

Abstract
MicroRNAs (miRNAs) are a growing class of small RNAs (about 22 nt) that play crucial regulatory roles in the genome by targeting mRNAs for cleavage or translational repression. Most of the identified miRNAs are highly conserved among species, indicating strong functional constraint on miRNA evolution. However, nonconserved miRNAs may contribute to functional novelties during evolution. Recently, an X-linked miRNA cluster was reported with multiple copies in primates but not in rodents or dog. Here we sequenced and compared this miRNA cluster in major primate lineages including human, great ape, lesser ape, Old World monkey, and New World monkey. Our data indicate rapid evolution of this cluster in primates including frequent tandem duplications and nucleotide substitutions. In addition, lineage-specific substitutions were observed in human and chimpanzee, leading to the emergence of potential novel mature miRNAs. The expression analysis in rhesus monkeys revealed a strong correlation between miRNA expression changes and male sexual maturation, suggesting regulatory roles of this miRNA cluster in testis development and spermatogenesis. We propose that, like protein-coding genes, miRNA genes involved in male reproduction are subject to rapid adaptive changes that may contribute to functional novelties during evolution.
MicroRNAs (miRNAs) are a family of small, noncoding RNAs important for a diverse range of biological functions (Lagos-Quintana et al. 2001; Lee and Ambros 2001; Bartel 2004; He and Hannon 2004; Plasterk 2006). Since most current computational methods for prediction of miRNA genes rely heavily on phylogenetic conservation of sequences, most research has focused on highly conserved miRNAs (Grad et al. 2003; Lim et al. 2003a, b; Berezikov et al. 2005; Legendre et al. 2005; Xie et al. 2005; Pang et al. 2006). However, nonconserved miRNAs represent a potentially important source of functional novelties during evolution. Recently, various nonconserved miRNAs have been discovered and experimentally verified in virus (Pfeffer et al. 2005) and human (Bentwich et al. 2005). Bentwich and colleagues identified two miRNA clusters in primates (human, chimpanzee, and rhesus monkey) that have more miRNA copies than do rodents and dog, implying miRNA family expansion during primate evolution (Bentwich et al. 2005). One of the two clusters is located on the X chromosome and contains 10 miRNAs, which were classified into seven different seeds (MIRN513, MIRN506, MIRN507, MIRN508, MIRN509, MIRN510, and MIRN514). These miRNAs are preferentially expressed in testis (Bentwich et al. 2005). However, the timing and functional significance of X-linked miRNA expansion is unknown.
To reconstruct the evolutionary history of this cluster, we screened bacterial artificial chromosome (BAC) libraries and sequenced the miRNA cluster in three nonhuman primates (siamang, Hylobates syndactylus; Yunnan snub-nosed monkey [Xu et al. 2004], Rhinopithecus bieti, and black-handed spider monkey [Qian et al. 2004], Ateles geoffroyi). These species represent three major lineages in primates (lesser ape, Old World monkey, and New World monkey), reflecting 45 million years of primate evolutionary history (Goodman et al. 1998). Sequence comparison combined with the available data in human, chimpanzee, and rhesus monkey indicates a complex evolutionary history of the cluster, in which rapid miRNA evolution was observed.
Results and Discussion
We reanalyzed the human sequences of the X-linked miRNA cluster and identified an additional five new adjacent miRNAs, which had not been reported previously. Two belong to MIRN513, another two to MIRN509, and one is highly similar to MIRN510, possibly a new seed (designated as MIRN510L pending experimental verification). Figure 1A shows the phylogenetic relationship of the 15 human precursor miRNA sequences. Interestingly, phylogenetic relationships between the miRNAs reflect physical proximity, suggesting miRNA cluster expansion by tandem duplication (Ohno 1970). Figure 1B shows the structure of the miRNA cluster in the six primate species compared. All six species contain all eight miRNA seeds. However, three miRNA seeds (MIRN513, MIRN509, and MIRN514) have different copy numbers among species. For example, MIRN514 has three copies in human, four copies in chimpanzee, but only one copy in the other primate species, indicating common miRNA duplications in primates. Phylogenetic analysis on MIRN514 indicates that duplications occurred independently in human and chimpanzee. Alternatively, it is possible that the topology of the tree could have arisen because of gene conversion (Supplemental Fig. 1).
Figure 1.

