

Summary
The Wingless (Wg)/Wnt signaling pathway regulates a myriad of developmental processes and its malfunction leads to human disorders including cancer. Recent studies suggest that casein kinase I (CKI) family members play pivotal roles in the Wg/Wnt pathway. However, genetic evidence for the involvement of CKI family members in physiological Wg/Wnt signaling events is lacking. In addition, there are conflicting reports regarding whether a given CKI family member functions as a positive or negative regulator of the pathway. Here we examine the roles of seven CKI family members in Wg signaling during Drosophila limb development. We find that increased CKIε stimulates whereas dominant negative or a null CKIε mutation inhibits Wg signaling. In contrast, inactivation of CKIα by RNA interference (RNAi) leads to ectopic Wg signaling. Interestingly, hypomorphic CKIε mutations synergize with CKIα RNAi to induce ectopic Wg signaling, revealing a negative role for CKIε. Conversely, CKIα RNAi enhances the loss-of-Wg phenotypes caused by CKIε null mutation, suggesting a positive role for CKIα. While none of the other five CKI isoforms can substitute for CKIα in its inhibitory role in the Wg pathway, several CKI isoforms including CG12147 exhibit a positive role based on overexpression. Moreover, loss of Gilgamesh (Gish)/CKIγ attenuates Wg signaling activity. Finally, we provide evidence that several CKI isoforms including CKIα and Gish/CKIγ can phosphorylate the Wg co-receptor Arrow (Arr), which may account, at least in part, for their positive roles in the Wg pathway.
Introduction
The Wnt family of secreted growth factors controls many key developmental processes, including cell proliferation, cell fate determination, tissue patterning, and planar cell polarity in a wide variety of organisms (Logan and Nusse, 2004). Mutations in Wnt signaling components lead to many types of cancers including colon and skin cancers (Moon et al., 2004). The Drosophila Wingless (Wg), a founding member of the Wnt family, controls embryonic segmental polarity and patterning of adult appendages such as wing, leg, and eye. Wg exerts its biological influence through the canonical Wnt/β-catenin pathway, which is evolutionarily conserved from invertebrates to vertebrates.
Genetic and biochemical studies in several organisms have suggested a model for Wnt/Wg signal transduction (Logan and Nusse, 2004). Binding of Wnt/Wg proteins to their cognate receptors, members of the Frizzled (Fz) family of seven transmembrane proteins, and co-receptors, LRP5/6/Arrow (Arr), activates a cytoplasmic signaling component Dishevelled (Dsh), which counteracts the activity of a destruction complex composed of Axin, APC, and the Ser/Thr kinase GSK3β/Shaggy (Sgg)/Zest White 3 (Zw3), leading to the accumulation and nuclear translocation of the transcriptional effector β-catenin/Armadello (Arm). β-catenin/Arm forms a complex with the DNA binding protein Lef1/TCF to activate Wnt/Wg target genes.
A cohort of studies have provided evidence that CKI family members participate in many aspects of the Wnt/Wg signaling pathway (Price, 2006). CKIε was first identified as a positive regulator of the canonical Wnt pathway (Peters et al., 1999; Sakanaka et al., 1999). Overexpression of CKIε in Xenopus embryos induced ectopic dorsal axis formation, activated Wnt-responsive genes, and rescued the axial formation of UV treated embryos. Dominant negative forms of CKIε and a pharmacological inhibitor of CKI blocked the responses to ectopic Wnt signaling in Xenopus. Biochemical and epistasis study suggested that CKIε binds Dsh and acts between Dsh and GSK3β (Peters et al., 1999; Sakanaka et al., 1999). In vivo and In vitro kinase assays showed that CKIε can phosphorylate Dsh and a pharmacological CKI inhibitor can block Wnt induced Dsh phosphorylation, suggesting that Dsh is a target of CKIε (Peters et al., 1999). However, the role of CKIε appears to be more complex than it was originally anticipated. For example, it has also been shown that CKIε interacts with Axin, and Axin-bound CKIε phosphorylates APC and modulates its ability to regulate β-catenin (McKay et al., 2001; Peters et al., 1999; Rubinfeld et al., 2001; Sakanaka et al., 1999). What makes the picture even more complicated is the finding that, in a reconstituted system of Xenopus extracts, CKIε can phosphorylate Tcf3 and enhance Tcf3-β-catenin association and β-catenin stability, implying that CKIε may also exert a positive influence downstream of GSK3β (Lee et al., 2001).
