

Abstract
The serine–threonine protein kinase Akt has been identified as an important mediator of cell survival able to counteract apoptotic stimuli. However, hibernation, a model of natural tolerance to cerebral ischemia, is associated with downregulation of Akt. We previously established a model of ischemic tolerance in a PC12 cell line and using this model we now addressed the question whether ischemic tolerance also downregulates Akt in PC12 cells. Kinetic studies showed decreased Akt phosphorylation in tolerized cells. Similarly, phosphorylated levels of three major targets of Akt and well-known proapoptotic factors, the glycogen synthase kinase 3 (GSK-3), a Forkhead family member, FoxO4, and the protein murine double minute 2 (MDM2), all inactivated upon phosphorylation by Akt, were decreased in preconditioned cells. In addition, pharmacological blockade of the phosphoinositide 3-kinase (PI3K)/Akt pathway reduced cell death induced by oxygen and glucose deprivation (OGD) and increased the protective effect of preconditioning (PC). Furthermore, decreasing availability of P-Akt by transfecting PC12 cells with constructs of inactive Akt also resulted in protection against OGD and potentiation of the protective effect of PC. Depending on the environment, GSK-3, FOXO-4, and MDM2 can trigger apoptotic responses or cell cycle arrest, and thus, in a situation of reduced energy, driving the cells into a state of quiescence might be neuroprotective. This work suggests that in the context of tolerance downregulation of Akt is beneficial.
Keywords: Akt, FoxO4, GSK-3, ischemic tolerance, MDM2, PC12 cells
Introduction
Preconditioning (PC) with a brief toxic stimulus allows the cells to acquire tolerance to a more severe insult. This general phenomenon known as tolerance has been reported in a wide variety of models. The molecular mechanisms associated with the acquisition of this tolerant state can be induced by diverse insults and are believed to be independent of the nature of insult. However, these mechanisms are still mainly unknown. To improve our knowledge of signaling cascades involved in the phenomenon of tolerance, we developed a model of ischemic tolerance in PC12 cells (Hillion et al, 2005). In this report, we also showed the contribution of apoptosis in cell death induced by oxygen and glucose deprivation (OGD) in PC12 cells and the capacity of 6 h of OGD PC to counteract cell death induced by 15 h of OGD 1 day later.
The serine–threonine protein kinase Akt has been shown to counteract apoptosis by phosphorylating and inactivating proapoptotic proteins such as glycogen synthase kinase 3 (GSK-3), the Bcl2 family member, BAD, Forkhead family members and procaspase-9. Direct targets of Akt also include the ubiquitin ligase protein murine double minute 2 (MDM2), responsible for the negative regulation of p53, a known proapoptotic transcription factor. The activation of Akt is dependent on its translocation to the plasma membrane and association via its pleckstrin homology domain with the phospholipids phosphatidyl-inositol-3,4,5-triphosphate [PI(3,4,5)P3], products of the conversion of phosphatidyl-inositol-4,5-biphosphate [PI(4,5)P2] by phosphoinositide 3-kinases (PI3K). Phosphorylation of Akt on Ser473 and Thr308 occurs after relocalization at the plasma membrane and is required for Akt enzymatic activation.
Although a decrease of Akt activity is expected to be proapoptotic, hibernation, a model of natural tolerance to cerebral ischemia (Frerichs and Hallenbeck, 1998) is associated with downregulation of Akt (Cai et al, 2004). Using our model of ischemic tolerance in PC12 cells, we addressed the question whether ischemic tolerance also downregulates Akt in PC12 cells. Our kinetic studies showed a reduction of Akt phosphorylation in tolerized cells. In addition, pharmacological blockade of the PI3K/Akt pathway reduced OGD-induced cell death and increased the protective effect of PC. Decreasing availability of P-Akt by transfecting PC12 cells with constructs of inactive Akt, with mutations of Thr308 and/or Ser473 to Ala, also resulted in protection against OGD and potentiation of the protective effect of PC.
Materials and methods
Oxygen and Glucose Deprivation and Preconditioning of PC12 Cells
PC12 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were plated at a density of 5 × 105 cells/well in six-well multiwell Biocoat plates precoated with poly-D-lysine (BD Biosciences, Bedford, MA, USA) and grown for 24 h in RPMI 1640 culture medium supplemented with 10% horse serum and 5% fetal bovine serum (FBS) (ATCC), ‘complete medium’, at 37°C in a normoxia + 5% CO2 atmosphere. For the induction of OGD, cells were washed twice in RPMI without glucose (Life Technologies, Carlsbad, CA, USA) switched to RPMI 1640 without glucose supplemented with 2% horse serum and 1% FBS (OGD medium) and placed in modular incubator chambers (Billups-Rothenberg, Del Mar, CA, USA). The chambers were flushed with a gas mixture of 95% N2/5% CO2 for 30 mins at room temperature at 3 L/min. After flushing, the chambers were sealed and placed at 37°C. Oxygen and glucose deprivation was performed for 15 h or for the indicated times (O2 levels 2% to 3%). For PC experiments, cells were grown for 24 h in complete medium. They were washed twice in RPMI without glucose, switched to OGD medium and subjected to 6 h of OGD with initial flushing as described above. Sister plates not subjected to OGD PC (sham preconditioned) were washed twice with RPMI without glucose and maintained in RPMI 1640 (glucose present) supplemented with 2% horse serum and 1% FBS. Sham-preconditioned and OGD-preconditioned cells were maintained in glucose-containing medium and normoxia for 24 h for the development of cellular tolerance. Subsequently, these cells were exposed to severe OGD for 15 h. In addition, ‘nonischemic’ controls were sham preconditioned and maintained in RPMI 1640 supplemented with 2% horse serum and 1% FBS, but were not exposed to OGD. To evaluate the influence of different cell proliferation rates in preconditioned versus non-preconditioned cells, sister plates were seeded at different cell densities and OGD-induced cell death was measured by lactate dehydrogenase (LDH) assay (data not shown). As expected, we observed decreased cell death with lower cell density. However, given the very small reduction in cell number in preconditioned cells compared with naïve cells (because of cell cycle, arrest during the 6 h of OGD), we also observed that the resulting effect on cell death is minimal in comparison to the effect of PC and only very partially accounts for the protection observed in preconditioned cells.