(Legend on next page)
In contrast, nonprimate mammalian species show varying numbers of seeds. In addition to the three previously identified miRNA seeds (orthologous to MIRN506, MIRN507, and MIRN508) (Bentwich et al. 2005), we identified two new miRNAs in dog (designated as cfa-miR-513 and cfa-miR-513) and one in rodents (designated as mmu-(rno)-miR-X). The cfa-miR-513 is orthologous to MIRN513 in primates (bootstrap value, 99%) and the cfa-miR-X and mmu-(rno-)miR-X are phylogenetically close to the lineage clustering MIRN509, MIRN510, MIRN510L, MIRN514 in primates (bootstrap value, 85%) (Fig. 1C). Analysis of amphibian, bird, and fish species failed to detect any homologs, implying that either this cluster emerged relatively recently or the miRNA sequences have diverged too far to recognize orthology.
Figure 1C shows the phylogenetic relationships among all the precursor miRNAs in primates, rodents, and dog. Again, as observed in the human miRNAs (Fig. 1A), the physical distances between miRNAs correlate to the phylogenetic relationships, supporting the proposed tandem duplications in primates.
To rule out the possibility that some of the putative miRNAs in primates might be pseudogenes, we tested the expression of three miRNAs with copy number variations (MIRN509, MIRN513, and MIRN514) in rhesus monkey testis. We detected the expression of all copies of the precursors (data not shown), suggesting that all the copies are probably functional. Hence, the difference of copy numbers among primate species may have dosage effects in regulating testis development and/or function. However, we cannot rule out the possibility that, despite their transcription, some miRNA copies may represent processed pseudogenes.
It is well known that copy number variations (CNVs) are common in human populations (Redon et al. 2006). To test whether the miRNAs with CNVs between species could also be polymorphic within human populations, we tested MIRN509 in three major continental populations (10 Chinese, 10 Caucasians, and 10 Africans) and all the three MIRN509 copies were detected in all the human samples, an implication of fixation of the duplicated miRNAs in human.
We also observed rapid sequence evolution of the miRNAs. In five miRNA members with confirmed primate orthologs (MIRN506, MIRN507, MIRN508, MIRN510 in all six species, and MIRN514 in four species), we observed a total of 77 sites with sequence substitutions in the precursors. The average substitution rate is 15.4 per miRNA, which is much higher than the rate reported by Berezikov et al. (2005) (2.27 per miRNA in 122 miRNAs based on species with an even larger sampling covering distantly related primate species). In Berezikov and colleagues’ study, only three of the 122 observed substitutions were located in the mature miRNA (i.e., most occurred in the terminal loop and at the ends of a precursor). In contrast, 15 out of 77 substitutions in the X-linked cluster were located within the mature miRNA (P = 7 × 10−5, two-tailed Fisher’s exact test) (Supplemental Fig. 2).
To confirm the rapid sequence evolution of the miRNAs within the X-linked cluster, we compared the substitution rates of the X-linked miRNAs (MIRN506, MIRN507, MIRN508, MIRN510 with confirmed orthologs) with that of 102 known intergenic miRNAs (data from http://mirnamap.mbc.nctu.edu.tw/). We calculated the between-species substitution rates (human vs. rhesus monkey) for the miRNA precursor (Kp) and the flanking genomic sequence (Kf) (presumably nonfunctional). The average Kf values are similar between the X-linked (0.064) and the intergenic miRNAs (0.053), an indication of nearly equal mutation rates. However, the X-linked miRNA cluster has a much larger average Kp (0.047) compared to the intergenic miRNAs (0.013) (P < 0.001, two-tailed Student's t-test), again supporting the proposed rapid evolution of the X-linked miRNA cluster. We also compared Kp with KTE between human and chimpanzee. KTE is the substitution rate of the transposable elements (30 in total) within the X-linked cluster shown to be neutral (Lunter et al. 2006). Three out of the eight Kp values are out of the 99% confidence interval of the KTE distribution, an indication of excess of substitutions over neutrality (Supplemental Fig. 3).
It should be noted that, despite the rapid sequence substitutions, the general secondary structures (hairpin and loop structures) are highly conserved in all the primate miRNAs, as reflected by the excess of compensatory substitutions over random expectation (Table 1) (single substitutions, P = 0.002, double substitutions, P = 0.05, two-tailed Fisher’s exact test), consistent with strong functional constraint on miRNA secondary structures. This substitution pattern suggests that compensatory mutations could be the mechanism of miRNA evolution, as shown in other functional RNA genes (Hancock et al.1988; Higgs 2000). In addition, the few observed deletions in the precursors did not affect the secondary structures (Berezikov et al. 2005).