The potential role of other CKI isoforms in Wnt signaling has also been examined in several systems. In an overexpression study using Xenopus embryonic explants, all other CKI isoforms, including α, β, γ, and δ, can activate Wnt signaling (McKay et al., 2001). All of these CKI isoforms with the exception of CKIγ can stimulate Dsh phosphorylation in cultured cells (McKay et al., 2001). However, subsequent studies provided evidence that CKIα plays a negative role in Wnt/Wg signaling that acts as a priming kinase for GSK3β-mediated phosphorylation of β-catenin/Arm (Amit et al., 2002; Liu et al., 2002; Yanagawa et al., 2002). Purification of the Axin-bound kinases that can prime GSK3β-mediated phosphorylation of β-catenin identified CKIα (Liu et al., 2002). RNAi knockdown of CKIα inhibited phosphorylation at Ser45 of β-catenin and subsequent phosphorylation by GSK3β, resulting in β-catenin stabilization (Liu et al., 2002). Consistent with the vertebrate results, CKIα RNAi of Drosophila embryos resulted in “naked cuticle”, a phenotype consistent with gain-of-Wg signaling (Liu et al., 2002). The possible role of CKIε as a priming kinase for β-catenin remained unclear. Overexpression of a dominant negative CKIε inhibited Axin-induced phosphorylation at Ser45 of β-catenin in 293 cells (Amit et al., 2002). In addition, RNAi knockdown of CKIε stabilized Arm in Drosophila S2+ cells, although the effect was less dramatic than CKIα RNAi knockdown (Yanagawa et al., 2002). On the other hand, RNAi knockdown of CKIε in 293T cells had no detectable effect on Ser45 phosphorylation and stability of β-catenin (Liu et al., 2002). It remains possible that CKIε plays a minor partially redundant role in β-catenin/Arm phosphorylation and the effect of its inactivation on β-catenin/Arm phosphorylation and degradation could have been masked by CKIα.
Although CKIα RNAi in Drosophila embryos resulted in phenotypes consistent with “gain-of-Wg” function, the recent finding that CKIα is also a negative regulator of the Hh pathway complicated the interpretation (Jia et al., 2005; Lum et al., 2003). Because Wg and Hh cross-regulate each other during embryonic development, the “gain-of-Wg” phenotype resulted from CKIα RNAi could be attributed to ectopic Hh signaling. To further investigate the physiological roles of the CKI family members in Wg signaling In vivo, we applied overexpression, dominant-negative, genetic mutations, and RNAi approaches to study the function of CKIε, CKIα and Gish/CKIγ in Drosophila wing development where Wg signaling is independent of Hh. We also assessed the potential roles of other CKI family members (Fig. 1; Morrison et al., 2000) in Wg signaling using overexpression assays.
Figure 1. CKI family in Drosophila.
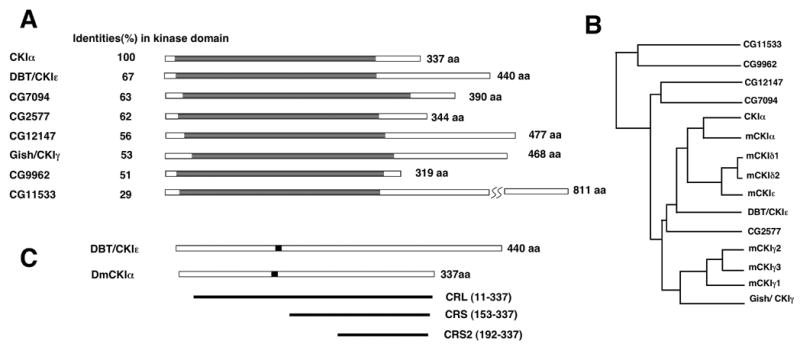
A. Schematic drawings of CKI family members in Drosophila with kinase domains depicted in grey. The percentage of amino acid identify in the kinase domains between CKIα and other CKI isoforms is indicated by the numbers to the left.
B. Family tree of Drosophila CKI isoforms and mouse CKI isoforms including mCKIα, ε, δ1-2, and γ1-3.
C. The solid lines indicate the CKIα coding regions targeted by CRL, CRS, and CRS2, respectively. The black boxes indicate the 30-nucleotide contiguous sequence shared between CKIα and DBT/CKIε.
