

Summary
The function of synaptotagmin as a Ca2+ sensor in neurotransmitter release involves Ca2+-dependent phospholipid binding to its two C2 domains, but this activity alone does not explain why Ca2+ binding to the C2B domain is more critical for release than Ca2+ binding to the C2A domain. Synaptotagmin also binds to SNARE complexes, which are central components of the membrane fusion machinery, and displaces complexins from the SNAREs. However, it is unclear how phospholipid binding to synaptotagmin is coupled to SNARE binding and complexin displacement. Using supported lipid bilayers deposited within microfluidic channels, we now show that Ca2+ induces simultaneous binding of synaptotagmin to phospholipid membranes and SNARE complexes, resulting in an intimate quaternary complex that we name SSCAP complex. Mutagenesis experiments show that Ca2+ binding to the C2B domain is critical for SSCAP complex formation and displacement of complexin, providing a clear rationale for the preponderant role of the C2B domain in release. This and other correlations between the effects of mutations on SSCAP complex formation and their functional effects in vivo suggest a key role for this complex in release. We propose a model whereby the highly positive electrostatic potential at the tip of the SSCAP complex helps to induce membrane fusion during release.
Abbreviations: synaptotagmin, SNAREs, Ca2+ binding, phospholipid binding, neurotransmitter release, microfluidic channels
Introduction
The release of neurotransmitters by synaptic vesicle exocytosis is a central event in interneuronal communication. Release is acutely triggered by Ca2+, exhibiting a fast, synchronous component that emerges in less than 0.5 ms after Ca2+ influx, and a slower, asynchronous component1. Synaptotagmin 1 acts as a Ca2+ sensor in fast release through its two C2 domains (the C2A and C2B domain)2–6, which form similar β-sandwich structures and bind multiple Ca2+ ions via loops at one tip of the β-sandwich7–11. Also critical for release are the SNARE proteins synaptobrevin/VAMP, syntaxin and SNAP-25, which are central components of the exocytotic machinery and form a tight four-helix bundle (the SNARE complex) through sequences called SNARE motifs12–14. Assembly of this complex brings the synaptic vesicle and plasma membranes together15 and is likely key for membrane fusion (reviewed in16–18). Complexins, small soluble proteins that bind tightly to SNARE complexes and are selectively required for normal synchronous release19, 20, appear to be involved at least in part in coupling synaptotagmin 1 and SNARE function21–23. However, the mechanism of this coupling remains unclear.
Both synaptotagmin 1 C2 domains bind to phospholipids in a Ca2+-dependent manner through their Ca2+-binding loops11, 24–28, and decreasing or increasing their apparent Ca2+ affinity in the presence of phospholipids leads to parallel changes in the Ca2+ sensitivity of release2, 4. Hence, there is little doubt that Ca2+-dependent phospholipid binding is crucial for synaptotagmin 1 function. However, a key observation that cannot be explained solely by this activity is that mutating the Ca2+-binding sites of the C2B domain strongly impairs release while disrupting Ca2+ binding to the C2A domain has much milder effects on release (note that analogous mutations in the C2A or C2B domain have only moderate effects on overall phospholipid binding because the intact C2 domain can still bind Ca2+-phospholipids)3, 29–32. A potential explanation for these findings was provided by reports of Ca2+-dependent synaptotagmin 1 oligomerization mediated by the C2B domain (see ref.33), but extensive evidence has shown that a fragment including both synaptotagmin 1 C2 domains (C2AB fragment) does not oligomerize in solution or on membranes25, 34, 35. A property that is more likely to underlie the preponderant role of the C2B domain in release is its ability to bind to two membranes simultaneously upon Ca2+ binding, which is not shared by the C2A domain and could cooperate with the SNAREs in promoting membrane fusion25. However, such cooperation could also involve direct synaptotagmin 1/SNARE interactions.
Many studies have described binding of synaptotagmin 1 to syntaxin, to SNAP-25, to syntaxin/SNAP-25 heterodimers and/or to SNARE complexes (reviewed in1, 5, 6), but it has been difficult to unravel which of these interactions are physiologically relevant. Thus, studies of which SNAREs bind to synaptotagmin 1, which sequences are involved in binding, and whether the interactions are promoted by Ca2+ have yielded contradictory data (e.g. refs.36–40). Moreover, synaptotagmin 1 and the SNAREs are sticky proteins that have been reported to bind to many different targets (e.g. more than 40 for syntaxin; ref.18), suggesting that at least some of the described synaptotagmin 1/SNARE interactions arise from these sticky properties. Nevertheless, Ca2+-dependent interactions between synaptotagmin 1 and SNARE complexes39–43 are attractive from a mechanistic point of view because synaptotagmin 1 acts at the Ca2+ triggering step of release and SNARE complexes are believed to contribute to membrane fusion during this last step. This view has been reinforced by the recent observation that synaptotagmin 1 can displace complexins from SNARE complexes in a Ca2+-dependent manner23, which, together with electrophysiological data23 and membrane fusion studies21, 22, suggests that such displacement may be crucial to trigger fusion.
A key question that arises is whether synaptotagmin 1 can bind simultaneously to phospholipids and SNARE complexes. Immunoprecipitated synaptotagmin 1/SNARE complexes were reported to bind to phospholipids41, but the synaptotagmin 1 fragment used in this study contained a mutation that prevents correct folding of the C2B domain34. Conversely, NMR studies demonstrated that phospholipids displace soluble SNARE complexes bound to the synaptotagmin 1 C2AB fragment44, although this result could arise because the SNARE complexes were not attached to membranes. Indeed, the binding mode of the C2AB-fragment on SNARE complexes attached to neutral membranes appears to be compatible with membrane binding43. The finding that synaptotagmin 1 enhances the rate of assembly of membrane-attached syntaxin/SNAP-25 heterodimers also provided evidence for simultaneous interactions of synaptotagmin 1 with phospholipids and SNAREs, leading to the proposal that, upon Ca2+ influx, synaptotagmin 1 induces formation of the heterodimers followed by SNARE complex formation45. However, the observed rate enhancement was rather modest (ca. 5-fold) and it seems unlikely that all of these events can occur in the fast time scale of release. Simultaneous binding of synaptotagmin 1 to SNAREs and phospholipids was also proposed to underlie enhancement of SNARE-dependent proteoliposome fusion by Ca2+-synaptotagmin 146, but the increase is also modest (2–4 fold) and is more severely impaired by disruption of Ca2+-binding to the C2A domain than to the C2B domain47, in sharp contrast with the dramatic increase in release induced by Ca2+ in vivo (ca. 18,000 fold; ref.4) and with the critical role of Ca2+-binding to the C2B domain for release3, 32.
To shed light on how the functions of synaptotagmin 1 and the SNAREs are coupled, we have studied interactions of the C2AB fragment with SNARE complexes anchored at single, negatively charged phospholipid bilayers that were deposited within microfluidic channels. In combination with other biophysical experiments, mutagenesis and complexin displacement assays, our data unambiguously show that the C2AB fragment forms an intimate, specific quaternary complex with Ca2+, phospholipids and SNARE complexes, which we refer to as SSCAP complex (for SNAREs, Synaptotagmin, Ca2+, Phospholipids). We also find that the effects of several point mutations on formation of the SSCAP complex correlate with their effects on exocytosis, and that the C2B domain plays a central role in formation of this complex, which can explain its preponderant role in release. A preliminary model of the SSCAP complex suggests a mechanism whereby the highly positive electrostatic potential created by the synaptotagmin 1 C2B domain and the C-terminus of the SNARE complex after fast displacement of complexin helps to induce membrane fusion and neurotransmitter release.
Results
Synaptotagmin 1 binds simultaneously to phospholipids and SNARE complexes
If Ca2+ can induce simultaneous binding of the synaptotagmin 1 C2AB fragment to negatively charged phospholipids and membrane-anchored SNARE complexes, it is expected that these complexes should increase the membrane affinity of the C2AB fragment. However, measuring such an increase directly is hindered by the very high affinity of the C2AB fragment for negatively charged phospholipids in the presence of Ca2+ (in the low nanomolar range; see refs.25, 48). As an alternative, we sought to design a partition experiment that could reveal whether the C2AB fragment binds preferentially to membranes containing SNARE complexes compared to protein-free membranes. Microfluidic channel technology provided a powerful tool for this purpose. As illustrated in the diagram of Figure 1a, we prepared a PDMS slab that, when sealed against a glass slide, yielded two exterior microchannels, a shorter parallel microchannel in the middle, and a perpendicular microchannel with a valving screw. Supported phospholipid bilayers lacking or containing reconstituted SNAREs complexes were deposited by the vesicle fusion method49 into the exterior microchannels with the valving screw closed to avoid mixing of the two bilayers (both membranes contained 41% POPC, 32% DPPE, 12% DOPS, 5% PI and 10% cholesterol [w/w], which approximates the lipid composition of synaptic membranes; see ref.50). We then added BODIPY-FL-labeled C2AB fragment with the valving screw open (the single native cysteine of the C2AB fragment, C277, was mutated to serine, and a V419C mutation was introduced to yield a single exposed cysteine where the probe was attached). After washing the microchannels with buffer, binding to the plane of the bilayers was monitored with a confocal microscope (Figures 1b–e).