Assessment of Cell Survival and Cell Death
Immediately after OGD, cell survival and cell death were assessed by release of LDH, with the LDH assay kit (Sigma-Aldrich, St Louis, MO, USA). Aliquots of culture medium were collected from sister wells for measurement of LDH leakage. For assessment of total LDH activity, cells were incubated with 100 μL of lysis solution/well for 30 mins at 37°C and lysates were centrifuged to remove cellular debris. Absorbance was read at 490 nm and LDH release was expressed as a percentage of the total LDH (cellular plus medium LDH), which represents the proportion of cell death caused by OGD.
Staining of Apoptotic Cells and Fluorescence-activated cell sorter Analysis
Cells were collected using a papain dissociation system (Worthington Biochemical Corporation, Lakewood, NJ, USA) and 106 cells were labeled with Hoechst 33342 and propidium iodide (PI) (Vybrant Apoptosis Assay Kit #5; Molecular Probes, Eugene, OR, USA), according to manufacturer’s specifications. The cells were analyzed using a dual-laser FACSVantage SE flow cytometer (Becton Dickinson, Mountain View, CA, USA). Propidium iodide signal was excited using a 488-nm laser light and the emission captured with a bandpass filter set at 613 ± 20 nm. Hoechst 33342 was excited using a 351-nm ultraviolet laser light and its emission captured with a bandpass filter set at 450 ± 20 nm. Cell Quest Acquisition and Analysis software (Becton Dickinson) was used to acquire and quantify the fluorescence signal intensities and to graph the data as bivariate dot density plots. In two color experiments with Hoechst and PI, no compensation was required. In three color experiments with enhanced green fluorescent protein (EGFP), Hoechst and PI, spectral overlap between EGFP and PI was electronically compensated.
Western Blots
Immediately after OGD, cells were washed once, lysed in lysis buffer containing 100 mmol/L Tris-Hcl, 40 mmol/L ethylenediaminetetraacetic acid and 2% sodium dodecyl sulfate (SDS), scraped, and boiled at 95°C for 5 mins. The lysates were then sonicated for 10 secs, boiled at 95°C for an additional 5 mins, and centrifuged for 5 mins at 4°C. Total protein extracts (30 μg) were boiled for 5 mins in Laemmli buffer and resolved by electrophoresis on a 10% SDS-polyacrylamide gel electrophoresis gel (Invitrogen, Carlsbad, CA, USA). Proteins were electrotransferred onto a polyvinylidine fluoride membrane (Invitrogen). Non-specific binding was blocked by incubation overnight in 20 mmol/L Tris, pH 7.6-buffered saline with 0.1% Tween 20 (TBST) containing 5% nonfat dried milk. Membranes were probed overnight with primary antibody against Akt, P-Akt, GSK-3, P-GSK-3, P-FoxO4, P-MDM2 (all from Cell Signaling Technology, Beverly, MA, USA) or against β-actin (Sigma-Aldrich, St Louis, MO, USA), diluted 1:1,000 in Primary Antibody Dilution Buffer (1:10,000 for β-actin antibodies) and washed three times for 5 mins in TBST. The blots were then incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) (1:2,000) (anti-mouse IgG, 1:5,000 dilution, for β-actin) and horseradish peroxidase-conjugated anti-biotin antibody (1:1,000) in blocking buffer for 1 h at room temperature, washed three times for 5 mins in TBST, and visualized with a chemiluminescence detection system (SuperSignal West Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA).
Construction of Akt Mutant Constructs
Wild-type and mutated mouse Akt1 complementary DNA (cDNA) fragments were cloned in the EGFP-N1 expression vector (Clontech) at the HindIII/BamHI sites, so they were expressed as fusion to the N-terminus of EGFP. Mutations (Thr308 and Ser473 to Ala) were introduced by a Quick Change XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and confirmed by sequencing. The DNA sequencing also ensured that no stop codon was in the joint area of the Akt fragments and EGFP and that they were in the same reading frame.
Transient Transfection
PC12 cells were plated at a density of 5 × 105 cells/well in six-well multiwell Biocoat plates precoated with poly-D-lysine (BD Biosciences) and grown for 24 h in complete medium at 37°C in a normoxia + 5% CO2 atmosphere. They were then subjected or not to 6 h OGD for PC in the conditions described above and returned to low serum medium for another 24 h. For transfection, cells were incubated overnight at 37°C in 1 mL low serum medium containing a mixture of Akt plasmid cDNA and Lipofectamine 2000 transfection reagent (Invitrogen) at a DNA:LP2000 ratio of 1:2 in Opti-MEM (Invitrogen). The transfection incubation solution was removed 12 h later and cells returned to normal medium.
Statistics
All values are given as mean ± s.e.m. Repeated measures analysis of variance (ANOVA) or bifactorial repeated measures ANOVA followed by Bonferroni’s post hoc tests (GraphPad Prism 4.0 software, San Diego, CA, USA) were used for the analysis of differences between cells receiving different treatments. For each experiment, the same initial cell preparation was split and analyzed under the different experimental conditions.