Materials and Methods
Mutations and transgenes
dco3, dco2, dcoP103, and dcole88 are hypomorphic, strong, and null allele of dco/dbt, (Zilian et al., 1999). gishe01759 is a strong allele of gish (Jia et al., 2005). dshV26is a null allele (Jiang and Struhl, 1996). hsp-flp1, hsp-CD2, hsp-Myc-GFP, MS1096, act>CD2>Gal4, ap-Gal4, omb-Gal4 have been described (Jiang and Struhl, 1995; Jiang and Struhl, 1998; Pignoni et al., 1997; Wang et al., 1999). The CKIα RNAi constructs CRL and CRS have been described (Jia et al., 2004; Jia et al., 2005). DN-DBT (K38 to R) and DN-XCKIε (K38 to R) have been described (Jia et al., 2005; Peters et al., 1999). N-terminal flag tagged CKI isoforms have been described (Jia et al., 2005). UAS-XCKIε-KD contained the coding sequence for the kinase domain of XCKIε (Peters et al., 1999) inserted into the pUAST vector. UAS-Sgg, UAS-DN-GSK3, UAS-DN-dFtz2, UAS-P35 have been described (Hay et al., 1994; Hazelett et al., 1998; Jia et al., 2002; Zhang and Carthew, 1998).
Genotypes for generating clones are as follow.
dco clones (with CRS2 coexpression): MS1096 UAS-FLP/UAS-P35; (CRS2/+); FRT82 dcole88/FRT82 hsp-CD2, y+M(3)w124.
dsh clones expressing XCKIε-KD: hsp-FLP hsp-Myc-GFP FRT101/dshV26FRT101; ap-Gal4/UAS- XCKIε-KD.
gish clones: MS1096 UAS-FLP; FRT82 gishe01759/FRT82 hsp-CD2, y+M(3)w124.
dco gish double mutant clones: MS1096 UAS-FLP/P35; FRT82 gishe01759dcole88/FRT82 hsp-CD2, y+M(3)w124.
Cell culture, transfection, immunoprecipitation, and western blot analysis
S2 cells were cultured in the Schneider’s Drosophila Medium (Invitrogen) with 10% fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml of Streptomycin. Transfection was carried out using Calcium Phosphate Transfection Kit (Specialty Media) according to manufacturer’s instructions. An ub-Gal4 construct was cotransfected with pUAST expression vectors for all the transfection experiments. 4 μg DNA for ub-Gal4 and 2 μg DNA for each pUAST expression vector were used in a typical transfection experiment. Immunoprecipitation and western blot analyses were performed using standard protocols. Antibodies used are: mouse αHA, F7 (Santa Cruz), mouse αFlag, M2 (Sigma).
Immunostaining of imaginal discs and cultured cells
Standard protocols for immunofluorescence staining of imaginal discs and S2 cells were used. Antibodies used in this study: mouse anti-Wg and anti-Arm (DSHB, University of Iowa); rabbit anti-Sc and rabbit anti-Vg (a gift from by Dr. S. Carroll); rat anti-Sen (a gift from Dr. Hellen); Rabbit anti-βGal (Cappel); mouse anti-Myc, 9E10 (Santa Cruz); mouse anti-Flag (Sigma); and mouse anti-CD2 (Jiang and Struhl, 1995).
Results
Misexpressing CKIε induces ectopic Wg signaling
To explore the function of CKIε in Drosophila limb development, we generated UAS transgenes expressing either the full-length form (FL) or the kinase domain (KD) of Xenopus CKIε (XCKIε) (Peters et al., 1999). In late third instar wild type wing imaginal discs, Wg is produced at the dorsal/ventral (D/V) compartment boundary of wing imaginal discs and high levels of Wg signaling activity induce the expression of several target genes including scute (sc), senseless (sen), and vestigial (vg) in cells flanking the D/V boundary (Fig. 2A, 3A–C; (Campuzano and Modolell, 1992; Nolo et al., 2001; Zecca et al., 1996). Overexpressing XCKIε-FL with a wing specific Gal4 driver, MS1096, caused ectopic sc expression in wing pouch region distant from the D/V boundary, suggesting that CKIε stimulates Wg signaling activity (Fig. 2B). Overexpressing XCKIε-KD resulted in more robust ectopic sc expression (Fig. 2C), consistent with a previous observation that XCKIε-KD is more potent in inducing Wnt responses than XCKIε-FL (Peters et al., 1999).