Figure 1.
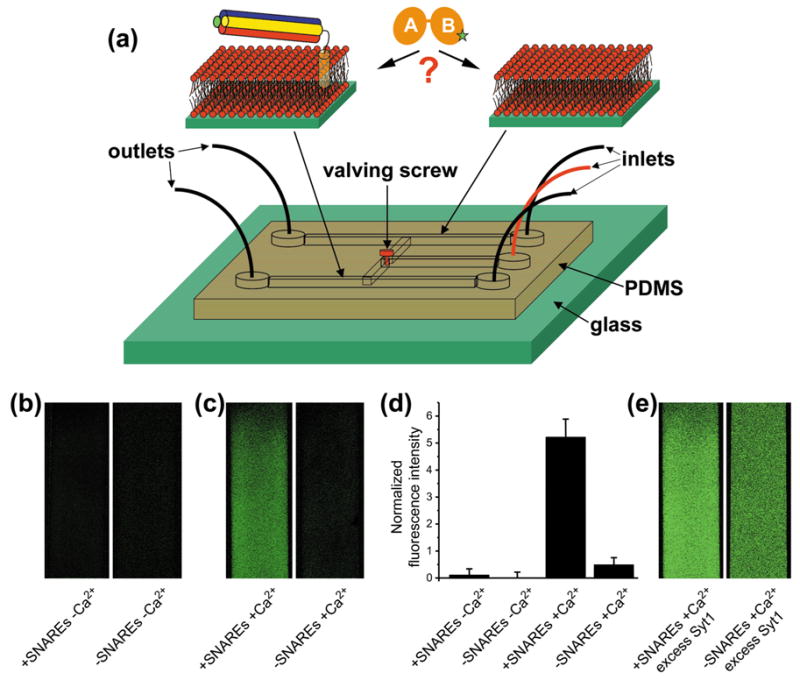
The C2AB fragment binds preferentially to membranes containing SNARE complexes. (a) Diagram summarizing the microfluidic channel experiments designed to test whether the C2AB fragment partitions preferentially to membranes containing reconstituted SNARE complexes formed by the syntaxin SNARE motif and TM region (yellow), the synaptobrevin SNARE motif (red) and the SNAP-25 N-terminal (blue) and C-terminal (green) SNARE motifs (molar protein to lipid ratio 1:1000). The V419C-C2AB fragment is represented by two orange elipses with a green star depicting the site labeled with BODIPY-FL. The same color coding is used in all figures. See text for further details. (b) Confocal micrographs of supported bilayers (41% POPC, 32% DPPE, 12% DOPS, 5% PI and 10% Cholesterol [w/w]) containing or lacking reconstituted SNARE complexes, which where deposited in the external microchannels of the PDMS slab (0.2 mm width). The micrographs were obtained after injecting 0.5 equivalents of BODIPY-FL labeled V419C-C2AB fragment (40 nM concentration) in buffer containing 1 mM EDTA through the central microchannel with the valved screw open, incubating for 1 hr and washing the microchannels with the same buffer. (c) Analogous experiments performed with buffer containing 1 mM Ca2+ and 0.5 equivalents of BODIPY-FL labeled V419C-C2AB fragment. (d) Quantitative analysis of the data shown in (b,c). Average fluorescence intensities were measured in each microchannel. The average intensity observed for the residual binding to SNARE-free membranes in the absence of Ca2+ (right panel in (b)) was then subtracted and used to normalize all resulting values. Error bars reflect standard deviations. (e) Partition experiment performed as in (c) but with 2 equivalents of BODIPY-FL labeled V419C-C2AB fragment.
When we added substoichiometric amounts of the C2AB fragment (0.5 equivalents with respect to the SNARE complex) in the absence of Ca2+, practically no binding to the membranes containing or lacking SNARE complexes was observed (Figure 1b,d). However, in analogous experiments performed in the presence of Ca2+, most of the C2AB fragment partitioned to the bilayer containing SNARE complexes (Figure 1c). Quantitative analysis of the fluorescence intensities revealed that the amount of C2AB fragment bound to the SNARE-containing bilayer was more than 10-fold higher than that bound to the SNARE-free bilayer (Figure 1d). Note that, since the molar SNARE complex to lipid ratio was 1:1000 and the membranes contained 17% negatively charged phospholipids, the partition coefficient between membrane sites containing SNARE complexes and sites containing negatively charged lipids is much higher than 10-fold. In additional experiments where we added an excess of C2AB fragment (2 equivalents with respect to the SNARE complex) in the presence of Ca2+, binding to both bilayers was observed but still with a clear preference for the bilayer containing SNARE complexes (Figure 1e), confirming that the C2AB fragment binds to SNARE-free bilayers under the conditions of these experiments but with lower affinity than to membranes containing SNARE complexes.
These results can almost certainly be attributed to simultaneous binding of the C2AB fragment to phospholipids and SNARE complexes, as the C2AB fragment binds with much higher affinity to the lipids than to soluble SNARE complexes23, 25, 44. To verify that the preference of the C2AB fragment for membranes containing SNARE complexes does not simply reflect binding to the SNARE complexes without C2AB fragment/membrane interactions, we performed fluorescence experiments using C2AB fragments labeled with an NBD probe at single cysteines that were introduced at the tip of one of the Ca2+-binding loops of the C2A domain (F234C) or the C2B domain (V304C). Note that NBD probes exhibit a large increase in fluorescence intensity when transferred from an aqueous to a hydrophobic environment51, and that biochemical and functional experiments indicated that the mutated residues insert into membranes during neurotransmitter release4; hence, the mutations are highly unlikely to perturb binding to SNARE complexes in vivo.
As described previously25, the fluorescence intensity of both NBD-labeled C2AB fragments in the presence of liposomes increased dramatically upon addition of Ca2+ due to insertion of the probes into the hydrophobic interior of the bilayer (Figure 2, black traces). Almost identical results were obtained when we performed analogous experiments using vesicles containing reconstituted SNARE complexes (Figure 2, red traces). In contrast, soluble SNARE complexes did not induce substantial increases in the fluorescence intensities of the NBD-labeled C2AB fragments in the presence or absence of Ca2+ (Figure 2; compare green and magenta traces with the black traces labeled PL - Ca2+). Together with the microchannel partition experiments (Figure 1), these results clearly show that Ca2+ induces simultaneous binding of the C2AB fragment to phospholipids and SNARE complexes, forming a quaternary SNARE-synaptotagmin-Ca2+-phospholipid (SSCAP) complex. These data also show that the Ca2+-binding loops of both C2 domains remain inserted into the bilayer upon formation of this complex.
Figure 2.
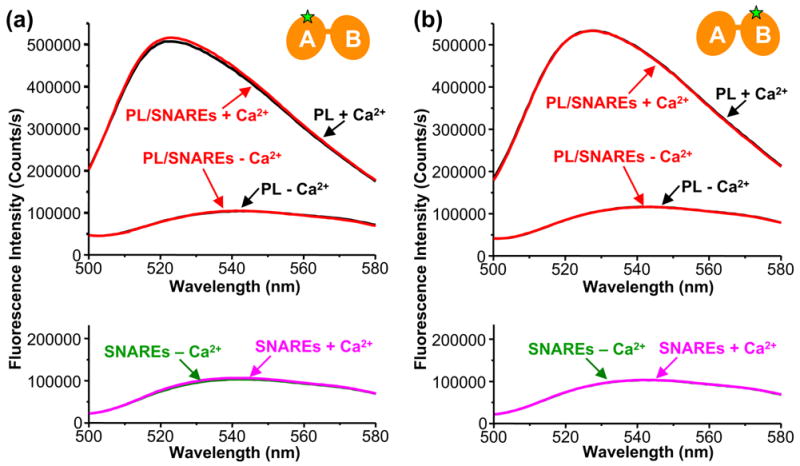
The Ca2+-binding loops of the synaptotagmin 1 C2 domains remain inserted into the membrane in the presence of reconstituted SNARE complexes. (a) Fluorescence spectra of NBD-F234C mutant C2AB fragment in the presence of phospholipid vesicles lacking (black traces) or containing (red traces) reconstituted SNARE complexes, acquired in 1 mM EDTA or 1 mM Ca2+. The bottom panel shows analogous spectra acquired in the presence soluble SNARE complexes and 1 mM EDTA (green) or 1 mM Ca2+ (magenta). (b) Analogous experiments performed with NBD-V304C mutant C2AB fragment. The presence of phospholipid vesicles does not perturb the fluorescence spectra of both isolated C2AB fragments in 1 mM EDTA25.