Table 1.
Substitutions observed in precursor miRNAs
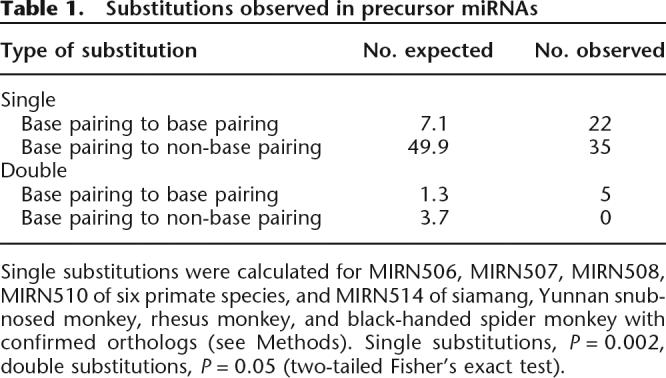
Single substitutions were calculated for MIRN506, MIRN507, MIRN508, MIRN510 of six primate species, and MIRN514 of siamang, Yunnan snub-nosed monkey, rhesus monkey, and black-handed spider monkey with confirmed orthologs (see Methods). Single substitutions, P = 0.002, double substitutions, P = 0.05 (two-tailed Fisher’s exact test).
Sequence substitutions may lead to the emergence of novel miRNAs. In MIRN513 and MIRN509, we observed sequence substitutions in the mature miRNAs both between species and between copies within species. Target gene prediction for MIRN513 (two human copies, one chimpanzee copy, and one rhesus copy) by miRanda (Enright et al. 2003) showed that the duplicated miRNAs target fewer genes than do the ancestral copy, and that many of the targets are novel (not targeted by the ancestral copy). This observation suggests that the new miRNA copies might be functionally more specialized (data not shown). Moreover, for MIRN508 and MIRN510, we observed lineage-specific substitutions important for target recognition (Bartel 2004; Brennecke et al. 2005) (human MIRN508 at site 16 of the mature miRNA; siamang MIRN508 at site 12; human and chimpanzee MIRN510 at site 6; chimpanzee MIRN510 at site 4). These sequence changes of miRNAs are possibly driven by natural selection on testis-expressed miRNAs, as previously found in protein-coding genes involved in male reproduction (Wyckoff et al. 2000; Swanson and Vacquier 2002; Dorus et al. 2004; Clark and Swanson 2005). It should be noted that current target prediction methods may suffer from a high false positive rate, and the miRanda algorithm used reported a success rate of ∼64% in Drosophila (Stark et al. 2005). Therefore, the suggested functional specialization of the miRNAs is yet to be tested with experimental data.
To understand the functional roles of the X-linked miRNA cluster, we tested the expression of seven miRNA seeds (MIRN510L was not tested) in testis of infant (1–2 yr, sexually immature) and adult rhesus monkeys (8–10 yr, sexually matured), using real-time quantitative PCR. All seven showed a significant expression reduction of the mature miRNAs in adults compared with infants (Fig. 2A). In particular, MIRN514 is only expressed in infants (Fig. 2B). Recent studies have shown that animal miRNAs can not only repress translation of target gene mRNAs, but also induce their degradation by nonperfect sequence recognition (Bagga et al. 2005; Lim et al. 2005). Hence, the observed reduced expression of the primate miRNAs in adults would allow development-stage-related functioning of the target genes during male sexual maturation. Thus the X-linked miRNA cluster likely plays regulatory role in primate testis development and spermatogenesis, and experimental data are needed to confirm the suggested regulatory role of this cluster.
Figure 2.

The expression analysis of the X-linked miRNAs in infant and adult rhesus monkeys. (A) The expression levels of the mature miRNAs in MIRN513, MIRN506, MIRN507, MIRN508, MIRN509, and MIRN510 showing obvious reductions in adult monkeys. The standard errors are indicated. (B) The electrophoretic gel analysis of MIRN514 expression. From left to right: lane 1, infant1; lane 2, infant2; lane 3, adult1; lane 4, adult2; lane 5, adult3; lane 6, adult4; M, molecular marker. Note: The Human Genome Nomenclature Committee–approved gene symbols currently follow the MIRN# convention (e.g., MIRN513).
