaying alkaline phosphatase (AP) activity using the substrate p-nitrophenyl phosphate. To analyze the dose-dependent binding of SIRPα mutants with CD47, purified CD47-AP (5μg/ml) was used to coat 96-well microtiter plate. After blocking, the wells were incubated with wild-type or mutant SIRP-Fc fusion proteins at varied concentrations for 45min. After washing, SIRP-Fc binding was detected using peroxidase conjugated goat anti-rabbit Fc antibody.
Binding of SIRP-Fc to epithelial monolayers and leukocytes
T84 intestinal epithelial monolayers cultured on permeable transwells were washed with PBS and then treated with the low calcium medium SEM (Sigma) for 30 min to open cell-cell contacts42. Leukocytes (HL-60) were in suspension. After blocking nonspecific protein binding with 1% BSA in PBS for 30 min, these cells were incubated with wild-type and mutant SIRP.IgV-Fc fusion proteins (5–10μg/ml each) for 1 hr. For control, the monolayers / cells were also incubated with anti-CD47 mAb C5D5 to label CD47. Monolayers were then washed and fixed with 3.7% paraformaldehyde followed by incubatioin with Alexa Fluor 488-conjugated goat anti-mouse/rabbit IgG (Molecular Probe) followed by washing. After mounting in ProLong antifading embedding solution (Molecular Probes), the monolayers / cells were analyzed by fluorescence or confocal microscopy. To test whether binding of SIRPα-Fc to cell surface was through CD47 expressing on epithelial cells, labeling experiments were performed as above except in the presence of soluble CD47-AP (10μg/ml).
Cell adhesion assays
To test the binding of cell adhesion / binding to wild-type and mutant SIRP fusion proteins, microtiter plate wells were coated with purified SIRPα-Fc and SIRPβ1-Fc fusion proteins followed by blocking with 1% BSA. Suspension of cells were preloaded with BCECF (Molecular Probes) 43 and then incubated with the fusion protein-coated wells at a density of 1–5x105/ per well for 30min at 10ºC. After gently washing, adherent cells were imaged with a microscope equipped with a CCD camera and the fluorescence intensity was measured with a fluorescence plate reader (BioTek,) with the excitation/emission wavelengths of 485/535nm.
Leukocyte transmigration assays
Leukocyte transmigration assays were performed using DMSO-differentiated, neutrophil-like HL-60 cells and well-established transwell setups following a procedure described in our previous publications 22; 25. To be brief, HL-60 cells (2.5x106) were applied into the upper chambers of the transwell setups in the presence or absence of soluble SIRP fusion proteins (40μg/ml) and mAbs (20μg/ml). Cell transmigration across collagen-coated transwell filters was initiated by adding chemoattractant fMLP (1μM) into the lower chamber and incubation at 37ºC. Cell migration into the lower chambers after 1hr was quantified by myeloperoxidase (MPO) assays 22; 25.
Protein sequence analysis of SIRPα extracellular IgV loop and computational homology modeling
The sequence alignment of the SIRPα (Bit) extracellular IgV domain with homologous IgV domains from several Ig superfamily proteins was performed by the program ClustalW 44 with the gap-open penalty and gap-extension penalty set at 10 and 0.1, respectively. The secondary structure elements, α helices and β sheets, in Bit IgV were predicted based on the consensus analysis using computational programs JPRED, PHD and PSIPRED 33. The homology modeling of the Bit IgV structure was constructed using the comparative structure modeling program MODELLER 45 and the homology-modeling server SWISS-MODEL 46; 47. Rat CD4 extracellular domain IgV structure was used as the template for Bit IgV modeling based on the available high resolution CD4 IgV (PDB code: 3cd4) 34 structures. Modeling was primarily based on the sequence alignment and secondary structure similarities with manual insertion of gaps to align the pairwise sequences according to the protein secondary structures.
Acknowledgments
We thank Ms. Ileana Soto for technique support during initiation of this work. We also thank Ms. Ranita A. Williams for proof reading of the manuscript. This work was supported, in part by NIH grants DK62894, a Beginning Grant-in-Aid (Y. Liu) and a Scientist Development Grant (K. Zen) from American Heart Association.