Structural analysis of interactions of the C2AB fragment with soluble SNARE complexes
Structural characterization of the quaternary SSCAP complex at high resolution by X-ray crystallography or NMR spectroscopy is hindered by the necessity of including membranes in the complex and by its large molecular weight. Attempts to study Ca2+-dependent interactions between the C2AB fragment and soluble SNARE complexes by NMR spectroscopy invariably led to precipitation even at 12 μM protein concentrations. This tendency to precipitate may arise from Ca2+-induced nonspecific interactions involving the Ca2+-binding loops of the C2AB fragment, which may bind to the highly acidic surface of the SNARE complex in the absence of membranes (see discussion). We reasoned that such non-specific interactions may be minimized in the absence of Ca2+ and that Ca2+-independent binding between soluble SNARE complexes and the C2AB fragment may reflect more faithfully the relevant interactions within the quaternary SSCAP complex, since the Ca2+-binding regions of the two C2 domains interact with the membrane in this complex (Figure 2). To facilitate analysis by NMR spectroscopy, we used a SNARE complex with a C-terminal truncation in the syntaxin SNARE motif (at residue 253) that renders the complex monomeric52 (we refer to this complex as ‘short SNARE complex’].
We first acquired TROSY-enhanced 1H-15N HSQC spectra of Ca2+-free 2H,15N-labeled C2AB fragment in the absence and presence of unlabeled short SNARE complex at 40 μM protein concentration as a compromise to obtain sufficient sensitivity while minimizing non-specific interactions. We previously showed that 1H-15N TROSY-HSQC spectra of the C2AB fragment contain two subsets of cross-peaks that precisely correspond to those of the isolated C2A and C2B domain because the two domains are flexibly linked25. Upon addition of the short SNARE complex to the 2H,15N-labeled C2AB fragment, the 1H-15N TROSY-HSQC cross-peaks from the C2B domain exhibited much more severe broadening than those from the C2A domain (Figures 3a,b). This observation shows that the short SNARE complex binds to the Ca2+-free C2AB-fragment at the concentrations used in these experiments [note that little binding was observed in the microchannel experiments (Figures 1(b,d)) because they were performed at much lower concentrations]. Moreover, these data show that binding is primarily mediated by the C2B domain, while the C2A domain remains flexibly linked to the C2B domain/SNARE complex. Mapping the significant cross-peak shifts and strongest broadening effects onto the structure of the C2B domain (Figure 3c) showed that strand 4 and its neighboring sequences are the most affected by binding to the short SNARE complex, but some perturbations are also observed in other regions of the C2B domain. These results are consistent with a previous report that a polybasic sequence in strand 4 of the C2B domain mediates Ca2+-independent SNARE complex binding53. The widespread perturbations observed in the 1H-15N TROSY-HSQC cross-peaks of the C2B domain may arise from an overall (although likely subtle) structural rearrangement induced by binding. It is also possible that several regions around the C2B domain in addition to the polybasic sequence may interact with SNARE complexes under the conditions of these experiments.
Figure 3.
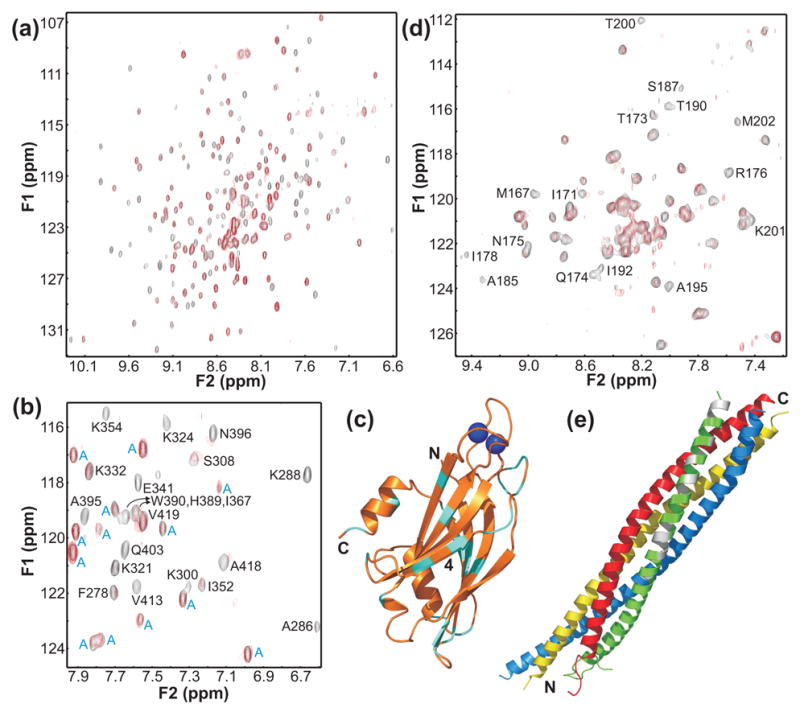
NMR analysis of Ca2+-independent interactions between synaptotagmin 1 and the SNARE complex. (a) Superposition of 1H-15N HSQC spectra of Ca2+-free 2H,15N-labeled C2AB fragment in the absence (black contours) and presence (red contours) of unlabeled short SNARE complex. (b) Expansions of the 1H-15N HSQC spectra shown in (a). Cross-peaks from the C2B domain are labeled with the residue number and one letter abbreviation, whereas cross-peaks from the C2A domain are labeled with a blue ‘A’. (c) Ribbon diagram of the C2B domain summarizing the most significant perturbations in the 1H-15N HSQC cross-peaks of the C2B domain caused by Ca2+-independent binding to the short SNARE complex. Residues corresponding to cross-peaks that exhibit significant shifts or have strong initial intensities and are broadened beyond detection upon binding are colored in cyan (disappearance of cross-peaks with weak initial intensities was not considered significant due to the overall broadening observed for most C2B domain cross-peaks). The two Ca2+ ions that bind to the C2B domain are shown as blue spheres to point out their binding sites, but the data were obtained in the absence of Ca2+. Strand 4 of the β-sandwich is labeled. (d) Superposition of 1H-15N TROSY-HSQC spectra of a sample of the short SNARE complex with the C-terminal SNAP-25 SNARE motif 2H,15N-labeled, acquired in 1 mM EDTA and the absence (black contours) or presence (red contours) of unlabeled C2AB fragment. Cross-peaks exhibiting the most significant broadening upon binding are labeled. The cross-peak assignments were described previously71. (e) Ribbon diagram of the SNARE complex with the SNAP-25 residues corresponding to severely broadened cross-peaks shown in white.
To gain insight into the region(s) of the SNARE complex involved in binding to the C2B domain, we acquired 1H-15N TROSY-HSQC spectra of a sample of the short SNARE complex with the C-terminal SNARE motif of SNAP-25 (SN25C) 2H,15N labeled, since some evidence suggested a functional interaction between this SNARE motif and synaptotagmin 140. Spectra acquired in the absence and presence of Ca2+-free unlabeled C2AB fragment (Figure 3d) showed that binding to the C2AB fragment induced selective broadening in subsets of cross-peaks that map to three regions of the SNARE complex (Figure 3e): i) an acidic central region that was previously implicated in Ca2+-independent synaptotagmin 1 binding54; ii) another acidic region close to the C-terminus that was implicated in Ca2+-dependent synaptotagmin 1 binding in a separate study40 and iii) a basic region at the very C-terminus. Hence these data are partially consistent with previous studies of synaptotagmin/SNARE complex interactions, but raise the possibility that these interactions may not be highly specific.
Structural determinants of quaternary SSCAP complex formation
During the course of this work, the finding that the C2AB-fragment binds to membrane-anchored SNARE complexes led us to test whether this interaction is compatible with binding of a complexin 1 fragment spanning its SNARE complex-interacting region (residues 26–83; see ref. 55). We found that the C2AB fragment displaces this complexin fragment from membrane-anchored SNARE complexes in the presence of Ca2+ (ref.23). Such displacement may be key to trigger membrane fusion during neurotransmitter release21–23, and most likely arises because of SSCAP complex formation. Hence, the complexin displacement assay that we developed provides a convenient means to study the structural determinants of quaternary SSCAP complex formation while mimicking a reaction that likely underlies, at least in part, the sequence of late events that leads to Ca2+-triggering of release. The assay uses a BODIPY-FL-labeled complexin fragment and supported phospholipid bilayers with reconstituted SNARE complexes that are deposited into single microfluidic channels. Binding of the complexin fragment to the plane of the bilayer, as well as release of the complexin fragment upon addition of synaptotagmin 1 fragments, is monitored with a confocal fluorescence microscope.
We first tested whether negatively charged phospholipids are required for complexin displacement. As shown previously23, the C2AB fragment quantitatively displaced complexin bound to the membrane anchored SNARE complexes in the presence of Ca2+, but not in its absence, when we used our standard membrane composition (Figure 4a, two left images). However, no complexin displacement was observed when the membranes lacked negatively charged phospholipids (Figure 4a, right image). This result shows that membrane binding and hence quaternary SSCAP complex formation is required for the C2AB fragment to displace complexin from SNARE complexes.