Previous genome-wide analysis identified a series of fast-evolving genomic regions or lineage-specific structure changes (deletions, insertions, and inversions) in human (Newman et al. 2005; Pollard et al. 2006; Prabhakar et al. 2006). Our data add another example by showing rapid evolution of a microRNA cluster in human and nonhuman primates. However, the previously identified fast-evolving regions do not include the X-linked microRNA cluster, which is probably due to the different strategies used since the previous studies were aimed to identify human-specific changes (Pollard et al. 2006; Prabhakar et al. 2006).
In conclusion, we report rapid evolution of the X-linked miRNA cluster in primates, with frequent copy number changes and sequence substitutions. This observation is consistent with the finding on a protein-coding gene cluster, i.e., PRAME, which is testis-expressed and has expansion of copy numbers in primates (Birtle et al. 2005). Hence, like protein-coding genes (Wyckoff et al. 2000; Swanson and Vacquier 2002; Dorus et al. 2004; Clark and Swanson 2005; Zhang et al. 2007), miRNA genes related with male reproduction are subject to adaptive changes due to selection, which may contribute to the emergence of functional novelties during evolution.
Methods
BAC library screening, sequence assembly, and sequence analysis
The pooled PCR-based method was used in screening the primate BAC libraries and identifying positive BAC clones. Primer sequences are shown in Supplemental Table 1. A total of seven positive BAC clones were detected, three of which (one for each species, 150 kb, 160 kb, 210 kb) were selected by end-sequencing, then full-length sequenced using shotgun sequencing method with 6× coverage at Beijing Genomics Institute, CAS. The sequences were aligned and assembled by phred/phrap/consed package (Ewing and Green 1998; Ewing et al. 1998; Gordon et al. 1998). The sequence gaps were filled by PCR-based sequencing on an ABI-3130 sequencer. The sequence quality of the putative miRNAs was checked by looking at the raw electro-morph data.
We reanalyzed the human X-linked cluster using BLASTN (E-value cutoff 10−4) with previously identified 10 miRNAs as query sequences. Paralogous relationship of the newly identified members of MIRN513 and MIRN509 in human was determined by phylogenetic analysis (neighbor-joining method, sequence similarity, 97%–99%; bootstrap values, 98%) (Nei 1987). We searched the homologs of the miRNAs in nonhuman primates with BLASTN (E-value cutoff 10−10), and all the 15 human miRNA precursors were used as query sequences. The orthologous miRNAs were identified based on reciprocal best hits. The orthologous miRNAs of rodent (mouse and rat) and dog were identified using multiple alignment data in the UCSC genome browser (the Vertebrate Multiz Alignment & Conservation track). The identified orthologous sequences were also supported by synteny check. We used the UCSC genome database (Hinrichs et al. 2006) in our sequence analysis, including the Human Mar. 2006 (hg18) assembly, the Chimp Mar. 2006 (panTro2) assembly, the Rhesus Jan. 2006 (rheMac2) assembly, the Dog May 2005 (canFam2) assembly, the Mouse Feb. 2006 (mm8) assembly, and the Rat Nov. 2004 (rn4) assembly. Gene sequences were aligned by ClustalX (Jeanmougin et al. 1998) with manual adjustment. Phylogenetic trees were reconstructed by using the NJ method in MEGA3 (Kumar et al. 2004) and evaluated by 1000 bootstrap replications. For calculation of between-species sequence substitutions, the orthologous precursor miRNA sequences were aligned by ClustalX (Jeanmougin et al. 1998). For MIRN506, 507, 508, 510, the orthologs were identified in all the six primate species. Due to multiple duplications, for MIRN514, there are only four species with unambiguous orthologs, and no orthologs from more than three species could be unambiguously identified for MIRN509, 513 (Berezikov et al. 2005).
For target prediction, the ancestral copy of MIRN513 was inferred using the core region of the within-species duplication units with dog MIRN513 as outgroup. Dotter (Sonnhammer and Durbin 1995) was used to find the duplication units of MIRN513, MIRN509, and MIRN514. Repeatmasker (http://repeatmasker.org) was used to find repeat elements.