Results
Preconditioning Downregulates Akt and Attenuates Oxygen and Glucose Deprivation-Induced Upregulation of Akt
Oxygen and glucose deprivation induced a strong increase in P-Akt expression in non-preconditioned cells. This increase was most prominent after 2 h of OGD and went back to basal values after 6 h of OGD. Preconditioning, that is, pre-exposure to 6 h of OGD one day before, significantly reduced basal P-Akt expression (Figure 1). Furthermore, although OGD markedly increased P-Akt expression, the levels were significantly lower than in non-preconditioned cells (Figure 1). Thus, a bifactorial repeated measures ANOVA showed a significant effect of the PC factor (two levels: PC + OGD versus OGD; P < 0.001, n = 5) and the time factor (four levels: 0, 2, 4, and 6 h of OGD; P < 0.001). Bonferroni’s post hoc test showed a significant difference between the preconditioned and non-preconditioned cells at 0, 2, and 4 h of OGD (Figure 1). As total Akt levels were unchanged (Figure 1), these results indicate that OGD and PC modified the phosphorylation state, and therefore the activity of Akt. In agreement with these results, similar effects of OGD and PC could be observed in the patterns of phosphorylation of GSK-3, FoxO4, and MDM2, major substrates of Akt. As for P-Akt, the levels of P-GSK-3 observed under both basal conditions and OGD were significantly lower in preconditioned compared with non-preconditioned cells (Figure 2). A bifactorial repeated measures ANOVA showed a significant effect of the PC factor (two levels: PC + OGD versus OGD; P < 0.001, n = 5) and of the time factor (four levels: 0, 2, 4, and 6 h of OGD; P < 0.001). Bonferroni’s post hoc test showed a significant difference between preconditioned and non-preconditioned cells at 0 and 2 h of OGD (Figure 2). Total GSK-3 levels remained unchanged in both conditions (OGD or PC + OGD). Basal levels of P-FoxO4 were barely detectable, but a very high increase in P-FoxO4 expression was observed during the first 2 h of OGD. Preconditioning significantly attenuated OGD-induced increase in FoxO4 expression (Figure 3). A bifactorial repeated measures ANOVA showed a significant effect of the PC factor (two levels: PC + OGD versus OGD; P < 0.001, n = 4) and of the time factor (four levels: 0, 2, 4, and 6 h of OGD; P < 0.001). Bonferroni’s post hoc test showed a significant difference between the preconditioned and non-preconditioned cells at 2 and 4 h of OGD (Figure 3). Finally, the levels of P-MDM2 observed under both basal conditions and OGD were significantly lower in preconditioned compared with non-preconditioned cells (Figure 4). A bifactorial repeated measures ANOVA showed a significant effect of the PC factor (two levels: PC + OGD versus OGD; P < 0.0001, n = 4) and of the time factor (four levels: 0, 2, 4, and 6 h of OGD; P < 0.05). Bonferroni’s post hoc test showed a significant difference between the preconditioned and non-preconditioned cells at 0 and 2 h of OGD (Figure 4). Our results suggest that PC induces downregulation of Akt (lower basal expression of P-Akt, P-GSK-3 and P-MDM2, without changes in nonphosphorylated Akt or GSK-3). This downregulation does not affect the ability of OGD to activate Akt, although the final P-Akt levels and, consequently, P-GSK-3, P-FoxO4, and P-MDM2 levels are significantly lower in preconditioned than in non-preconditioned cells.
Figure 1.
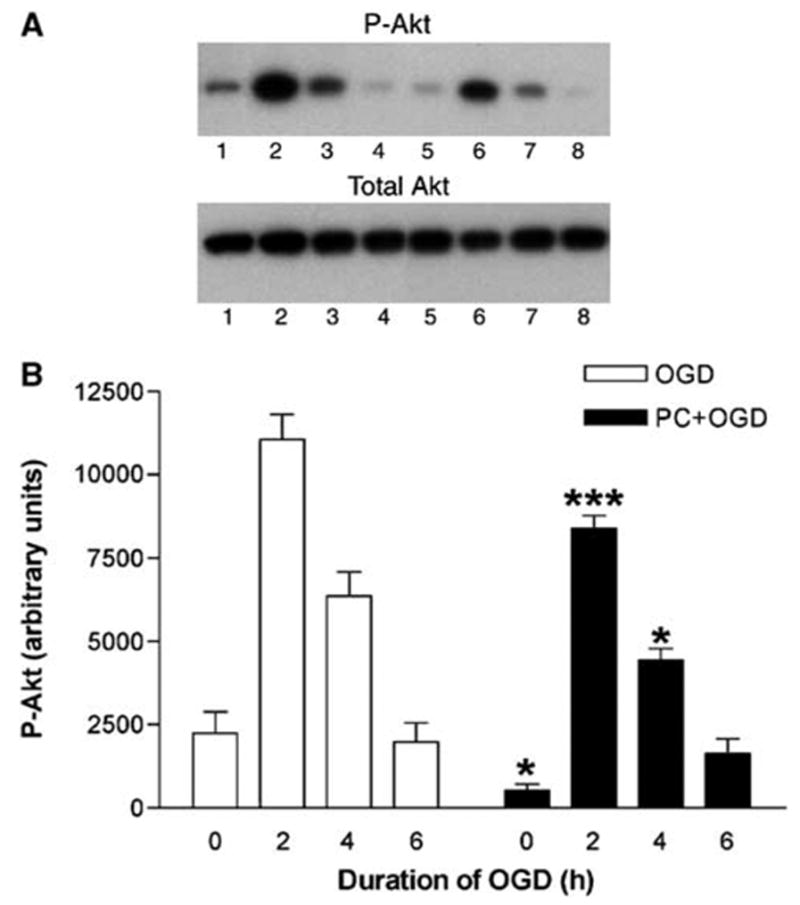
Effect of preconditioning on P-Akt and total Akt levels in PC12 cells exposed to oxygen and glucose deprivation (OGD). (A) Representative Western blots of P-Akt and total Akt levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (lanes 1, 2, 3, and 4, and lanes 5, 6, 7, and 8, respectively). (B) Quantitative densitometric analysis of the effect of preconditioning on P-Akt levels in PC12 cells exposed to OGD. Results represent means + s.e.m. (n = 6) of P-Akt levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (OGD and PC + OGD, respectively). A significant difference was obtained among the values of preconditioned and non-preconditioned cells during 0, 2, and 4 h of OGD. *P < 0.05 and ***P < 0.001, compared with OGD values; bifactorial repeated measures analysis of variance.
Figure 2.