Figure 4.
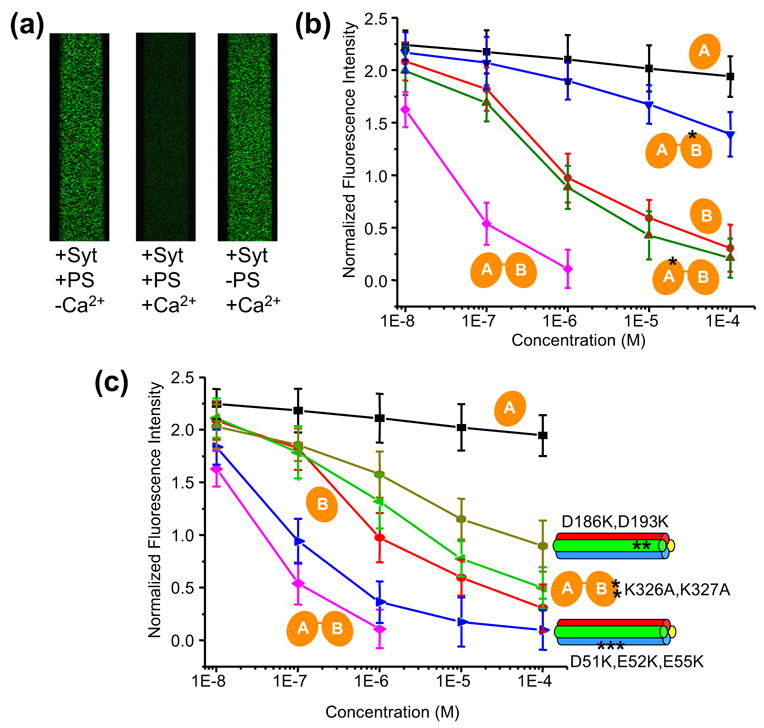
Structural determinants of SSCAP complex formation. (a) Confocal micrographs of supported bilayers containing reconstituted SNARE complexes deposited within microfluidic channels (width: 0.2 mm). The bilayers were loaded with 50 nM BODIPY-FL-labeled complexin fragment (residues 26–83) and washed with standard buffer containing 1 μM C2AB fragment (+Syt) and 1 mM EDTA (-Ca2+) or 1 mM Ca2+ (+Ca2+). The membranes contained 41% POPC, 32% DPPE, 12% DOPS, 5% PI and 10% Cholesterol [w/w] (two left images) or 58% POPC, 32% DPPE, and 10% Cholesterol [w/w] (right image). (b),(c) Displacement of BODIPY-FL-labeled complexin fragment from membrane anchored wild-type and mutant SNARE complexes upon addition of different concentrations of wild-type and mutant C2AB fragments. Experiments were performed as in (a) in 1 mM Ca2+ with membranes containining 41% POPC, 32% DPPE, 12% DOPS, 5% PI and 10% Cholesterol [w/w]. Average fluorescence intensities measured under each condition were normalized to a control in which the fluorescently labeled complexin fragment was added to a supported bilayer lacking SNARE complexes. Error bars represent SEMs derived from three separate measurements. In (b) all experiments were performed with wild type SNARE complexes and additions of wild type C2AB fragment (magenta), C2A domain (black) or C2B domain (red), or mutant C2AB fragments bearing mutations that disrupt Ca2+- binding to the C2A domain (D178N, green) or the C2B domain (D309N, blue). In (c), experiments were performed with wild type SNARE complexes and C2AB fragment bearing the K326A,K327A mutation in the polybasic region of the C2B domain (green), or with wild type C2AB fragment and SNARE complexes bearing mutations in acidic residues of SNAP-25 (D51K,E52K,E55K blue; D186K,D193K, light green); the data obtained with wild type SNARE complexes and the C2AB fragment (magenta), C2A domain (black) or C2B domain (red) are also shown in this diagram for comparative purposes.
We next monitored the displacement of complexin as a function of the concentration of different synaptotagmin 1 fragments in the presence of Ca2+. The wild type C2AB fragment displaced complexin efficiently at submicromolar concentrations and the isolated C2B domain was also able to displace complexin, albeit at higher concentrations (Figure 4b). However, the C2A domain was basically unable to displace complexin even at 100 μM concentration. A C2AB fragment with a mutation that disrupts Ca2+ binding to the C2A domain (C2A*B) was as effective in displacing complexin as the isolated C2B domain, whereas disrupting Ca2+ binding to the C2B domain in the C2AB fragment (C2AB*) almost abolished its ability to displace complexin (Figure 4b). These results are consistent with a model whereby the C2B domain binds directly to the SNAREs in the SSCAP complex, displacing complexin, while the C2A domain plays an auxiliary role by cooperating with the C2B domain in phospholipid binding, thereby increasing indirectly the overall affinity of the quaternary SSCAP complex and the ability of the C2B domain to displace complexin (see Figure 5). Importantly, these results correlate with the much stronger impairment of neurotransmitter release caused by disruption of Ca2+ binding to the C2B domain compared to disruption of Ca2+ binding to the C2A domain3, 29–32. Moreover, since the C2AB* mutant should still bind to the membrane through the C2A domain25, the strong effect of the mutation in the C2B domain Ca2+ binding sites shows that formation of the SSCAP complex is not simply a result of membrane localization but requires Ca2+-dependent binding of the C2B domain to phospholipids. These observations suggest that the SSCAP complex involves intimately correlated contacts of the C2B domain with the lipids and the SNAREs.
Figure 5.
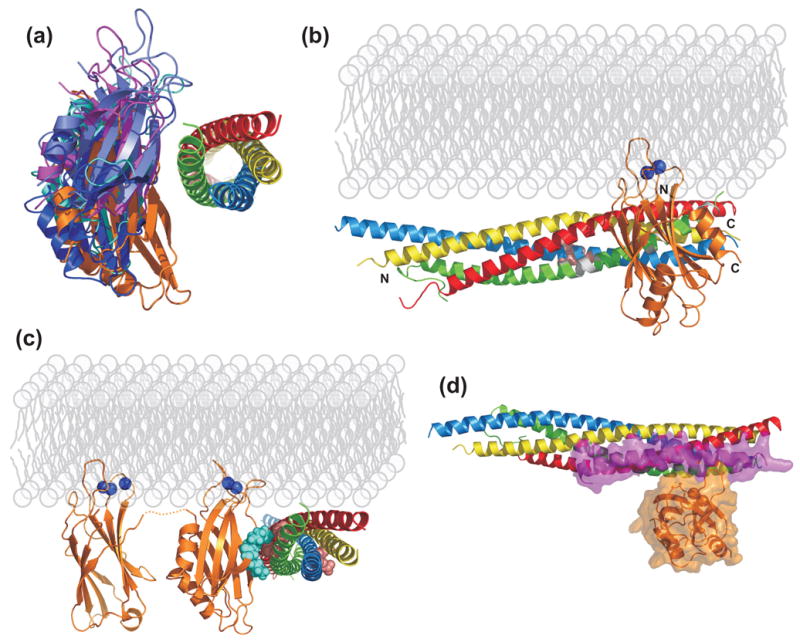
Model of the SSCAP complex. (a) Ribbon diagrams of the five top HADDOCK models. The five models were superimposed using the coordinates of the SNARE complex to illustrate the variability in the relative orientation of the C2B domain. (b) Ribbon diagram of the HADDOCK model that is most similar to the model obtained with FTDOCK, in an orientation rotated 90° with respect to that of (a). The two Ca2+ ions that bind to the C2B domain, which were not included in the modeling calculations, are represented as blue spheres, and the membrane phospholipids are indicated in light gray to help visualizing the relative locations of all the components of the SSCAP complex. N and C denote the N- and C-termini of the C2B domain and the SNARE complex. The residues of the SNAP-25 SNARE motif corresponding to cross-peaks that were perturbed by C2AB fragment binding in our NMR experiments (Figure 3) are colored in white. (c) Ribbon diagram of the same HADDOCK model shown in B but in a different orientation and including the C2A domain to illustrate how it emerges from the N-terminus of the C2B domain away from the SNARE complex. The linker sequence between the two C2 domains (four residues) is represented by a dashed orange curve. Residues that were mutated in our complexin displacement assays of Figure 4C are shown as spheres: K326 and K327 of the C2B domain are in blue; D51, E52, E55, D186 and D193 of SNAP-25 are in pink. D51, E522 and E55 are also shown in (B). (d) Superposition of the crystal structure of complexin bound to the SNARE complex52 with the model of the C2B domain/SNARE complex shown in (b). Partially transparent surfaces of complexin and the C2B domain are shown to illustrate the limited overlap between their binding sites on the SNARE complex. The color-coding for the SNAREs is the same as in Figure 1(a), synaptotagmin is in orange and complexin in pink. The models were rendered with PyMol (DeLano Scientific, San Carlos, CA).