Figure 2. CKIε induce ectopic Wg signaling activity.
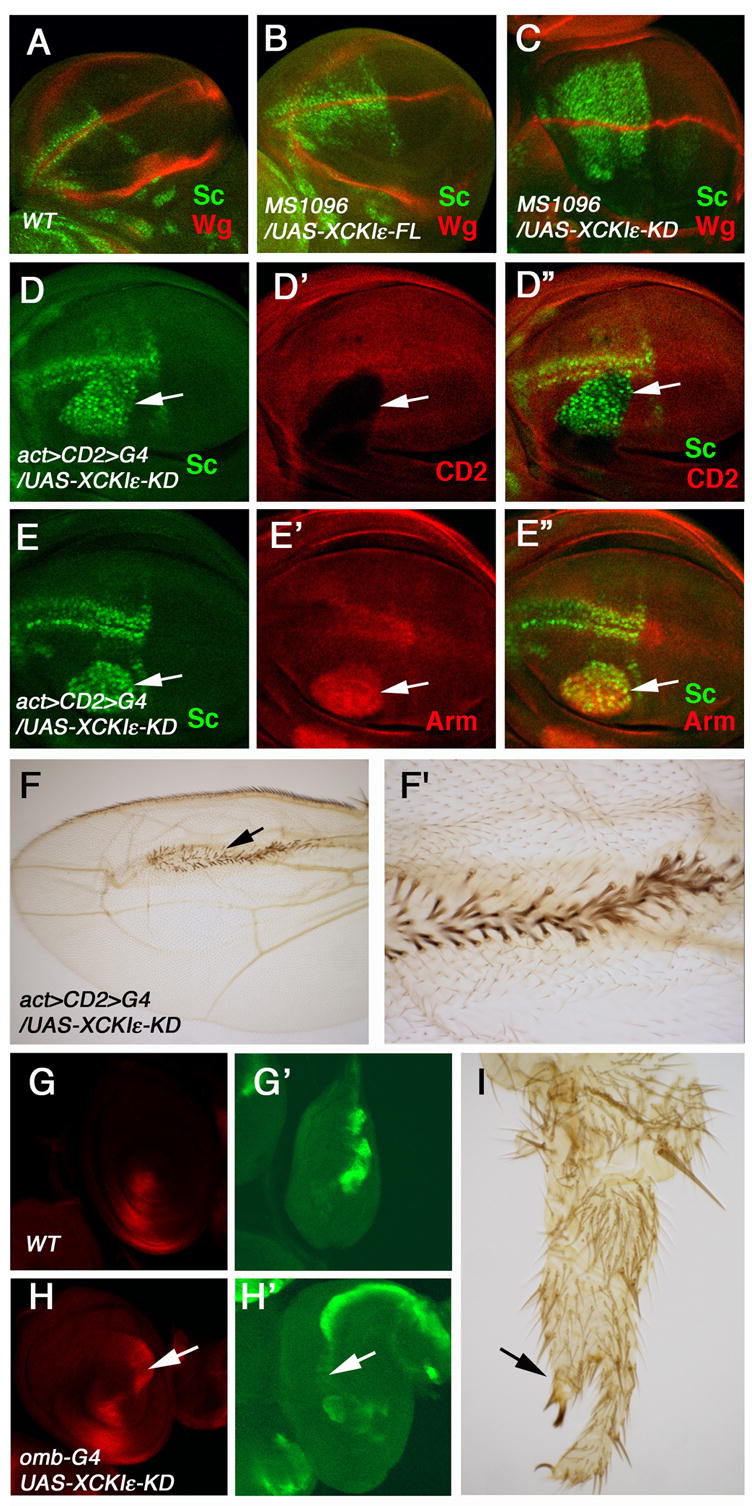
(A–C) Late third instar wild type wing disc (A), and wing disc expressing the full-length XCKIε (B) or the kinase domain (KD) of XCKIε (C) with MS1096 were immunostained with anti-Wg (red) and anti-Sc (green) antibodies. Overexpressing XCKIε or its kinase domain induces ectopic expression of sc without affecting wg expression. All wing discs shown in this and following figures were oriented with anterior to the left and ventral at the top.
(D-D″) Late instar wing disc containing a flip-out clone expressing XCKIε-KD with act-Gal4 was immunostained to show Sc (green) and CD2 (red) expression.