Calculation of compensatory substitutions
The secondary structures of the pre-miRNAs were predicted by mfold (Mathews et al. 1999; Zuker 2003). The miRNA sequences and secondary structures were provided in Supplemental Figure 4. The pre-miRNA sequences were divided into four parts as described before (Han et al. 2006). We considered two classes of changes in the stem region: substitutions that change one pair of complementary bases to another pair of complementary bases (e.g., C–G to U–G) and substitutions that change one pair of complementary bases to a pair of noncomplementary bases, or vice versa (e.g., C–G to C–C). For a particular site in the stem region, a single substitution refers to change of either of the two corresponding bases, and a double substitution refers to changes of both bases. The probability of a single substitution or a double substitution converting one pair of complementary bases to another pair of complementary bases was provided before (Dixon and Hillis 1993). The expected values of substitutions were calculated following the method of Dixon and Hillis (1993). We aligned the orthologs or paralogs of the X-linked miRNAs from the six primate species by ClustalX. For single substitution analysis, only the confirmed orthologs were used to avoid multiple counts of shared mutations between species in paralogs. The single substitution pattern in paralogs is listed in Supplemental Table 2, which is similar to the result of the orthologs. Both the orthologs and paralogs were used in calculating double substitutions (multiple counts were removed) to test if compensatory substitutions are overrepresented.
Calculation of the substitution rates of precursor miRNAs (Kp) and flanking genomic sequences (Kf)
For calculation of Kf, the 5′ and 3′ flanking sequences (500 bp each side, 1000 bp in total) of the pre-miRNAs were used. Due to the close proximity of MIRN506 and MIRN507 in the genome (142 bp), only 642 bp were used. A total of 108 known intergenic human miRNAs (physical distance >500 bp from each other) (http://mirnamap.mbc.nctu.edu.tw/) were used to blast the rhesus monkey genome (rheMac2), and 102 rhesus orthologs were identified. The sequences were then aligned by ClustalX. Kimura 2-parameter model was used to calculate substitution rates of the precursor miRNAs (Kp) and flanking genomic sequences (Kf) (Kimura 1980).
Calculation of K of transposable elements (TE)
Due to the variable and complex nature of this region, it is difficult to collect enough orthologous TEs in distantly related species (such as between human and rhesus monkey). With the use of siamang as outgroup, we identified 30 TE sequences within the sequenced X-linked cluster in human and chimpanzee. The orthology was determined by three criteria: TE classification, reciprocal best hits, and synteny. Kimura 2-parameter model was used to calculate substitution rates (Kimura 1980). By One-Sample Kolmogorov-Smirnov Test, the normal distribution of KTE was confirmed.
Target gene prediction
Human, chimpanzee, and rhesus monkey 3′UTRs are from Ensembl BioMart. For each gene, we only choose one transcript. If a gene has more than one transcript, the longest one is chosen for analyses. miRanda (Enright et al. 2003) was used to identify miRNA binding sequences in the 3′UTR sequences. Due to the rapid substitution nature of this cluster, we did not use the conservation criterion because it was shown that the nonconserved target sites also mediated repression (Farh et al. 2005). The parameters used for scanning are S:90, ΔG: −17 kcal/mol, and -shuffle.
Real-time quantitative PCR analysis of miRNA expression
The method used to quantify the expression level of mature miRNAs was described previously (Raymond et al. 2005). The LNA primers were purchased from Proligo LCC. The potential dimers of GS-primer and L-primer were evaluated by Autodimer (Vallone and Butler 2004). All primers are listed in Supplemental Table 3. RNA was extracted in duplicates from three infant and three adult rhesus testis samples using TRIzol (Invitrogen). After DNase treatment, a mixture of GS-primer and GAPDH specific primer was used for reverse-transcription by Omniscript Reverse Transcriptase (QIAGEN). The sequences of each miRNA amplicon were determined by TA-cloning (Takara). Quantitative RT-PCR by SYBR green (Bio-Rad) for each cDNA was run in triplicates in MJ Opticon. We calculated the relative amounts of miRNAs by the comparative Ct (2−ΔΔCt) method (Livak and Schmittgen 2001). The target miRNA values were normalized based on the internal control of GAPDH. Each Ct value used for the calculation was the mean obtained from each cDNA in triplicates. Statistical analysis was done with Student's t-test or t-test for unequal variances.
Precursor miRNAs detection
The list of primers is provided in Supplemental Table 1. Total RNA was isolated by TRIzol. After DNase treatment, we used GS-primer for reverse transcription. The PCR amplicons were validated using gel electrophoresis, purified using MinElute PCR Purification Kits (QIAGEN), and subcloned with TA-cloning vectors. Thirty randomly chosen positive clones were sequenced for each pre-miRNA.