Since our NMR experiments and previous data53 indicate that the polybasic region in strand 4 of the C2B domain may be involved in SNARE complex binding (Figures 3a–c), we also performed the complexin displacement assay with a C2AB fragment containing a double Lys to Ala mutation in the middle of strand 4 (K326A,K327A) that has been widely used in previous studies50, 53, 56, 57. This mutation considerably decreased but did not abolish the ability of the C2AB fragment to displace complexin (Figure 4c), which correlates with the partial but not complete impairment of neurotransmitter release caused by this mutation50, 57 and shows that the polybasic region of the C2B domain participates in SSCAP complex formation. We also studied the effects of mutations in two acidic regions of SNAP-25 that have previously been implicated in synaptotagmin 1 binding40, 54. One mutation (D51K,E52K,E55K) is in the middle of the SNARE complex, adjacent to the central region of the SNAP-25 C-terminal SNARE motif that was perturbed by the C2AB fragment in our NMR experiments (Figures 3d,e; see also Figures 5b,c). The other mutation (D186K,D193K) is closer to the C-terminus of the SNARE complex, in a region of the SNAP-25 C-terminal SNARE motif that was also implicated in C2AB fragment binding by our NMR data (Figures 3d,e and 5c). The mutations did not decrease the amount of complexin bound to the SNARE complexes in the absence of C2AB fragment (Figure 4c), showing that they did not prevent SNARE complex assembly. Importantly, the wild type C2AB fragment was almost equally effective in displacing complexin from the D51K,E52K,E55K mutant SNARE complexes as from the wild type SNARE complexes (practically within experimental error), whereas the D186K,D193K mutation strongly impaired such displacement (Figure 4c). These results show that the acidic region containing D186 and D193 at the C-terminus of SNAP-25 participates in binding to the C2AB fragment within the SSCAP complex, correlate with the impairment of exocytosis caused by mutations in this region40, and suggest that membrane anchoring of the SNARE complexes increases the specificity of C2AB fragment/SNARE complex interactions.
Model of the SCCAP complex
To gain further insight into the structure of the quaternary SSCAP complex, we investigated how the C2B domain may dock onto the SNARE complex using the HADDOCK program58 and imposing proximity between the C2B domain polybasic region and residues D186,D193 of SNAP-25 as the sole restraints. Ribbon diagrams representing the top five models of the orientation of the C2B domain with respect to the SNARE complex yielded by HADDOCK are shown in Figure 5a. Interestingly, while there is an expected variability in the orientations observed in the five models given the paucity of restraints, the Ca2+-binding loops of the C2B domain pointed in the same direction in all models (toward the top in Figure 5a). In addition, one of the HADDOCK models (in orange in Figure 5a; shown from a different view in Figure 5b) was similar to the top model yielded by parallel calculations performed with FTDOCK59 (data not shown). Clearly, these models must be considered preliminary, but they all share in common three interesting features.
First, the C2A domain is oriented away from the SNARE complex and cannot participate in SNARE binding in this orientation (Figure 5c), but it can contribute to the cooperative assembly of the SSCAP complex by binding to Ca2+-phospholipids. This feature correlates with the moderate effects of disrupting Ca2+ binding to the C2A domain on complexin displacement (Figure 4b) and neurotransmitter release29–31. Second, a paradox emerged from several lines of evidence suggesting that complexin is released from SNARE complexes upon Ca2+ influx21–23, since the off rate of complexin from membrane-attached SNARE complexes (2.5 s−1; ref.43) is much slower than the time scale of fast release (< 0.5 ms); our model of the SSCAP complex suggests a speculative solution for this apparent contradiction. Thus, complexin binds to the SNARE complex forming a long, antiparallel α-helix that only contacts the SNAREs through its C-terminal half (residues 48–70)52, and superpositions of our HADDOCK models with the crystal structure of the complexin/SNARE complex52 reveal only a small overlap between the C2B domain and residues 48–70 of complexin, as well as a more significant overlap with the N-terminal part of the complexin helix (e.g. Figure 5d). This observation suggests that thermal fluctuations in the N-terminal part of the helix, which has some flexibility52, may allow initial contact of the C2B domain with the SNARE complex and subsequent displacement of the C-terminal half of the complexin helix. This mechanism could occur at a much faster rate than mechanisms involving dissociation of complexin from the SNARE complex followed by synaptotagmin 1 binding. The third interesting feature of our model is that a highly positive face of the C2B domain is adjacent to the highly basic C-terminus of the SNARE complex. Hence, the strongly positive electrostatic potential generated in this region of the SSCAP complex could be critical to help bending the membranes to initiate membrane fusion, or to help opening the fusion pore if the vesicle and plasma membranes were already hemifused before Ca2+ influx (Figure 6).
Figure 6.
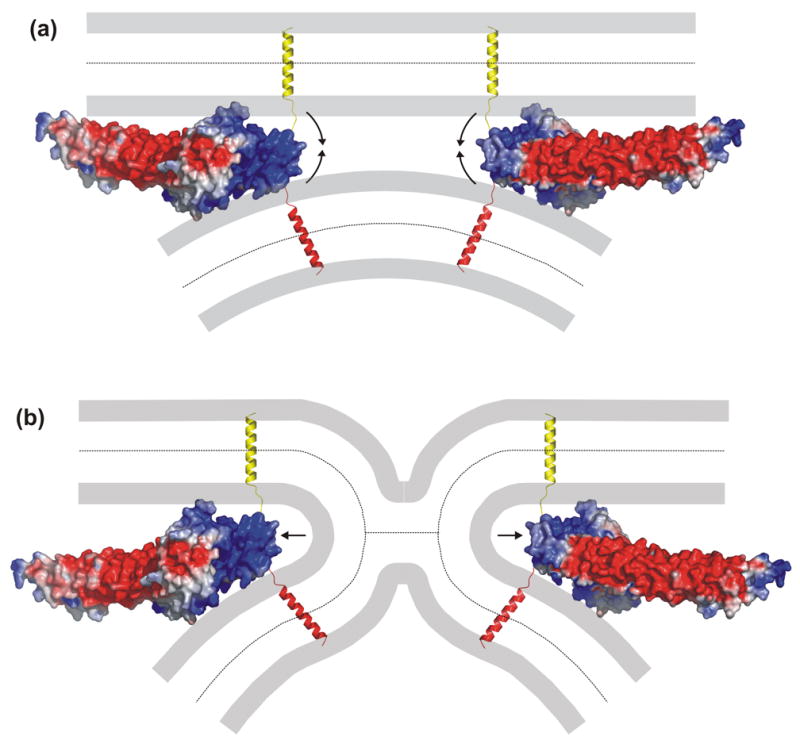
Models illustrating how the SSCAP complex could trigger neurotransmitter release. (a,b) The diagrams show the surface electrostatic potential of two copies of the modeled C2B domain/SNARE complex and how the highly positive potential at the tips of the complexes could help to bend the membranes to initiate membrane fusion (a) or could help opening the fusion pore (b). The model on the left was slightly rotated to better show the highly positive surface at the tip. The model on the right is rotated 180° around the vertical axis with respect to that on the left. The TM regions of syntaxin and synaptobrevin are in yellow and red, respectively. The electrostatic surface potential was computed with GRASP76. The models were rendered with PyMol (DeLano Scientific, San Carlos, CA). The electrostatic potential is contoured at the 5 kT/e level, with red denoting negative potential and blue denoting positive potential.
Discussion
The role of synaptotagmin 1 as a Ca2+ sensor in fast neurotransmitter release and the importance of Ca2+-dependent phospholipid binding for synaptotagmin 1 function are well established1–6, but the basis for the preponderant role of the C2B domain versus the C2A domain in release was not understood. Evidence for a role of synaptotagmin 1/SNARE interactions in release was also reported (e.g. refs.40, 42, 54), yet there were contradictory data on these interactions and it was unclear how they might be coupled with Ca2+-phospholipid binding to synaptotagmin 1. The results described here now shed light on these questions, showing that synaptotagmin 1 forms an intimate quaternary complex with the SNAREs, Ca2+ and phospholipids, and suggesting that membrane-anchoring of SNARE complexes increases the specificity of their interactions with synaptotagmin 1. The correlation between the effects of mutations in synaptagmin 1 and SNAP-25 on SSCAP complex formation and those of similar mutations on exocytosis strongly suggests that this complex plays a critical role in exocytosis and provides a clear rationale for the preponderant role of the C2B domain in release. Furthermore, our preliminary model of the SSCAP complex suggests plausible mechanisms for how this complex may trigger exocytosis.