XCKIε expressing cells, marked by the lack of CD2 expression, activate Sc expression cell autonomously (arrows).
(E-E″) Late instar wing disc carrying a clone expressing XCKIε-KD with act-Gal4 was immunostained to show the expression of Sc (green) and Arm (red).
XCKIε-KD expressing cells stabilize Arm and activate Sc.
(F, F′) Low (F) and high (F′) magnification view of an adult wing carrying a clone expressing XCKIε-KD. XCKIε-KD induces ectopic wing margin bristles forming on the wing blade.
(G, G′, H, H′) Late third instar wild type leg disc (G, G′) and leg disc expressing XCKIε-KD with omb-Gal4 were immunostained to show wg (red) and dpp-lacZ (green) expression. Overexpressing XCKIε-KD suppresses dpp-lacZ and induces ectopic wg expression in the dorsal region of the leg disc (arrows in H, H′).
(I) Adult leg expressing XCKIε-KD with omb-Gal4. A secondary leg (arrow) branches out from the dorsal side of the primary leg. Note the craw points to the ventral side.
Figure 3. Loss of CKIε inhibits Wg signaling activity.
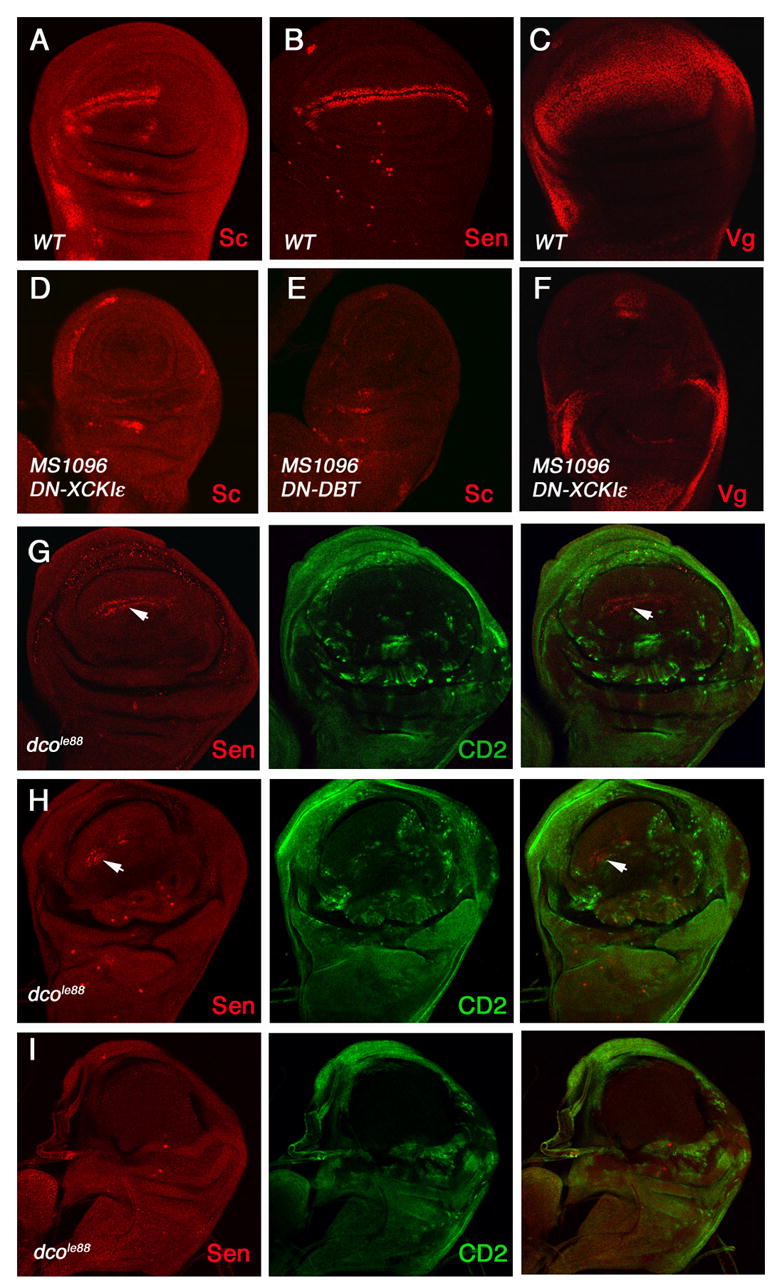
(A–C) Wild type wing discs immunostained to show the expression of Sc (A), Sen (B) and Vg (C).