Primers for detection of MIRN509 copy numbers are listed in Supplemental Table 1.
Acknowledgments
We thank Scott W. Roy for extensive discussion and critical reading of the manuscript, Ayelet Chajut for kindly providing sequence data of mouse and dog, and Chris Raymond for technical suggestion on miRNA quantification. We also thank Xiao-jing Yu, Yin-qiu Wang, Ao-lei Niu, Cheng-hong Liao, and Ruo-lin Yang for their help in this study. We thank Xiao-na Fan and Hui Zhang for their technical assistance. This study was supported by grants from the Chinese Academy of Sciences (KSCX1-YW-R-34), the National Natural Science Foundation of China (30630013, 30525028), the Natural Science Foundation of Yunnan Province of China, and the National 973 project of China (2006CB701506).
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. EF466135–EF466137.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6146507
References
- Bagga S., Bracht J., Hunter S., Massirer K., Holtz J., Eachus R., Pasquinelli A.E., Bracht J., Hunter S., Massirer K., Holtz J., Eachus R., Pasquinelli A.E., Hunter S., Massirer K., Holtz J., Eachus R., Pasquinelli A.E., Massirer K., Holtz J., Eachus R., Pasquinelli A.E., Holtz J., Eachus R., Pasquinelli A.E., Eachus R., Pasquinelli A.E., Pasquinelli A.E. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bentwich I., Avniel A., Karov Y., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Avniel A., Karov Y., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Karov Y., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Aharonov R., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Gilad S., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Barad O., Barzilai A., Einat P., Einav U., Meiri E., Barzilai A., Einat P., Einav U., Meiri E., Einat P., Einav U., Meiri E., Einav U., Meiri E., Meiri E., et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Berezikov E., Guryev V., de van Belt J., Wienholds E., Plasterk R.H., Cuppen E., Guryev V., de van Belt J., Wienholds E., Plasterk R.H., Cuppen E., de van Belt J., Wienholds E., Plasterk R.H., Cuppen E., Wienholds E., Plasterk R.H., Cuppen E., Plasterk R.H., Cuppen E., Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Birtle Z., Goodstadt L., Ponting C., Goodstadt L., Ponting C., Ponting C. Duplication and positive selection among hominin-specific PRAME genes. BMC Genomics. 2005;6:120. doi: 10.1186/1471-2164-6-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J., Stark A., Russell R.B., Cohen S.M., Stark A., Russell R.B., Cohen S.M., Russell R.B., Cohen S.M., Cohen S.M. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark N.L., Swanson W.J., Swanson W.J. Pervasive adaptive evolution in primate seminal proteins. PLoS Genet. 2005;1:e35. doi: 10.1371/journal.pgen.0010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon M.T., Hillis D.M., Hillis D.M. Ribosomal RNA secondary structure: Compensatory mutations and implications for phylogenetic analysis. Mol. Biol. Evol. 1993;10:256–267. doi: 10.1093/oxfordjournals.molbev.a039998. [DOI] [PubMed] [Google Scholar]
- Dorus S., Evans P.D., Wyckoff G.J., Choi S.S., Lahn B.T., Evans P.D., Wyckoff G.J., Choi S.S., Lahn B.T., Wyckoff G.J., Choi S.S., Lahn B.T., Choi S.S., Lahn B.T., Lahn B.T. Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat. Genet. 2004;36:1326–1329. doi: 10.1038/ng1471. [DOI] [PubMed] [Google Scholar]
- Enright A., John B., Gaul U., Tuschl T., Sander C., Marks D., John B., Gaul U., Tuschl T., Sander C., Marks D., Gaul U., Tuschl T., Sander C., Marks D., Tuschl T., Sander C., Marks D., Sander C., Marks D., Marks D. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B., Green P., Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B., Hillier L., Wendl M.C., Green P., Hillier L., Wendl M.C., Green P., Wendl M.C., Green P., Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Farh K.K.-H., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P., Lim L.P., Burge C.B., Bartel D.P., Burge C.B., Bartel D.P., Bartel D.P. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Goodman M., Porter C.A., Czelusniak J., Page S.L., Schneider H., Shoshani J., Gunnell G., Groves C.P., Porter C.A., Czelusniak J., Page S.L., Schneider H., Shoshani J., Gunnell G., Groves C.P., Czelusniak J., Page S.L., Schneider H., Shoshani J., Gunnell G., Groves C.P., Page S.L., Schneider H., Shoshani J., Gunnell G., Groves C.P., Schneider H., Shoshani J., Gunnell G., Groves C.P., Shoshani J., Gunnell G., Groves C.P., Gunnell G., Groves C.P., Groves C.P. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol. Phylogenet. Evol. 1998;9:585–598. doi: 10.1006/mpev.1998.0495. [DOI] [PubMed] [Google Scholar]