The SSCAP complex
Synaptotagmin 1-induced increases in the rates of SNARE-mediated liposome fusion46 and of formation of membrane-anchored syntaxin-SNAP-25 heterodimers45 provided some evidence for simultaneous synaptotagmin 1-SNARE-phospholipid interactions, but the increases were rather modest and their physiological significance was unclear (see Introduction). Because interactions of synaptotagmin 1 with both phospholipids and SNARE complexes could be crucial for neurotransmitter release, we designed a series of experiments to rigorously test whether such interactions can occur simultaneously. The preferential partition of the C2AB fragment to membranes containing SNARE complexes (Figure 1), the observation that the C2 domain Ca2+ binding loops remain inserted into such membranes (Figure 2), and the finding that negatively charged phospholipids are required for displacement of complexin from SNARE complexes by the C2AB fragment (Figure 4a), unambiguously establish that synaptotagmin 1 forms a tight quaternary complex with Ca2+, phospholipids and SNARE complexes. Moreover, our NBD fluorescence data and mutagenesis study of complexin displacement lead to a model of the SSCAP complex that allows rationalization of a variety of functional data and formulation of a number of interesting predictions.
The finding that the C2B-domain can bind simultaneously to two membranes yielded a potential basis for its preponderant role in release and suggested a model whereby the highly positive electrostatic potential of the C2B-domain could bend the membranes to initiate fusion25. However, it was unclear how this action could be coordinated with the proposed function of the SNAREs in helping to induce membrane fusion. The critical role of Ca2+-binding to the C2B-domain for SSCAP complex formation (Figure 4b) now suggests an additional basis for the key function of the C2B-domain in release that incorporates interactions of synaptotagmin 1 with the SNAREs, thus providing a clear mechanism to couple their function. It is actually plausible that the C2B-domain interacts with the SNARE complex through the side of the β-sandwich and with both membranes through the Ca2+-binding loops and the opposite tip of the β-sandwich, which is positively charged (Figure 6a; see also ref.25). Note that the previously proposed role of the C2B domain in bending the membranes to initiate fusion25, which has been supported by theoretical calculations60, could be played in a similar fashion and even more efficiently by the SSCAP complex, as the positive charge of the C-termini of the SNAREs adds to that of the C2B domain to form a continuous, highly positive surface (Figure 6a). This surface is located at the tip of the SSCAP complex that is expected to be close to both membranes and is thus in an ideal position to perform this action. In an alternative model that assumes that the synaptic vesicle and plasma membranes are hemifused before Ca2+ influx, this positive surface could also provide an attractive force on the membranes to open the fusion pore, as proposed previously for the role of the C2B domain6, 60 (Figure 6b).
The partial disruption of SSCAP complex formation caused by the K326A,K327A mutation in the C2B domain polybasic region (Figure 4c) provides another correlation with functional data, as this mutation leads to partial impairment of release in vivo50, 57. The polybasic region had been implicated in many interactions25, 50, 53, 56, 61, likely because of its promiscuity in the absence of the correct target, but it was unclear which was this target. An intriguing possibility was that the polybasic region could participate directly in phospholipid binding, but it was difficult to envisage how this region and the Ca2+-binding loops could contact one or even two membranes simultaneously25, 50. In contrast, it is easy to visualize how the polybasic region can contact the SNARE complex while the Ca2+ binding loops insert into one membrane (Figures 5b,c). Moreover, the participation of the polybasic region in SNARE binding within the SSCAP complex correlates with our NMR analysis in solution (Figure 3) and bodes well with the acidic nature of the C-terminal region of SNAP-25 involved in SSCAP complex formation (Figure 4c). The strong disruption of the SSCAP complex caused by mutations in this C-terminal region of SNAP-25 and the parallel reduction of Ca2+-triggered exocytosis in PC12 cells induced by similar mutations in this region40 provide yet another correlation supporting a key role for the SSCAP complex in exocytosis. Our data also yield a clear basis for the severe block of release caused by botulinum neurotoxin (BoNT) E62, which cleaves SNAP-25 at residues 180–181 and hence removes the SNAP-25 sequence that is key for synaptotagmin 1 binding in our model. BoNT A cleaves SNAP-25 at residues 197–198, leaving the synaptotagmin 1 binding region intact, and causes a milder inhibition of release that can be overcome by increasing Ca2+ concentrations63. These findings likely arise from destabilization of the C-terminus of the SNARE complex caused by the cleavage and from a tight cooperativity in SSCAP complex assembly. This cooperativity is reflected in the fact that SSCAP complex formation is strongly hampered by either disrupting Ca2+-binding to the C2B domain (Figure 4b) or by a lack of SNARE membrane anchoring44, and could arise from lipid binding at the basic SNARE complex-C2B domain interface and/or from conformational changes in the C2B domain induced by Ca2+-phospholipid binding that may be required for SNARE complex binding.
Influence of membranes on synaptotagmin 1-SNARE complex interactions
The diverse Ca2+-independent interactions between the C2B domain and soluble SNARE complexes revealed by our NMR data are weak, as they are barely detectable at low μM concentrations44. However, the observed perturbations in the C2B domain polybasic region and the SNAP-25 sequence bearing D186 and D193 (Figure 3) suggest that the C2B domain-SNARE interactions present in the SSCAP complex may be reproduced at least in part with soluble SNARE complexes. These interactions are likely strengthened in the SSCAP complex because the proper orientation to establish them is favored by anchoring the SNARE complex to the membrane and by Ca2+-induced insertion of the C2B domain Ca2+-binding loops into the bilayer. In turn, the resulting orientation of the C2B domain and the SNARE complex with respect to the bilayer likely hinders alternative binding modes, including interactions of the central regions of the SNAP-25 SNARE motifs with the C2B-domain that were revealed by our NMR data (Figure 3e) and a previous study54. Thus, the membrane appears to increase the specificity of binding of synaptotagmin 1 to the SNAREs by dictating their proper orientation.
Since the C2-domain Ca2+-binding regions interact with the membranes in the SSCAP complex (Figure 2), it is unclear why Ca2+ increases the affinity of synaptotagmin 1 for soluble SNARE complexes23, 39–41, 44 and why this increase is reduced by mutations in D186 and D193 of SNAP-2540. These findings could be explained if SNARE complex binding requires Ca2+-induced conformational changes in the C2 domains, but the question that still arises is: why soluble SNARE complexes bound to the C2AB fragment are displaced by phospholipid vesicles in the presence of Ca2+ (ref.44)? It is likely that this competition arises because, upon binding of the C2AB fragment to the membrane, interactions with soluble SNARE complexes are weakened by repulsion of the membrane with the abundant negative charges on the surface of the SNARE complex (see Figure 5e) and by steric clashes with the bilayer caused by thermal motions of the highly elongated four-helix bundle. Membrane anchoring of the SNARE complex may be crucial to overcome these hindrances and thus for the intimate nature of the SSCAP complex.
Our model suggests that the C2A domain does not contact the SNARE complex but contributes to the stability of the SSCAP complex by binding to Ca2+-phospholipids (Figure 5c). This feature provides a sound basis for the limited effects of disrupting Ca2+ binding to the C2A domain on SSCAP complex formation (Figure 4b) and for the auxiliary (rather than critical) role of Ca2+ binding to the C2A domain for release29–31. This feature and our overall model of the SSCAP complex also correlate with single molecule FRET studies of C2AB fragment binding to SNARE complexes anchored on neutral membranes, which revealed few contacts between the C2A-domain and the SNARE complex and suggested that the binding mode was compatible with C2AB-fragment/membrane interactions43. Indeed, the SSCAP complex may have been formed under the conditions used in this study, as some degree of Ca2+-dependent membrane binding to the C2AB fragment was observed despite the fact that the membranes contained only PC43.
Our model and the single molecule FRET results contrast with the observations that the C2A domain binds to syntaxin in a Ca2+-dependent manner38, 64, 65 and plays a dominant role in Ca2+-dependent binding of synaptotagmin 1 to syntaxin38, SNAP-2539 and SNARE complexes41 in solution. The findings that the Ca2+-binding loops of both C2 domains may be implicated in Ca2+-dependent binding to SNAP-2566, and that the C2A domain uses the same surface (i.e. the Ca2+-binding loops) to bind to phospholipids24, to the cytoplasmic region of syntaxin and to its N-terminal Habc domain65, 67, raise the possibility that Ca2+-dependent syntaptotagmin 1-SNARE interactions may be at least in part non-specific in solution. Thus, the Ca2+-binding regions of the C2 domains become highly positively charged upon Ca2+ binding9, 11, 65 and appear to be avid to bind to negatively charged protein surfaces in the absence of their natural targets, i.e. the membranes. Note that synaptotagmin 1-SNARE complex interactions are highly sensitive to the ionic strength23 and the SNARE complex is also promiscuous, as shown by its tendency to self-associate14, 52.