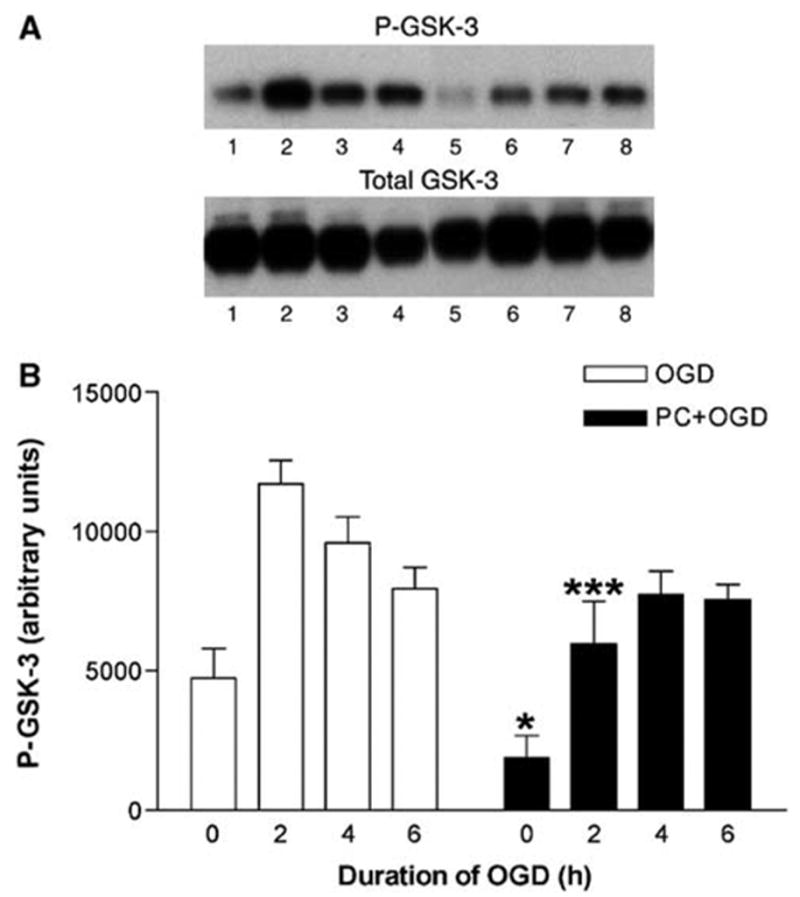
Effect of preconditioning on P-glycogen synthase kinase 3 (GSK-3) and total GSK-3 levels in PC12 cells exposed to oxygen and glucose deprivation (OGD). (A) Representative Western blots of P-GSK-3 and total GSK-3 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (lanes 1, 2, 3, and 4, and lanes 5, 6, 7, and 8, respectively). (B) Quantitative densitometric analysis of the effect of preconditioning on P-GSK-3 levels in PC12 cells exposed to OGD. Results represent means + s.e.m. (n = 6) of P-GSK-3 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (OGD and PC + OGD, respectively). A significant difference was obtained among the values of preconditioned and non-preconditioned cells during 0, and 2 h of OGD. *P < 0.05 and ***P < 0.001, compared with OGD values; bifactorial repeated measures analysis of variance.
Figure 3.

Effect of preconditioning on P-FoxO4 in PC12 cells exposed to oxygen and glucose deprivation (OGD). (A) Representative Western blot of P-FoxO4 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (lanes 1, 2, 3, and 4, and lanes 5, 6, 7, and 8, respectively). Reblotting with anti-β-actin was performed as loading control. (B) Quantitative densitometric analysis of the effect of preconditioning on P-FoxO4 levels in PC12 cells exposed to OGD. Results represent means + s.e.m. (n = 4) of P-FoxO4 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (OGD and PC + OGD, respectively). A significant difference was obtained among the values of preconditioned and non-preconditioned cells during 2 and 4 h of OGD. *P < 0.05 and ***P < 0.001, compared with OGD values; bifactorial repeated measures analysis of variance.
Figure 4.
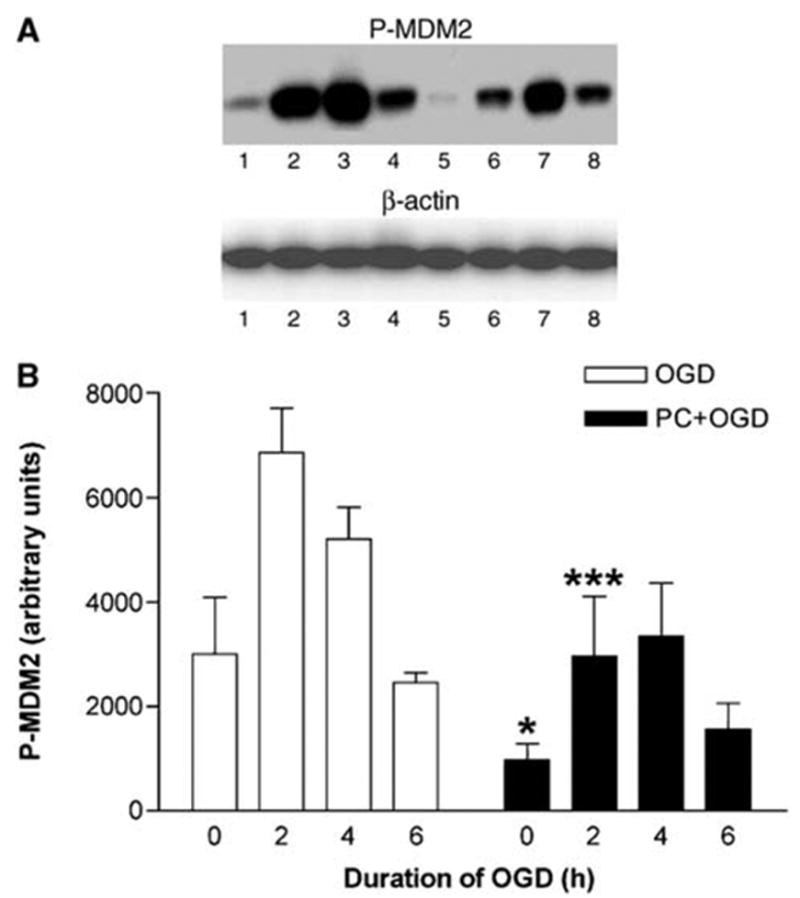
Effect of preconditioning on P-murine double minute 2 (MDM2) in PC12 cells exposed to oxygen and glucose deprivation (OGD). (A) Representative Western blot of P-MDM2 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (lanes 1, 2, 3, and 4, and lanes 5, 6, 7, and 8, respectively). Reblotting with anti-β-actin was performed as loading control. (B) Quantitative densitometric analysis of the effect of preconditioning on P-MDM2 levels in PC12 cells exposed to OGD. Results represent means + s.e.m. (n = 4) of P-MDM2 levels in cells exposed to OGD for 0, 2, 4, and 6 h without and with preconditioning (OGD and PC + OGD, respectively). A significant difference was obtained among the values of preconditioned and non-preconditioned cells during 0 and 2 h of OGD. *P < 0.05 and ***P < 0.001, compared with OGD values; bifactorial repeated measures analysis of variance.