- Gordon D., Abajian C., Green P., Abajian C., Green P., Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Grad Y., Aach J., Hayes G.D., Reinhart B.J., Church G.M., Ruvkun G., Kim J., Aach J., Hayes G.D., Reinhart B.J., Church G.M., Ruvkun G., Kim J., Hayes G.D., Reinhart B.J., Church G.M., Ruvkun G., Kim J., Reinhart B.J., Church G.M., Ruvkun G., Kim J., Church G.M., Ruvkun G., Kim J., Ruvkun G., Kim J., Kim J. Computational and experimental identification of C. elegans microRNAs. Mol. Cell. 2003;11:1253–1263. doi: 10.1016/s1097-2765(03)00153-9. [DOI] [PubMed] [Google Scholar]
- Han J., Lee Y., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Lee Y., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N., Cho Y., Zhang B.T., Kim V.N., Zhang B.T., Kim V.N., Kim V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Hancock J.M., Tautz D., Dover G.A., Tautz D., Dover G.A., Dover G.A. Evolution of the secondary structures and compensatory mutations of the ribosomal RNAs of Drosophila melanogaster. Mol. Biol. Evol. 1988;5:393–414. doi: 10.1093/oxfordjournals.molbev.a040501. [DOI] [PubMed] [Google Scholar]
- He L., Hannon G.J., Hannon G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Higgs P. RNA secondary structure: Physical and computational aspects. Q. Rev. Biophys. 2000;33:199–253. doi: 10.1017/s0033583500003620. [DOI] [PubMed] [Google Scholar]
- Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., Diekhans M., Furey T.S., Harte R.A., Hsu F., Furey T.S., Harte R.A., Hsu F., Harte R.A., Hsu F., Hsu F., et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmougin F., Thompson J.D., Gouy M., Higgins D.G., Gibson T.J., Thompson J.D., Gouy M., Higgins D.G., Gibson T.J., Gouy M., Higgins D.G., Gibson T.J., Higgins D.G., Gibson T.J., Gibson T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998;23:403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;V16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M., Tamura K., Nei M., Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut R., Lendeckel W., Tuschl T., Rauhut R., Lendeckel W., Tuschl T., Lendeckel W., Tuschl T., Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lee R.C., Ambros V., Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Legendre M., Lambert A., Gautheret D., Lambert A., Gautheret D., Gautheret D. Profile-based detection of microRNA precursors in animal genomes. Bioinformatics. 2005;21:841–845. doi: 10.1093/bioinformatics/bti073. [DOI] [PubMed] [Google Scholar]
- Lim L.P., Glasner M.E., Yekta S., Burge C.B., Bartel D.P., Glasner M.E., Yekta S., Burge C.B., Bartel D.P., Yekta S., Burge C.B., Bartel D.P., Burge C.B., Bartel D.P., Bartel D.P. Vertebrate microRNA genes. Science. 2003a;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Lim L.P., Lau N.C., Weinstein E.G., Abdelhakim A., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P., Lau N.C., Weinstein E.G., Abdelhakim A., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P., Weinstein E.G., Abdelhakim A., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P., Abdelhakim A., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P., Rhoades M.W., Burge C.B., Bartel D.P., Burge C.B., Bartel D.P., Bartel D.P. The microRNAs of Caenorhabditis elegans. Genes & Dev. 2003b;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M., Bartel D.P., Linsley P.S., Johnson J.M., Linsley P.S., Johnson J.M., Johnson J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lunter G., Ponting C.P., Hein J., Ponting C.P., Hein J., Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput. Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D.H., Sabina J., Zuker M., Turner D.H., Sabina J., Zuker M., Turner D.H., Zuker M., Turner D.H., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Newman T.L., Tuzun E., Morrison V.A., Hayden K.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E., Tuzun E., Morrison V.A., Hayden K.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E., Morrison V.A., Hayden K.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E., Hayden K.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E., McGrath S.D., Rocchi M., Eichler E.E., Rocchi M., Eichler E.E., Eichler E.E. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005;15:1344–1356. doi: 10.1101/gr.4338005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. Springer-Verlag; New York: 1970. [Google Scholar]