(D–F) Wing discs expressing dominant negative (DN) XCKIε (D, F) or DBT (E) with MS1096 were immunostained to show the expression of Sc (D, E) or Vg (F).
(G–I) Wing discs carrying dbt null (dcole88) clones induced in the Minute background were immunostained to show the expression of Sen (red) and CD2 (green). dbt mutant cells lack CD2 expression. Wg-induced sen expression alone the D/V boundary is diminished in these discs. Arrows indicate residual Sen expression (G, H).
Overexpressing XCKIε did not affect wg expression at D/V boundary (Fig. 2B, C), suggesting that XCKIε acts in Wg-responding cells downstream of Wg synthesis and transport. As a further support, we found that flip-out clones expressing XCKIε-KD accumulated high levels of Armadillo (Arm) and ectopically activated sc in a cell autonomous fashion (Fig. 2D-D″, 2E-E″). Consistent with the notion that increased XCKIε induces Wg signaling activity, XCKIε-KD-expressing cells on the wing blade formed ectopic sensory bristles normally induced by high levels of Wg signaling activity along the wing margin (Fig. 2F–F′).
In leg discs, Hh induces neighboring anterior-ventral cells to express wg and low levels of dpp and anterior-dorsal cells to express high levels of dpp (Fig. 2G-G′). The asymmetric expression of wg and dpp is maintained by mutual antagonism between Wg and Dpp signaling pathways (Jiang and Struhl, 1996). For example, ectopic Wg signaling suppresses dpp expression and promotes wg expression in dorsal cells, which creates ectopic juxtaposition of high levels of Wg and Dpp, leading to the formation of a supernumerary leg (Jiang and Struhl, 1996). We found that misexpression of XCKIε-KD in leg discs repressed dpp expression and promoted ectopic wg expression in dorsal cells (Fig. 2H-H′), leading to the formation of a supernumerary leg branching out from the dorsal side of the primary leg (Fig. 2I). Taken together, these observations suggest that CKIε positively regulates Wg signaling in Drosophila limb development.
Dominant negative CKIε and dbt null mutation inhibit Wg signaling
To determine whether CKIε is required for Wg signaling in Drosophila limb development, we generated UAS transgenes expressing dominant negative (DN) forms of XCKIε and DBT (see Materials and Methods). Consistent with the Xenopus studies, misexpression of DN-XCKIε or DN-DBT in wing discs using MS1096 resulted in a blockage of Wg signaling as revealed by the loss of expression of Wg responsive genes including sc and vg (Fig. 3D–F), suggesting that CKIε is required for transducing Wg signal.
It is possible that DN-CKIε not only blocks CKIε but also interferes with other CKI isoforms. To further clarify the role of CKIε in Wg signaling, we turned to genetic mutations in Drosophila CKIε, named as double time (dbt) or disc overgrown (dco) for its role in regulating circadian rhythms and imaginal disc growth, respectively (Price et al., 1998; Zilian et al., 1999). We analyzed several dbt/dco alleles including hypomorphic and strong alleles: dco3, dco2, and dcoP103, and a null allele, dcole88 (Zilian et al., 1999). Wg signaling, as monitored by sc or sen expression, is normal in wing discs carrying dco3 clones or in several transheterozygotic combinations including dco3/dco2, dco3/dcoP103, and dco3/dcole88 (Fig. 6F; data not shown), suggesting that partial loss of DBT/CKIε does not significantly affect Wg signaling.
Figure 6. dbt mutations synergize with CKIα RNAi to induce ectopic Wg signaling activity.