Pharmacological Blockade of Phosphoinositide 3-Kinase/Akt Pathway Induces Tolerance to Oxygen and Glucose Deprivation and Potentiates Preconditioning-Induced Tolerance to Oxygen and Glucose Deprivation
Next, we investigated whether PC-induced down-regulation of Akt played a role in PC-induced tolerance to OGD. As observed earlier (Hillion et al, 2005), PC significantly increased cell viability after 15 h of OGD 1 day later. Analysis of cell death by LDH release showed that PC significantly decreased cell death induced by 15 h of OGD 1 day later (Figure 5; PC + OGD versus OGD; repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.01; n = 6). Exposure to the PI3K inhibitor LY294002 (10 μmol/L) significantly decreased OGD-induced LDH release and significantly potentiated the protective effect of PC (Figure 5; OGD versus LY + OGD and PC + OGD versus PC + LY + OGD; repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.05 and P < 0.001, respectively). In a previous report, we showed the predominance of apoptosis in OGD-induced cell death (Hillion et al, 2005). Indeed, flow cytometry analysis of cells labeled with specific markers for apoptosis, Annex-in V, and Hoechst 33342, and of DNA content, revealed the important contribution of apoptosis. Immunocytochemistry of caspase-3, a central executioner in the apoptotic process, further confirmed the activation of apoptotic pathways in OGD-induced PC12 cell death. In this study, all markers, including Hoescht 33342 in combination with PI, gave the same qualitative results. The nuclei of early apoptotic cells can be stained with nucleic acid dyes such as the fluorophore Hoechst 33342 but not with PI, a cell-impermeant dye that stains cells with compromised membrane integrity such as necrotic or late apoptotic cells. When used in combination, Hoechst 33342 and PI allow mixed populations of viable, early apoptotic, late apoptotic, and necrotic cells to be accurately distinguished and quantified based on their differential staining patterns (Hamel et al, 1996). Consistent with our pevious report (Hillion et al, 2005), fluorescence-activated cell sorter (FACS) analysis of cells stained with Hoechst 33342 and PI showed apoptosis as the main process involved in OGD-induced cell death in PC12 cells. After 15 h of OGD, there was about a 40% reduction in the number of viable cells, 10-fold increase in the number of apoptotic cells and a twofold increase in necrotic cells compared with control (Figure 6). Preconditioning significantly attenuated the effect of OGD, by increasing (about 15%) the number of viable cells and by decreasing (about 25%) the number of apoptotic cells (Figure 6; PC + OGD versus OGD; repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.001 and P < 0.05, respectively; n = 5). Exposure of cells to LY294002 (10 μmol/L) significantly increased the number of viable cells compared with untreated OGD (LY + OGD versus OGD; repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.05). Furthermore, LY294002 (10 μmol/L) significantly potentiated the protective effect of PC after 15 h of OGD (Figure 6; PC + OGD versus PC + LY + OGD; repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.01 and P < 0.05, respectively). Therefore, pharmacological blockade of the PI3K/Akt pathway with LY294002 was cytoprotective and potentiated PC-induced tolerance to OGD.
Figure 5.
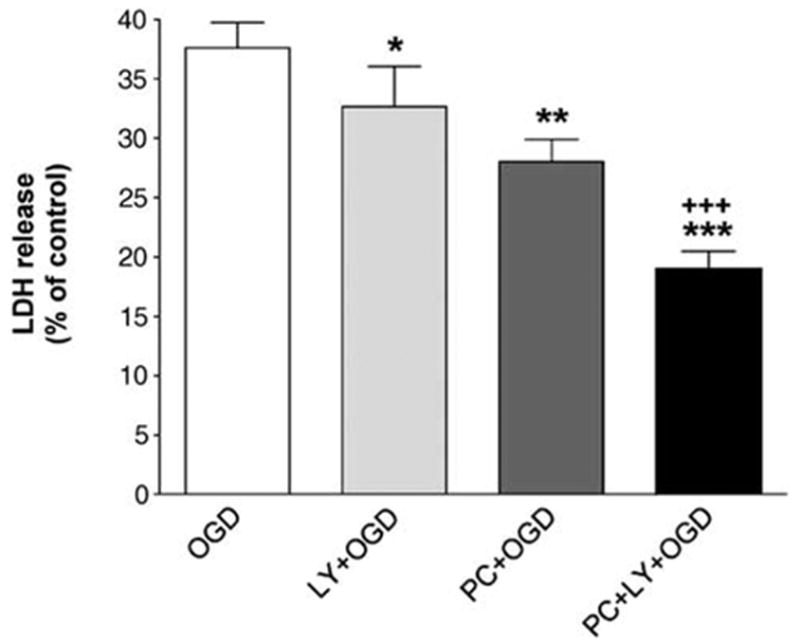
Effect of the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 and preconditioning on oxygen and glucose deprivation (OGD)-induced cell death in PC12 cells. Cell death is shown as means + s.e.m. (n = 6) of lactate dehydrogenase (LDH) release (in % of control). Both preconditioning (PC) and LY294002 (10 μmol/L) significantly decreased cell death induced by 15 h of OGD and LY294002 significantly potentiated the protective effect of PC. *P < 0.05 and ***P < 0.001, compared with OGD values; +++P < 0.001 compared with PC + OGD; repeated measures analysis of variance.
Figure 6.

Effect of the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 and preconditioning on apoptosis oxygen and glucose deprivation (OGD)-induced apoptosis in PC12 cells. Results represent means + s.e.m. (n = 5) of the percentage of total of viable, early apoptotic (E apop), medium–late apoptotic (M–L apop) and necrotic (necro.) cells quantified by fluorescence-activated cell sorter (FACS) analysis using propidium iodide (PI) and Hoechst 33342 labeling. Preconditioning significantly increased the percentage of viable cells and decreased the percentage of apoptotic cells compared with OGD alone. LY294002 (10 μmol/L) significantly increased the number of viable cells compared with OGD and significantly potentiated the protective effect of PC. *P < 0.05 and ***P < 0.001, compared with OGD values; +P < 0.05 and + +P < 0.001, compared with PC + OGD; repeated measures analysis of variance.