- Pang K.C., Frith M.C., Mattick J.S., Frith M.C., Mattick J.S., Mattick J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006;22:1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., van Dyk L.F., Ho C.K., Shuman S., Chien M., Ho C.K., Shuman S., Chien M., Shuman S., Chien M., Chien M., et al. Identification of microRNAs of the herpesvirus family. Nat. Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Plasterk R.H. Micro RNAs in animal development. Cell. 2006;124:877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Pollard K.S., Salama S.R., King B., Kern A.D., Dreszer T., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Salama S.R., King B., Kern A.D., Dreszer T., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., King B., Kern A.D., Dreszer T., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Kern A.D., Dreszer T., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Dreszer T., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Katzman S., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Siepel A., Pedersen J.S., Bejerano G., Baertsch R., Pedersen J.S., Bejerano G., Baertsch R., Bejerano G., Baertsch R., Baertsch R., et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2:e168. doi: 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S., Noonan J.P., Paabo S., Rubin E.M., Noonan J.P., Paabo S., Rubin E.M., Paabo S., Rubin E.M., Rubin E.M. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- Qian Y., Jin L., Su B., Jin L., Su B., Su B. Construction and characterization of bacterial artificial chromosome library of black-handed spider monkey (Ateles geoffroyi) Genome. 2004;47:239–245. doi: 10.1139/g03-122. [DOI] [PubMed] [Google Scholar]
- Raymond C.K., Roberts B.S., Garrett-Engele P., Lim L.P., Johnson J.M., Roberts B.S., Garrett-Engele P., Lim L.P., Johnson J.M., Garrett-Engele P., Lim L.P., Johnson J.M., Lim L.P., Johnson J.M., Johnson J.M. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA. 2005;11:1737–1744. doi: 10.1261/rna.2148705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Fiegler H., Shapero M.H., Carson A.R., Chen W., Shapero M.H., Carson A.R., Chen W., Carson A.R., Chen W., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer E.L., Durbin R., Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M., Brennecke J., Bushati N., Russell R.B., Cohen S.M., Bushati N., Russell R.B., Cohen S.M., Russell R.B., Cohen S.M., Cohen S.M. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Swanson W.J., Vacquier V.D., Vacquier V.D. The rapid evolution of reproductive proteins. Nat. Rev. Genet. 2002;3:137–144. doi: 10.1038/nrg733. [DOI] [PubMed] [Google Scholar]
- Vallone P.M., Butler J.M., Butler J.M. AutoDimer: A screening tool for primer-dimer and hairpin structures. Biotechniques. 2004;37:226–231. doi: 10.2144/04372ST03. [DOI] [PubMed] [Google Scholar]
- Wyckoff G.J., Wang W., Wu C.I., Wang W., Wu C.I., Wu C.I. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403:304–309. doi: 10.1038/35002070. [DOI] [PubMed] [Google Scholar]
- Xie X., Lu J., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M., Lu J., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M., Lindblad-Toh K., Lander E.S., Kellis M., Lander E.S., Kellis M., Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.L., Qian Y.P., Nie W.H., Chi J.X., Yang F.T., Su B., Qian Y.P., Nie W.H., Chi J.X., Yang F.T., Su B., Nie W.H., Chi J.X., Yang F.T., Su B., Chi J.X., Yang F.T., Su B., Yang F.T., Su B., Su B. Construction, characterization and chromosomal mapping of bacterial artificial chromosome (BAC) library of Yunnan snub-nosed monkey (Rhinopithecus bieti) Chromosome Res. 2004;12:251–262. doi: 10.1023/b:chro.0000021946.13556.40. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Zhang F., Chen X., Wang Y., Wang W., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Zhang F., Chen X., Wang Y., Wang W., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Chen X., Wang Y., Wang W., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Wang Y., Wang W., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Wang W., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Lin A.A., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Cavalli-Sforza L.L., Jin L., Huo R., Sha J., Jin L., Huo R., Sha J., Huo R., Sha J., Sha J., et al. Genetic variations, rapid evolution and clinical association of the human spermatogenesis related gene NYD-SP12. J. Mol. Evol. 2007 (in press) [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]