The abbreviations used are
- SIRP
signal regulatory protein
- IgV
immunoglobulin variable domain
- AP
alkaline phosphatase
- HBSS
Hank's balanced salt solution
- HBSS(−)
Hank's balanced salt solution devoid of Ca2+ and Mg2+
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Y, Soto I, Tong Q, Chin A, Buhring HJ, Wu T, Zen K, Parkos CA. SIRPbeta1 is expressed as a disulfide-linked homodimer in leukocytes and positively regulates neutrophil transepithelial migration. J Biol Chem. 2005;280:36132–40. doi: 10.1074/jbc.M506419200. [DOI] [PubMed] [Google Scholar]
- 2.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells [see comments] Science. 2000;288:2051–4. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 3.Saginario C, Sterling H, Beckers C, Kobayashi R, Solimena M, Ullu E, Vignery A. MFR, a putative receptor mediating the fusion of macrophages. Mol Cell Biol. 1998;18:6213–23. doi: 10.1128/mcb.18.11.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Vries HE, Hendriks JJ, Honing H, De Lavalette CR, van der Pol SM, Hooijberg E, Dijkstra CD, van den Berg TK. Signal-regulatory protein alpha-CD47 interactions are required for the transmigration of monocytes across cerebral endothelium. J Immunol. 2002;168:5832–9. doi: 10.4049/jimmunol.168.11.5832. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Buhring HJ, Zen K, Burst SL, Schnell FJ, Williams IR, Parkos CA. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J Biol Chem. 2002;277:10028–36. doi: 10.1074/jbc.M109720200. [DOI] [PubMed] [Google Scholar]
- 6.Chen TT, Brown EJ, Huang EJ, Seaman WE. Expression and activation of signal regulatory protein alpha on astrocytomas. Cancer Res. 2004;64:117–27. doi: 10.1158/0008-5472.can-3455-2. [DOI] [PubMed] [Google Scholar]
- 7.Kapoor GS, Kapitonov D, O'Rourke DM. Transcriptional regulation of signal regulatory protein alpha1 inhibitory receptors by epidermal growth factor receptor signaling. Cancer Res. 2004;64:6444–52. doi: 10.1158/0008-5472.CAN-04-0256. [DOI] [PubMed] [Google Scholar]
- 8.Seiffert M, Cant C, Chen Z, Rappold I, Brugger W, Kanz L, Brown EJ, Ullrich A, Buhring HJ. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood. 1999;94:3633–43. [PubMed] [Google Scholar]
- 9.van den Berg TK, van Beek EM, Buhring HJ, Colonna M, Hamaguchi M, Howard CJ, Kasuga M, Liu Y, Matozaki T, Neel BG, Parkos CA, Sano S, Vignery A, Vivier E, Wright M, Zawatzky R, Barclay AN. A nomenclature for signal regulatory protein family members. J Immunol. 2005;175:7788–9. doi: 10.4049/jimmunol.175.12.7788. [DOI] [PubMed] [Google Scholar]
- 10.Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. 1997;386:181–6. doi: 10.1038/386181a0. [DOI] [PubMed] [Google Scholar]
- 11.Olcese L, Cambiaggi A, Semenzato G, Bottino C, Moretta A, Vivier E. Human killer cell activatory receptors for MHC class I molecules are included in a multimeric complex expressed by natural killer cells. J Immunol. 1997;158:5083–6. [PubMed] [Google Scholar]
- 12.Dietrich J, Cella M, Seiffert M, Buhring HJ, Colonna M. Cutting edge: signal-regulatory protein beta 1 is a DAP12-associated activating receptor expressed in myeloid cells. J Immunol. 2000;164:9–12. doi: 10.4049/jimmunol.164.1.9. [DOI] [PubMed] [Google Scholar]
- 13.Tomasello E, Cant C, Buhring HJ, Vely F, Andre P, Seiffert M, Ullrich A, Vivier E. Association of signal-regulatory proteins beta with KARAP/DAP-12. Eur J Immunol. 2000;30:2147–56. doi: 10.1002/1521-4141(2000)30:8<2147::AID-IMMU2147>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Jiang P, Lagenaur CF, Narayanan V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J Biol Chem. 1999;274:559–62. doi: 10.1074/jbc.274.2.559. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, O'Connor MB, Mandell KJ, Zen K, Ullrich A, Buhring HJ, Parkos CA. Peptide-mediated inhibition of neutrophil transmigration by blocking CD47 interactions with signal regulatory protein alpha. J Immunol. 2004;172:2578–85. doi: 10.4049/jimmunol.172.4.2578. [DOI] [PubMed] [Google Scholar]