Altogether, these arguments suggest that, although the divide-and-conquer approach has yielded important clues about synaptotagmin 1-SNARE interactions, experiments that lack one or more of the components of the SSCAP complex may yield misleading results. In particular, the membranes appear to play an important role in favoring the proper orientation of synaptotagmin 1 and the SNARE complex, and in preventing alternative interactions between the highly charged surfaces of these molecules. Moreover, the requirement of negatively charged membranes for complexin displacement from SNARE complexes by the C2AB fragment (Figure 4a) indicates that membrane binding strengthens the correct C2AB fragment-SNARE complex interactions. As explained above, our model of the SSCAP complex suggests a speculative mechanism for rapid displacement of complexin by synaptotagmin 1 that could occur in the fast time scale of release and does not require initial dissociation of complexin. Such fast displacement may also be facilitated by the unusually fast speed of Ca2+-dependent binding of synaptotagmin 1 to membranes48. Clearly, the validity of these and other predictions made above will require further experimentation and challenging structural analyses of the SNARE complex at or near atomic resolution. It should also be kept in mind that our study of the SSCAP complex still represents a divide-and-conquer approach, as it used fragments from a limited number of components from the release apparatus. For instance, flotation experiments using full-length Drosophila complexin and reconstituted SNARE complexes containing full-length rat syntaxin have suggested that complexin may remain bound to the SNARE complex in the presence of the C2AB-fragment22, which may arise because of interactions involving syntaxin and complexin sequences outside the fragments used here. Moreover, our model does not explain the finding that mutating D232 of the C2A-domain disrupts two of its three Ca2+-binding sites9 but enhances the Ca2+ sensitivity of release31, 68; Ca2+ independent interactions of the C2A-domain involving this aspartate residue may underlie this unexpected result, but this hypothesis remains to be tested. Nevertheless, the numerous correlations between the results described here and functional data suggest that the interactions underlying SSCAP complex formation and complexin release in our experiments reproduce in part key interactions involved in neurotransmitter release.
Microfluidic channels in the study of protein interactions on membranes
In retrospective, the importance of including membranes in studies of interactions between proteins involved in neurotransmitter release is not surprising given the very nature of this process. Microfluidic channel technology, which has already been shown to be very useful for biological studies69, was instrumental for the design of the experiments that revealed the formation of the SSCAP complex (Figure 1) and the analysis of complexin displacement by synaptotagmin 1 (Figure 4 and ref.23). Particularly useful features of this approach to study interactions on membrane surfaces are that it allows high surface-to-volume ratios for easy access and favorable partition of the reagents to the bilayers, as well as easy delivery of reagents and quick, efficient removal of unbound materials. These features and the small reaction volumes involved facilitate the performance of multiple experiments in a relatively short time using limited amounts of reagents that may be scarce or expensive. Thus, it can be anticipated that this and similar approaches using microfluidic channels can be applied to investigate a wide variety of biological processes that occur on membrane surfaces.
Materials and methods
Recombinant proteins
The following recombinant fragments were expressed and purified as described previously: rat synaptotagmin 1 C2A domain (residues 140–267)8, C2B domain11, C2AB fragment (residues 140–421) and D178N-C2AB, C277S,F234C-C2AB, C277S,V304C-C2AB, D309N-C2AB, K326A,K327A-C2AB and C277S,V419C-C2AB mutants25; rat syntaxin 1A residues 191–25352 and 183–28870; rat synaptobrevin 2 residues 1–96 and 29–9352; human SNAP-25 full length71 and residues 11–82 and 141–20352; and rat complexin 1 V61C mutant (residues 26–83)23. The D51K,E52K,E55K and D186K,D193K mutations in human SNAP-25 were generated with QuickChange site-directed mutageneis kit (Stratagene), and the corresponding recombinant proteins were expressed and purified as described for WT SNAP-2571. Uniform 2H,15N-labeling was performed by growing E. coli BL21(DE3) in minimal medium made with D2O as the solvent and using 15NH4Cl as the sole nitrogen source. Labeling of proteins with NBD or BODIPY-FL was performed as described23, 25.
Preparation of supported proteolipid bilayers and microfluidic channels
Purified syntaxin 1A(183–288), synaptobrevin 2(1–96) and full-length SNAP-25 were used to assemble SNARE complexes in the presence of 1% β-OG. Liposomes (100 nm diameter) containing 41% POPC, 32% DPPE, 12% DOPS, 5% PI and 10% Cholesterol [w/w] were prepared by extrusion through 0.8 μm polycarbonate membranes, and SNARE complexes were reconstituted into these preformed liposomes in standard buffer (25 mM HEPES pH7.4, 100 mM KCl, 0.1 mM EGTA, 0.3mM TCEP) as described previously70 but with a 1:1000 protein:lipid ratio. Microfluidic channels of 200 um width, 200 um height and 2.5 cm length were formed using standard soft lithography techniques72. Briefly, a layer of negative photoresist (SU-8, micrometers to ~1 mm thick) was spin-coated onto silicon wafers and exposed to UV light through a high-resolution transparency photomask of the designed pattern. The resist was then reflowed using a previously reported procedure73 and developed to produce silicon wafer masters for molding channels. The masters were silanized with a vapor of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane for 3 h. PDMS (Sylgard 184, Dow Corning) prepolymer (300μm-4 mm thick) was cast on silicon masters and cured thermally (65 °C). Cured PDMS stamps were cut out and peeled away from the silicon master, and inlets and outlets were drilled with an 18 gauge blunt needle. Coverslips were plasma-oxidized with a glow discharge unit for 3 min under vacuum. PDMS stamps were then firmly pressed down against the coverslips to form a leak-tight but reversible seal. The microchannel system for the partition experiments (Figure 1) used a torque-actuated screw valve (TWIST valve)74 to switch on and off the connection between the channels because it is easy to fabricate and operate. To fabricate the valve, a hole of about 4mm in diameter was drilled to about 2–3mm deep. A smaller hole was drilled right above the channel to hold the screw in place; the diameter of the hole was similar to that of the screws (about 1 mm). The device was plasma oxidized for 1 min to render the PDMS hydrophilic and prevent polyurethane from flowing onto the device. The screw was then pushed into the smaller hole until it made contact with the underlying PDMS. Polyurethane (NOA81) was filled around the screw and cured by UV for 6–8 min. The whole device was then incubated at 60 °C for 6 h to improve the adhesion between the polyurethane and PDMS. Lanes of supported bilayers containing or lacking SNARE complexes were formed within the microfluidic channels using the vesicle fusion method49.
Partition experiments
Liposomes (1 mM) containing or lacking SNARE complexes were deposited into the exterior channels (see Figure 1) with the screw valve closed. After deposition and removal of excess liposomes, the valve was opened to connect all the microchannels. BODIPY-FL labeled C277S,V419C-C2AB fragment (40 nM) was injected through the middle inlet in standard buffer containing 1 mM EDTA or 1 mM Ca2+. After incubating for 1 hr and washing the microchannels with the same buffer, BODIPY-FL fluorophores that remained bound to the membranes were imaged on a Leica (Wetzlar, Germany) confocal fluorescence microscope (TCS SP2) using a HC PL FLUOTAR 10x, 0.3 numerical aperture confocal scanning objective, and a 488 nm argon laser excitation with a BODIPY-FL filter set.
NBD fluorescence experiments
NBD fluorescence-emission measurements (480–580 nm) were performed at 25°C on a Photon Technology Incorporated (PTI, Lawrenceville, NJ) spectrofluorometer with excitation at 478 nm using 0.1 μM NBD-C277S,F234C-C2AB or NBD-C277S,V304C-C2AB fragments dissolved in standard buffer containing 1 mM EDTA or 1 mM CaCl2, and 0.1 mg/ml of liposomes lacking or containing SNARE complexes reconstituted as described above (1:1000 P:L ratio) for the microchannel experiments. Analogous experiments were performed in the absence of lipids with 0.125 μM soluble SNARE complexes (the syntaxin(183–288) fragment was replaced with a syntaxin(180–264), which lacks the TM region).
NMR Spectroscopy
1H-15N HSQC spectra were acquired as described52 at 27 °C on a Varian INOVA800 spectrometer with samples dissolved in 20 mM Tris (pH 7.4), 100 mM NaCl, 1mM EDTA, 0.3mM TCEP. Short SNARE complexes were assembled and purified as described52 using the following fragments: syntaxin 1A(191–253), synaptobrevin 2(29–93)52, SNAP-25(11–82) and SNAP-25(141–203).
Complexin displacement assays
The experiments were performed in standard buffer containing 1 mM Ca2+ (unless indicated otherwise) basically as described23. Briefly, supported bilayers containing assembled WT or mutant SNARE complexes deposited within single microchannels were incubated with 50 nM BODIPY-FL labeled V61C-complexin 1(26–83) for 15 min, and unbound complexin was washed out. An unlabeled synaptotagmin 1 fragment at the desired concentration was then added and incubated for 10 min, followed by a wash with buffer. Control experiments where the complexin fragment was added to supported bilayers lacking reconstituted SNAREs were used to measure background fluorescent that might result from non-specific binding of the complexin fragment to the bilayers. BODIPY-FL fluorophores that remained bound to the membranes were imaged with a confocal microscope as described for the partition experiments and quantified with Image J (NIH, MD).