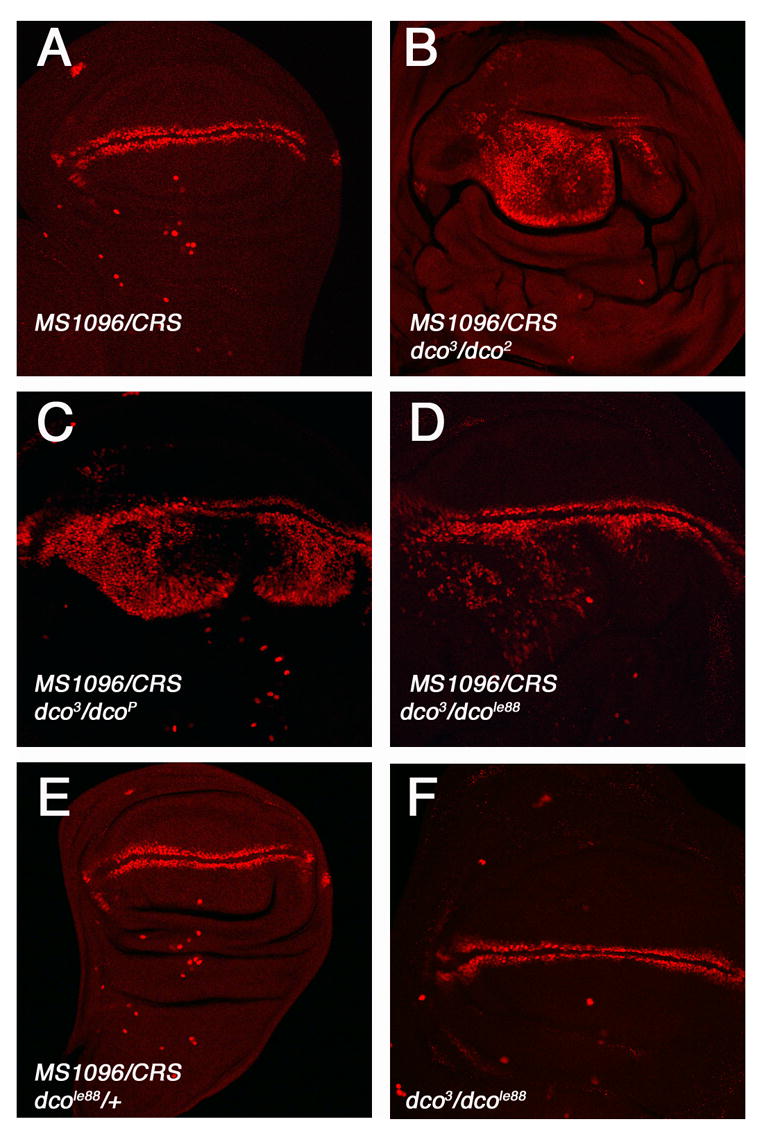
(A–F) Late third instar wing discs of the indicated genotypes were immunostained to show sen expression. Expressing one copy of CRS in wild type (A) or dbt heterozygous discs (E) did not cause any discernable change in sen expression. In contrast, CRS induced ectopic sen expression in several combinations of dbt transheterozygotes: dco3/dco2 (B), dco3/dcoP (C), and dco3/dcole88 (D). dco3/dcole88 transheterozygotic wing discs exhibited normal sen expression (F).
dcole88 mutants die at late embryonic and early larval stages, and dcole88 mutant cells have a growth deficit so that early induced dcole88 mutant clones tend to be eliminated during larval growth (Jia et al., 2005; Zilian et al., 1999). To increase the frequency of dcole88 mutant clones, we used MS1096 to express UAS-FLP as a stable source of flipase in a Minute background (Jia et al., 2005), which gave dco− cells a growth advantage over neighboring dco+ Minute cells (Morata and Ripoll, 1975). In addition, the baculovirus cell death inhibitor P35 was coexpressed to prevent the death of dcole88 mutant cells (Hay et al., 1994). Under such conditions, wing discs contain dcole88 mutant clones that occupy most of the wing pouch region (Fig. 3G–3I). Most wing discs (30/38) exhibited diminished Wg signaling as indicated by reduced expression of sen and vg (Fig. 3G–3H, 8C). The remaining discs (8/38) exhibited a more complete loss of Wg target gene expression (Fig. 3I). dcole88 mutant discs generated under these conditions expressed Hh target genes along the A/P boundary (Jia et al., 2005), suggesting that loss of Wg target gene expression is due to the positive role of CKIε in Wg signaling rather than the “sickness” of dcole88 mutant discs. The residual Wg signaling activity in some dcole88 mutant discs could be due to the perdurance of CKIε activity coupled with compensation by other CKI isoforms (see below).
Figure 8. Positive role of Gish/CKIγ and CKIa in the Wg pathway.