- 16.Seiffert M, Brossart P, Cant C, Cella M, Colonna M, Brugger W, Kanz L, Ullrich A, Buhring HJ. Signal-regulatory protein alpha (SIRPalpha) but not SIRPbeta is involved in T-cell activation, binds to CD47 with high affinity, and is expressed on immature CD34(+)CD38(−) hematopoietic cells. Blood. 2001;97:2741–9. doi: 10.1182/blood.v97.9.2741. [DOI] [PubMed] [Google Scholar]
- 17.Vernon-Wilson EF, Kee WJ, Willis AC, Barclay AN, Simmons DL, Brown MH. CD47 is a ligand for rat macrophage membrane signal regulatory protein SIRP (OX41) and human SIRPalpha 1. European Journal of Immunology. 2000 Aug;30:2130–7. doi: 10.1002/1521-4141(2000)30:8<2130::AID-IMMU2130>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 19.Latour S, Tanaka H, Demeure C, Mateo V, Rubio M, Brown EJ, Maliszewski C, Lindberg FP, Oldenborg A, Ullrich A, Delespesse G, Sarfati M. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J Immunol. 2001;167:2547–54. doi: 10.4049/jimmunol.167.5.2547. [DOI] [PubMed] [Google Scholar]
- 20.Hope JC, Sopp P, Collins RA, Howard CJ. Differences in the induction of CD8+ T cell responses by subpopulations of dendritic cells from afferent lymph are related to IL-1 alpha secretion. J Leukoc Biol. 2001;69:271–9. [PubMed] [Google Scholar]
- 21.Brooke G, Holbrook JD, Brown MH, Barclay AN. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol. 2004;173:2562–70. doi: 10.4049/jimmunol.173.4.2562. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Merlin D, Burst SL, Pochet M, Madara JL, Parkos CA. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem. 2001;276:40156–66. doi: 10.1074/jbc.M104138200. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113 ( Pt 13):2363–74. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- 24.Parkos CA, Colgan SP, Liang TW, Nusrat A, Bacarra AE, Carnes DK, Madara JL. CD47 mediates post-adhesive events required for neutrophil migration across polarized intestinal epithelia. Journal of Cell Biology. 1996;132:437–50. doi: 10.1083/jcb.132.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zen K, Reaves TA, Soto I, Liu Y. Response to genistein: Assaying the activation status and chemotaxis efficacy of isolated neutrophils. J Immunol Methods. 2006;309:86–98. doi: 10.1016/j.jim.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Veillette A, Thibaudeau E, Latour S. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. Journal of Biological Chemistry. 1998;273:22719–28. doi: 10.1074/jbc.273.35.22719. [DOI] [PubMed] [Google Scholar]
- 27.Adams S, van der Laan LJ, Vernon-Wilson E, Renardel de Lavalette C, Dopp EA, Dijkstra CD, Simmons DL, van den Berg TK. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. Journal of Immunology. 1998;161:1853–9. [PubMed] [Google Scholar]
- 28.Mi ZP, Jiang P, Weng WL, Lindberg FP, Narayanan V, Lagenaur CF. Expression of a synapse-associated membrane protein, P84/SHPS-1, and its ligand, IAP/CD47, in mouse retina. J Comp Neurol. 2000;416:335–44. [PubMed] [Google Scholar]
- 29.Yan HX, Wang HY, Zhang R, Chen L, Li BA, Liu SQ, Cao HF, Qiu XH, Shan YF, Yan ZH, Wu HP, Tan YX, Wu MC. Negative regulation of hepatocellular carcinoma cell growth by signal regulatory protein alpha1. Hepatology. 2004;40:618–28. doi: 10.1002/hep.20360. [DOI] [PubMed] [Google Scholar]
- 30.Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med. 2001;193:855–62. doi: 10.1084/jem.193.7.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaughn DE, Bjorkman PJ. The (Greek) key to structures of neural adhesion molecules. Neuron. 1996;16:261–73. doi: 10.1016/s0896-6273(00)80045-8. [DOI] [PubMed] [Google Scholar]