Computational docking of the C2B domain to the SNARE complex
The models were generated using HADDOCK58 in conjunction with CNS75. The SNARE complex from the crystal structure of the complexin/SNARE complex (PDB accession code 1KIL)52 and the solution structure of the synaptotagmin 1 C2B domain (PDB accession code 1K5W)11 with the two Ca2+ ions removed were used as starting structures. Ambiguous interaction restraints (AIRs) were used to force proximity between K326,K327 of the C2B domain and D186,D193 of SNAP-25 with a 2Å upper distance limit (the maximum distance between any atom of an active residue of one molecule to any atom of an active or passive residue of the second molecule). Passive residues were chosen as those that were surface neighbors of the active residues within 6Å and that have a high level of solvent accessibility (>50%). Rigid body energy minimization generated an initial set of 1000 rigid-body docking solutions, which were sorted by the intermolecular energy of each complex (sum of van der Waals, electrostatic, AIRs).
Acknowledgments
We thank Thomas C. Südhof for fruitful discussions. This work was supported by Welch Foundation grant I-1304 and NIH grants NS40944 (to J.R.) and GM65364 (to George M. Whitesides, Harvard University).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 3.Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418:340–344. doi: 10.1038/nature00846. [DOI] [PubMed] [Google Scholar]
- 4.Rhee JS, Li LY, Shin OH, Rah JC, Rizo J, Sudhof TC, Rosenmund C. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci USA. 2005;102:18664–18669. doi: 10.1073/pnas.0509153102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai J, Chapman ER. The C2 domains of synaptotagmin--partners in exocytosis. Trends Biochem Sci. 2004;29:143–151. doi: 10.1016/j.tibs.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Rizo J, Chen X, Arac D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 8.Shao X, Davletov BA, Sutton RB, Sudhof TC, Rizo J. Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science. 1996;273:248–251. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 9.Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? . EMBO J. 1998;17:3921–3930. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao X, Fernandez I, Sudhof TC, Rizo J. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry. 1998;37:16106–16115. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Sudhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 c(2)b-domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- 12.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 13.Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 14.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 15.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 16.Rizo J, Sudhof TC. Snares and munc18 in synaptic vesicle fusion. Nat Rev Neurosci. 2002;3:641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT. Structure and function of SNARE and SNARE-interacting proteins. Q Rev Biophys. 2005:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- 18.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 19.McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 20.Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 21.Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- 22.Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Rizo J, Sudhof TC. Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry. 1998;37:12395–12403. doi: 10.1021/bi9807512. [DOI] [PubMed] [Google Scholar]
- 25.Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 26.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 27.Frazier AA, Roller CR, Havelka JJ, Hinderliter A, Cafiso DS. Membrane-bound orientation and position of the synaptotagmin I C2A domain by site-directed spin labeling. Biochemistry. 2003;42:96–105. doi: 10.1021/bi0268145. [DOI] [PubMed] [Google Scholar]
- 28.Rufener E, Frazier AA, Wieser CM, Hinderliter A, Cafiso DS. Membrane-bound orientation and position of the synaptotagmin C2B domain determined by site-directed spin labeling. Biochemistry. 2005;44:18–28. doi: 10.1021/bi048370d. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Chacon R, Shin OH, Konigstorfer A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Sudhof TC, Rosenmund C. Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci. 2002;22:8438–8446. doi: 10.1523/JNEUROSCI.22-19-08438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- 31.Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 32.Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, He Y, Bai J, Ji SR, Tucker WC, Chapman ER, Sui SF. Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proc Natl Acad Sci USA. 2003;100:2082–2087. doi: 10.1073/pnas.0435872100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ubach J, Lao Y, Fernandez I, Arac D, Sudhof TC, Rizo J. The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry. 2001;40:5854–5860. doi: 10.1021/bi010340c. [DOI] [PubMed] [Google Scholar]
- 35.Garcia RA, Forde CE, Godwin HA. Calcium triggers an intramolecular association of the C2 domains in synaptotagmin. Proc Natl Acad Sci USA. 2000;97:5883–5888. doi: 10.1073/pnas.100127197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C, Davletov BA, Sudhof TC. Distinct Ca2+ and Sr2+ binding properties of synaptotagmins. Definition of candidate Ca2+ sensors for the fast and slow components of neurotransmitter release. J Biol Chem. 1995;270:24898–24902. doi: 10.1074/jbc.270.42.24898. [DOI] [PubMed] [Google Scholar]
- 37.Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- 38.Kee Y, Scheller RH. Localization of synaptotagmin-binding domains on syntaxin. J Neurosci. 1996;16:1975–1981. doi: 10.1523/JNEUROSCI.16-06-01975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerona RR, Larsen EC, Kowalchyk JA, Martin TF. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TF. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/s0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 41.Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- 42.Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion Pore Dynamics Are Regulated by Synaptotagmin*t-SNARE Interactions. Neuron. 2004;41:929–942. doi: 10.1016/s0896-6273(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 43.Bowen ME, Weninger K, Ernst J, Chu S, Brunger AT. Single-molecule studies of synaptotagmin and complexin binding to the SNARE complex. Biophys J. 2005;89:690–702. doi: 10.1529/biophysj.104.054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arac D, Murphy T, Rizo J. Facile detection of protein-protein interactions by one-dimensional NMR spectroscopy. Biochemistry. 2003;42:2774–2780. doi: 10.1021/bi0272050. [DOI] [PubMed] [Google Scholar]
- 45.Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- 46.Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 47.Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci USA. 2005;102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brian AA, McConnell HM. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc Natl Acad Sci USA. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, Shin OH, Rhee JS, Arac D, Rah JC, Rizo J, Sudhof T, Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- 51.Crowley KS, Reinhart GD, Johnson AE. The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell. 1993;73:1101–1115. doi: 10.1016/0092-8674(93)90640-c. [DOI] [PubMed] [Google Scholar]
- 52.Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Sudhof TC, Rizo J. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- 53.Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B. Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem. 2004;279:12574–12579. doi: 10.1074/jbc.M310710200. [DOI] [PubMed] [Google Scholar]
- 54.Rickman C, Jimenez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B. Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell. 2006;17:283–294. doi: 10.1091/mbc.E05-07-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, Rizo J, Fasshauer D. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem. 2000;275:19808–19818. doi: 10.1074/jbc.M002571200. [DOI] [PubMed] [Google Scholar]
- 56.Chapman ER, Desai RC, Davis AF, Tornehl CK. Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. J Biol Chem. 1998;273:32966–32972. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- 57.Mackler JM, Reist NE. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J Comp Neurol. 2001;436:4–16. [PubMed] [Google Scholar]
- 58.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 59.Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 60.Zimmerberg J, Akimov SA, Frolov V. Synaptotagmin: fusogenic role for calcium sensor? . Nat Struct Mol Biol. 2006;13:301–303. doi: 10.1038/nsmb0406-301. [DOI] [PubMed] [Google Scholar]
- 61.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 62.Pellizzari R, Rossetto O, Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos Trans R Soc Lond B Biol Sci. 1999;354:259–268. doi: 10.1098/rstb.1999.0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keller JE, Neale EA. The role of the synaptic protein snap-25 in the potency of botulinum neurotoxin type A. J Biol Chem. 2001;276:13476–13482. doi: 10.1074/jbc.M010992200. [DOI] [PubMed] [Google Scholar]
- 64.Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Sudhof TC. Ca(2+)-dependent and -independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 65.Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron. 1997;18:133–142. doi: 10.1016/s0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- 66.Wang P, Wang CT, Bai J, Jackson MB, Chapman ER. Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner. J Biol Chem. 2003;278:47030–47037. doi: 10.1074/jbc.M306728200. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez I, Ubach J, Dulubova I, Zhang X, Sudhof TC, Rizo J. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell. 1998;94:841–849. doi: 10.1016/s0092-8674(00)81742-0. [DOI] [PubMed] [Google Scholar]
- 68.Pang ZP, Shin OH, Meyer AC, Rosenmund C, Sudhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006;26:12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sia SK, Whitesides GM. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis. 2003;24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 70.Chen X, Arac D, Wang TM, Gilpin CJ, Zimmerberg J, Rizo J. SNARE-Mediated Lipid Mixing Depends on the Physical State of the Vesicles. Biophys J. 2006;90:2062–2074. doi: 10.1529/biophysj.105.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen X, Tang J, Sudhof TC, Rizo J. Are neuronal SNARE proteins Ca2+ sensors? . J Mol Biol. 2005;347:145–158. doi: 10.1016/j.jmb.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 72.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 73.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 74.Weibel DB, Kruithof M, Potenta S, Sia SK, Lee A, Whitesides GM. Torque-actuated valves for microfluidics. Anal Chem. 2005;77:4726–4733. doi: 10.1021/ac048303p. [DOI] [PubMed] [Google Scholar]
- 75.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 76.Nicholls A, Sharp KA, Honig B. Protein Folding and Association - Insights from the Interfacial and Thermodynamic Properties of Hydrocarbons. Proteins-Structure Function and Genetics. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]