Transfection of Inactive Akt Mutants Induces Tolerance to Oxygen and Glucose Deprivation and Potentiates Preconditioning-Induced Tolerance to Oxygen and Glucose Deprivation
Downregulation of Akt activation was also induced by transient transfection of several inactive mutants of Akt, with substitution of Ser473 and/or Thr308 by alanine (TA, AS, and AA). Fluorescence-activated cell sorter analysis allowed the separation of transfected (green fluorescent protein (GFP) + ) and nontransfected cells (GFP−). We could observe a small increase in sensitivity to OGD in transfected cells compared with previous experiments because of Lipofectamine-induced cytotoxicity. In cells transfected with wild-type Akt (TS/GFP + ) and their controls (TS/GFP−), OGD induced a 60% to 70% reduction in the number of viable cells (Figure 7A). The effect of OGD was significantly reduced in cells transfected with the AS and the AA mutant (AS/GFP + versus AS/GFP− and AA/GFP + versus AA/GFP−; bifactorial repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.05 in both cases; n = 6). A trend for a counteraction of OGD effect was also observed with the TA mutant (Figure 7A). In cells transfected with wild-type Akt (TS/GFP + ) and their controls (TS/GFP−), PC attenuated the effect of 15 h of OGD 2 days later (increase of about 20% in the number of viable cells compared with non-preconditioned cells; Figure 7B). Transfection with any of the mutant Akt constructs (TA-GFP +, AS-GFP +, or AA-GFP + ) significantly potentiated the protective effect of PC (TA-GFP−, AS-GFP−, or AA-GFP−) with about 40% to 50% increase in the number of viable cells compared with non-preconditioned cells (TA/GFP + versus TA/GFP−, AS/GFP + versus AS/GFP−, and AA/GFP + versus AA/ GFP−; bifactorial repeated measures ANOVA with Bonferroni’s post hoc test: P < 0.05, P < 0.05, and P < 0.01, respectively) (Figure 7B). Therefore, down-regulation of Akt by either blockade of PI3K/Akt pathway or transfection of inactive Akt mutants induced partial tolerance to OGD and potentiated PC-induced tolerance to OGD, strongly suggesting that downregulation of Akt is a mechanism involved in the induction of ischemic tolerance in PC12 cells.
Figure 7.
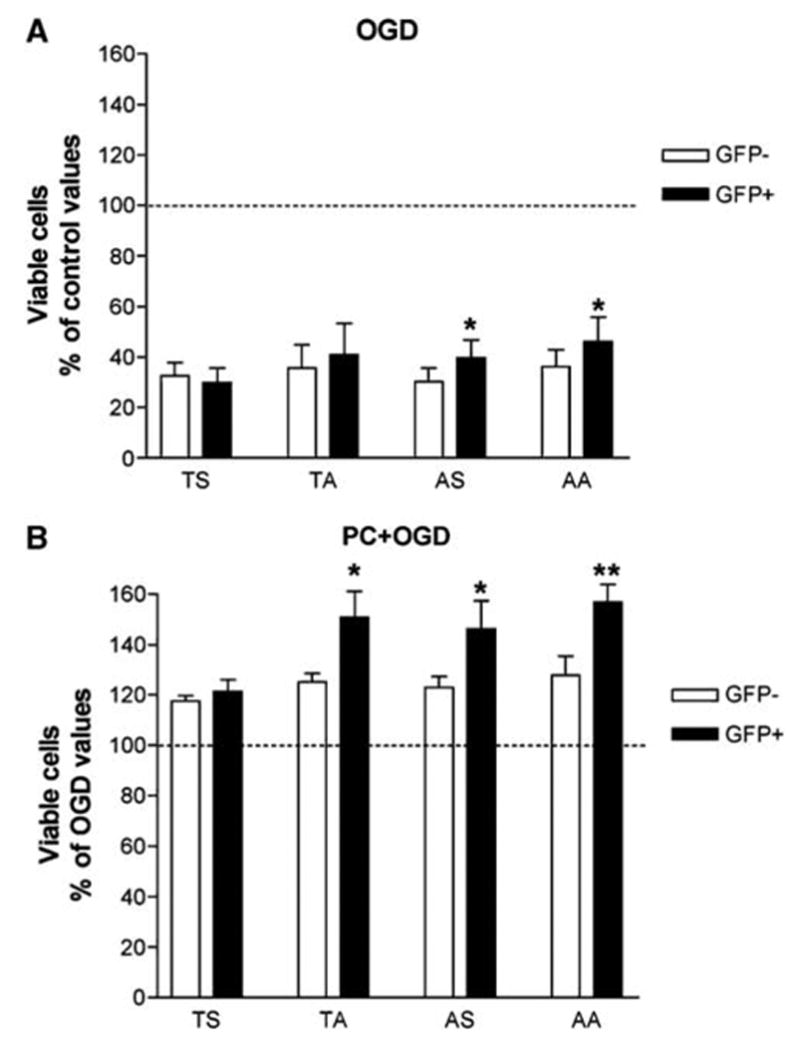
Effect of transfection of inactive Akt mutants and preconditioning on oxygen and glucose deprivation (OGD)-induced apoptosis in PC12 cells. Results represent means + s.e.m. (n = 6) of the percentage of total of viable PC12 cells (% of control or OGD values) obtained by fluorescence-activated cell sorter (FACS) analysis using propidium iodide (PI) and Hoechst 33342 labeling. Four different transfections, with either the wild-type Akt (TS) or Akt mutants were analyzed. In the mutants, an alanine was substituted for Ser473 (TA) or Thr308 (AS) or for both residues (AA). The wild-type (TS) and the mutated Akt constructs (TA, AS, and AA) were fused to the N-terminus of green fluorescent protein (GFP) and transiently transfected in PC12 cells. Fluorescence-activated cell sorter analysis allowed the separation of transfected (GFP + ) and nontransfected cells (GFP−). (A) Oxygen and glucose deprivation-induced decrease in viable cells was significantly counteracted by transfection with AS and AA (*P < 0.05 compared with GFP− cells; bifactorial repeated measures analysis of varainve (ANOVA)). (B) Preconditioning counteraction of OGD-induced decrease in viable cells was significantly potentiated by transfection with TA, AS, and AA (*P < 0.05 and **P < 0.01, compared with nontransfected GFP− cells; bifactorial repeated measures analysis of variance).