- 32.Bodian DL, Jones EY, Harlos K, Stuart DI, Davis SJ. Crystal structure of the extracellular region of the human cell adhesion molecule CD2 at 2.5 A resolution. Structure. 1994;2:755–66. doi: 10.1016/s0969-2126(94)00076-x. [DOI] [PubMed] [Google Scholar]
- 33.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 34.Garrett TP, Wang J, Yan Y, Liu J, Harrison SC. Refinement and analysis of the structure of the first two domains of human CD4. J Mol Biol. 1993;234:763–78. doi: 10.1006/jmbi.1993.1625. [DOI] [PubMed] [Google Scholar]
- 35.Withka JM, Wyss DF, Wagner G, Arulanandam AR, Reinherz EL, Recny MA. Structure of the glycosylated adhesion domain of human T lymphocyte glycoprotein CD2. Structure. 1993;1:69–81. doi: 10.1016/0969-2126(93)90009-6. [DOI] [PubMed] [Google Scholar]
- 36.Osborn L, Vassallo C, Browning BG, Tizard R, Haskard DO, Benjamin CD, Dougas I, Kirchhausen T. Arrangement of domains, and amino acid residues required for binding of vascular cell adhesion molecule-1 to its counter-receptor VLA-4 (alpha 4 beta 1) J Cell Biol. 1994;124:601–8. doi: 10.1083/jcb.124.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–51. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed N, Weidemann MJ. Interaction of reactive nitrogen and oxygen intermediates in HL60 and dimethylsulphoxide-differentiated HL60 cells. Leuk Res. 1996;20:271–9. doi: 10.1016/0145-2126(95)00145-x. [DOI] [PubMed] [Google Scholar]
- 39.Woo CH, Yoo MH, You HJ, Cho SH, Mun YC, Seong CM, Kim JH. Transepithelial migration of neutrophils in response to leukotriene B4 is mediated by a reactive oxygen species-extracellular signal-regulated kinase-linked cascade. J Immunol. 2003;170:6273–9. doi: 10.4049/jimmunol.170.12.6273. [DOI] [PubMed] [Google Scholar]
- 40.Liu SQ, Alkema PK, Tieche C, Tefft BJ, Liu DZ, Li YC, Sumpio BE, Caprini JA, Paniagua M. Negative regulation of monocyte adhesion to arterial elastic laminae by signal regulatory protein alpha and Src homology 2 domain-containing protein-tyrosine phosphatase-1. J Biol Chem. 2005;280:39294–301. doi: 10.1074/jbc.M503866200. [DOI] [PubMed] [Google Scholar]
- 41.Hofman P, Piche M, Far DF, Le Negrate G, Selva E, Landraud L, Alliana-Schmid A, Boquet P, Rossi B. Increased Escherichia coli phagocytosis in neutrophils that have transmigrated across a cultured intestinal epithelium. Infection. 68:449–55. doi: 10.1128/iai.68.2.449-455.2000. & Immunity 2000 Feb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zen K, Babbin BA, Liu Y, Whelan JB, Nusrat A, Parkos CA. JAM-C is a component of desmosomes and a ligand for CD11b/CD18-mediated neutrophil transepithelial migration. Mol Biol Cell. 2004;15:3926–37. doi: 10.1091/mbc.E04-04-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zen K, Liu Y, Cairo D, Parkos CA. CD11b/CD18-dependent interactions of neutrophils with intestinal epithelium are mediated by fucosylated proteoglycans. J Immunol. 2002;169:5270–8. doi: 10.4049/jimmunol.169.9.5270. [DOI] [PubMed] [Google Scholar]
- 44.Aiyar A. The use of CLUSTAL W and CLUSTAL X for multiple sequence alignment. Methods Mol Biol. 2000;132:221–41. doi: 10.1385/1-59259-192-2:221. [DOI] [PubMed] [Google Scholar]
- 45.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 46.Peitsch MC. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem Soc Trans. 1996;24:274–9. doi: 10.1042/bst0240274. [DOI] [PubMed] [Google Scholar]
- 47.Peitsch MC, Herzyk P, Wells TN, Hubbard RE. Automated modelling of the transmembrane region of G-protein coupled receptor by Swiss-model. Receptors Channels. 1996;4:161–4. [PubMed] [Google Scholar]