Discussion
Ischemic PC is a well-known phenomenon able to afford robust protection against injury to various organs. However, the compensatory survival mechanisms activated after a sublethal insult are still poorly understood. In this paper, we have examined the role of the PI3K/Akt pathway in OGD-induced tolerance in PC12 cells. Our data show that OGD induces an early upregulation of Akt phosphorylation. This is in agreement with previous reports where Akt phosphorylation was found to increase during hypoxia in PC12 cells (Alvarez-Tejado et al, 2001; Beitner-Johnson et al, 2001) or at the onset of middle cerebral artery occlusion in mice (Shibata et al, 2002). Similarly, in the dorsocaudal brainstem of the rat, hypoxia was associated with time-dependent increases in phosphorylated Akt (Simakajornboon et al, 2001). We then analyzed phosphorylation levels of Akt in preconditioned and non-preconditioned cells between 0 and 6 h of OGD. We found that PC for 6 h followed by reoxygenation for 24 h decreased the levels of phosphorylated Akt. Furthermore, although OGD markedly increased P-Akt expression in preconditioned cells, the levels were significantly lower than in non-preconditioned cells. Evidence has accumulated over the past decade consistently identifying Akt as an important mediator of cell survival able to counteract apoptotic stimuli (Marte and Downward, 1997; Brunet et al, 1999). However, Akt plays a key role in a broad spectrum of other essential cellular functions, therefore its mechanisms of action are multiple and complex and much remains to be elucidated (Kandel and Hay, 1999; Franke et al, 2003; Scheid and Woodgett, 2003). Thus, the role of Akt must be evaluated in a given model and given environment. In the context of ischemic tolerance, studies in cardiac or liver PC suggest a protective role for the PI3K/Akt pathway (Mocanu et al, 2002; Armstrong, 2004; Carini et al, 2004). In brain ischemic tolerance where few data are available, it is unclear whether activation of Akt contributes (Yano et al, 2001) or not (Namura et al, 2000) to PC-driven neuroprotection. Recently, Nakajima et al (2004) compared levels of phosphorylated Akt during reperfusion after focal cerebral ischemia in the rat and found that these levels were lower in the preconditioned than in the non-preconditioned animals. Hibernating animals can sustain for a long time a profound reduction in brain blood flow without any brain injury, thus making hibernation a natural model of ischemic tolerance (Frerichs and Hallenbeck, 1998). Akt phosphorylation and activity were found to be downregulated in the brain and other organs of hibernating ground squirrels (Cai et al, 2004).
We then investigated whether lower Akt phosphorylation levels correlate with a reduction in Akt activity and thus examined phosphorylation levels of several important downstream targets of Akt and proapoptotic factors. In fact, a significant decrease in phosphorylation levels of the protein kinase GSK3 and the transcription factor FoxO4 was observed in tolerized cells. Downregulation of Akt activity was further confirmed by examining levels of phosphorylated MDM2, another substrate of Akt. Compared with naïve cells, cells preconditioned 24 h earlier, exhibited lower levels of P-MDM2, both under basal conditions and after OGD. Akt-mediated phosphorylation of MDM2 allows subsequent ubiquitination and degradation of the p53 tumor suppressor protein whose activation is known to be proapoptotic (Ogawara et al, 2002; Feng et al, 2004). Phosphorylation of GSK3 and FoxO4 by Akt inactivates their function (van Weeren et al, 1998; Kops et al, 2002; Cross et al, 2000; Rena et al, 1999; Brazil et al, 2004) and promotes cell survival. Hence, a decrease in the phosphorylation levels of GSK3, FoxO4, and MDM2 is in favor of a reduced activation of cell survival pathways. Therefore, tolerance-induced increase in cell survival is expected to be associated with an increase rather than a reduction in Akt activation. However, in the model described here a decrease in Akt activation is associated with a reduction in cell death, suggesting that too much activation of Akt is harmful. In agreement, a recent report indicates that genetic inactivation of one of the three isoforms of Akt (Akt1) protects against ischemic brain injury (Alkayed et al, 2005). An explanation for these findings is that besides their well-documented role in apoptosis, GSK3, p53 as well as the FoxO superfamily of transcription factors through their multiple targets exert an essential regulatory influence on main cellular functions such as cell proliferation and growth arrest (Cohen and Frame, 2001; Jope and Johnson, 2004; Arden and Biggs, 2002; Tran et al, 2003). Thus, depending on the environment, these proteins can trigger apoptotic responses or cell cycle arrest, and, in a situation of stress, driving the cells into a state of quiescence might be neuroprotective. Indeed, the involvement of forkhead proteins in longevity was first shown in the dauer larva of the nematode Caenorhabditis elegans, where activation of the worm FoxO transcription factor, DAF-16, is necessary for the formation of the dauer larva stage under conditions of starvation and crowding (Ogg et al, 1997). In mammals, FoxO protein family was shown to be essential for the long-term survival of quiescent cells and to play an important role in cell cycle arrest (Burgering and Kops, 2002; Kops et al, 2002; Cunningham et al, 2004). Similarly, Imae et al (2003) reported that FoxO protein levels were increased by fasting, suggesting that in conditions of food scarcity, activation of FoxO transcription factors may be essential for survival. Furthermore, FoxO3a, by inducing DNA repair at the G2 to M checkpoint, was found to play an important role in resistance to stress (Tran et al, 2002).
In view of a potential deleterious effect of a strong activation of Akt during OGD, we tested whether pharmacological blockade of Akt would be protective against OGD-induced cell death. Inhibition of the PI3K/Akt pathway with LY294002, a specific inhibitor of PI3K, during OGD was found to be protective in PC12 cells. Similarly, in cells preconditioned for 6 h, exposure to LY294002 during OGD 24 h later induced a potentiation of PC-induced cytoprotection. These data support a beneficial impact of Akt inhibition in an environment low in energy. This finding was further confirmed in a last set of experiments where PC12 cells were transfected with different constructs of Akt cDNA, with mutations of Thr308 and/or Ser473 to Ala and survival to OGD analyzed. Transfection with either mutant Akt cDNA, but not with wild-type Akt cDNA, significantly potentiated PC-induced protection. Taken together, these data indicate that, in contrast to what could be expected, Akt down-regulation is an important mechanism of tolerance-induced protection. However, similarly to the regulation of apoptosis that involves different protein kinase pathways (Cross et al, 2000), acquisition of tolerance is an effect of a complex network of signals that cooperate to provide the cells with resistance to a severe insult. In a recent study, crosstalk between kinases showed to provide compensation to ensure protection of PC (Hausenloy et al, 2004). One should also keep in mind that PC allows moderate activation of molecular pathways, which may prevent their overactivation during subsequent lethal stimuli.
In conclusion, the results obtained with a model of ischemic tolerance in PC12 cells suggest that overactivation of the antiapoptotic serine–threonine kinase Akt is detrimental rather than beneficial, confirming the alteration of cell responses after OGD PC. Future research will focus on deciphering the biochemical pathways responsible for the development of tolerance to generate novel opportunities for drug discovery and drug-based therapies.
Footnotes
This research was supported by the Intramural Research Program of the NIH, NINDS.
References
- Alkayed NJ, Murphy SJ, Graham SM, Hurn PD. Role of Akt1 in estrogen-mediated neuroprotection. J Cereb Blood Flow Metab. 2005;25:S51. [Google Scholar]
- Alvarez-Tejado M, Naranjo-Suarez S, Jimenez C, Carrera AC, Landazuri MO, Del Peso L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells—protective role in apoptosis. Proc Natl Acad Sci USA. 2001;276:22368–74. doi: 10.1074/jbc.M011688200. [DOI] [PubMed] [Google Scholar]
- Arden KC, Biggs WH., III Regulation of the FoxO family of transcription factors by phosphatidylinositol-3 kinase-activated signaling. Arch Biochem Biophys. 2002;403:292–8. doi: 10.1016/s0003-9861(02)00207-2. [DOI] [PubMed] [Google Scholar]
- Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Rust RT, Hsieh TC, Milhorn DE. Hypoxia activates Akt and induces phosphorylation of GSK-3 in PC12 cells. Cell Signal. 2001;13:23–7. doi: 10.1016/s0898-6568(00)00128-5. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling—AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–42. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control—long live Forkheads. Trends Biochem Sci. 2002;27:352–60. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Cai D, McCarron RM, Hallenbeck JM. Cloning and characterization of a forkhead transcription factor gene, FoxO1a, from thirteen-lined ground squirrel. Gene. 2004;343:203–9. doi: 10.1016/j.gene.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Carini R, Grazia De Cesaris M, Splendore R, Baldanzi G, Nitti MP, Alchera E, Filigheddu N, Domenicotti C, Pronzato MA, Graziani A, Albano E. Role of phosphatidylinositol 3-kinase in the development of hepatocyte preconditioning. Gastroenterology. 2004;127:914–923. doi: 10.1053/j.gastro.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cross TG, Scheel-Toellner D, Henriquez NV, Deacon E, Samon M, Lord JM. Serine/threonine protein kinases and apoptosis. Exp Cell Res. 2000;256:34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- Cunningham MA, Zhu Q, Hammond JM. FoxO1a can alter cell cycle progression by regulating the nuclear localization of p27kip in granulosa cells. Mol Endocrinol. 2004;18:1756–67. doi: 10.1210/me.2004-0071. [DOI] [PubMed] [Google Scholar]
- Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279:35510–7. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis—size matters. Oncogene. 2003;22:8983–98. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- Frerichs KU, Hallenbeck JM. Hibernation in ground squirrels induces state and species-specific tolerance to hypoxia and aglycemia—an in vitro study in hippocampal slices. J Cereb Blood Flow Metab. 1998;18:168–75. doi: 10.1097/00004647-199802000-00007. [DOI] [PubMed] [Google Scholar]
- Hamel W, Dazin P, Israel MA. Adaptation of a simple flow cytometric assay to identify different stages during apoptosis. Cytometry. 1996;25:173–81. doi: 10.1002/(SICI)1097-0320(19961001)25:2<173::AID-CYTO6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Mocanu MM, Yellon DM. Cross-talk between the survival kinases during early reperfusion—its contribution to ischemic preconditioning. Cardiovasc Res. 2004;63:305–12. doi: 10.1016/j.cardiores.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Hillion JA, Takahashi K, Maric D, Ruetzler C, Barker JL, Hallenbeck JM. Development of an ischemic tolerance model in a PC12 cell line. J Cereb Blood Flow Metab. 2005;25:154–62. doi: 10.1038/sj.jcbfm.9600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imae M, Fu Z, Yoshida A, Noguchi T, Kato H. Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J Mol Endocrinol. 2003;30:253–62. doi: 10.1677/jme.0.0300253. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–29. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Marte BM, Downward BM. PKB/Akt—connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–8. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Bell RM, Yellon DM. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J Mol Cell Cardiol. 2002;34:661–8. doi: 10.1006/jmcc.2002.2006. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Iwabuchi S, Miyakazi H, Okuma Y, Kuwabara M, Nomura Y, Kawahara K. Preconditioning prevents ischemia-induced neuronal death through persistent Akt activation in the penumbra region of the rat brain. J Vet Med Sci. 2004;66:521–7. doi: 10.1292/jvms.66.521. [DOI] [PubMed] [Google Scholar]
- Namura S, Nagata I, Kikuchi H, Andreucci M, Alessandrini A. Serine-threonine protein kinase Akt does not mediate ischemic tolerance after global ischemia in the gerbil. J Cereb Blood Flow Metab. 2000;20:1301–5. doi: 10.1097/00004647-200009000-00004. [DOI] [PubMed] [Google Scholar]
- Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–50. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–9. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–12. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamawaki T, Sasaki T, Hattori H, Hamada J, Fukuuchi Y, Okano H, Miura M. Upregulation of Akt phosphorylation at the early stage of middle cerebral artery occlusion in mice. Brain Res. 2002;942:1–10. doi: 10.1016/s0006-8993(02)02474-5. [DOI] [PubMed] [Google Scholar]
- Simakajornboon N, Szerlip NJ, Gozal E, Anonetapipat JW, Gozal D. In vivo PDGF beta receptor activation in the dorsocaudal brainstem of the rat prevents hypoxia-induced apoptosis via activation of Akt and BAD. Brain Res. 2001;895:111–8. doi: 10.1016/s0006-8993(01)02054-6. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE 2003. 2003:RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–6. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;19:173–83. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]