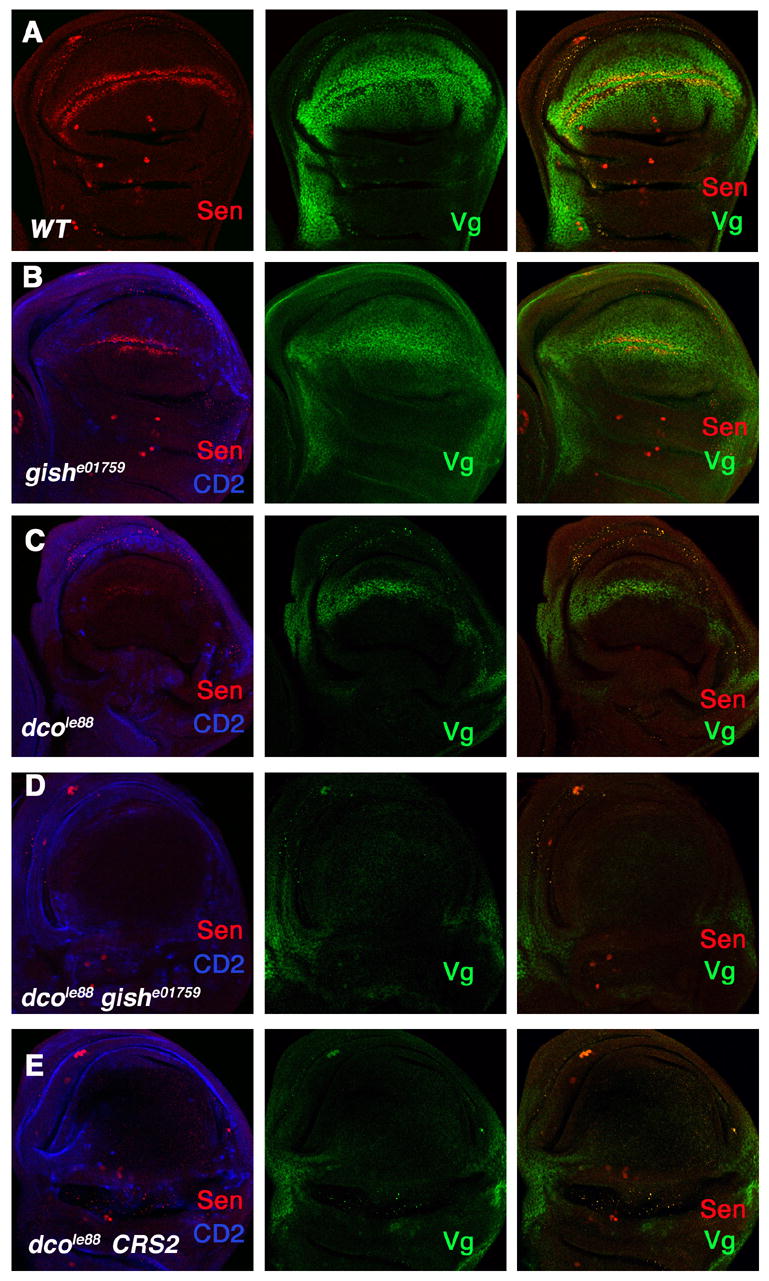
Wild type wing disc (A), wing discs carrying clones for gishe01759 (B), dcole88 (C), or both gishe01759 and dcole88 (D), or wing disc carrying dcole88 mutant clones and expressing CRS2 (E) were immunostained to show the expression of Sen (red), Vg (green), and CD2 (blue). Mutant clones are recognized by the lack of CD2 expression (blue in B–E). Of note, mutant clones were induced by FLP/FRT-mediated mitotic recombination in the Minute background. P35 was expressed in discs carrying dcole88 mutant clones (C–E).
CKIε regulates the Wg pathway at multiple levels
To determine where CKIε acts in the pathway to promote Wg signaling activity, we carried out genetic epistasis experiments, taking advantage of the observation that XCKIε confers constitutive Wg signaling activity. Overexpression of a dominant negative form of Frizzled 2 (DN-dFz2; Zhang and Carthew, 1998) blocked Wg signaling and resulted in loss of sc expression (Fig. 4F). However, co-expression of XCKIε-KD with DN-dFz2 reversed the phenotype and led to ectopic sc expression (Fig. 4G), which is similar to misexpression of XCKIε-KD alone (Fig. 4E), suggesting that CKIε acts downstream of dFz2.
Figure 4. CKIε acts at multiple levels in the Wg pathway.
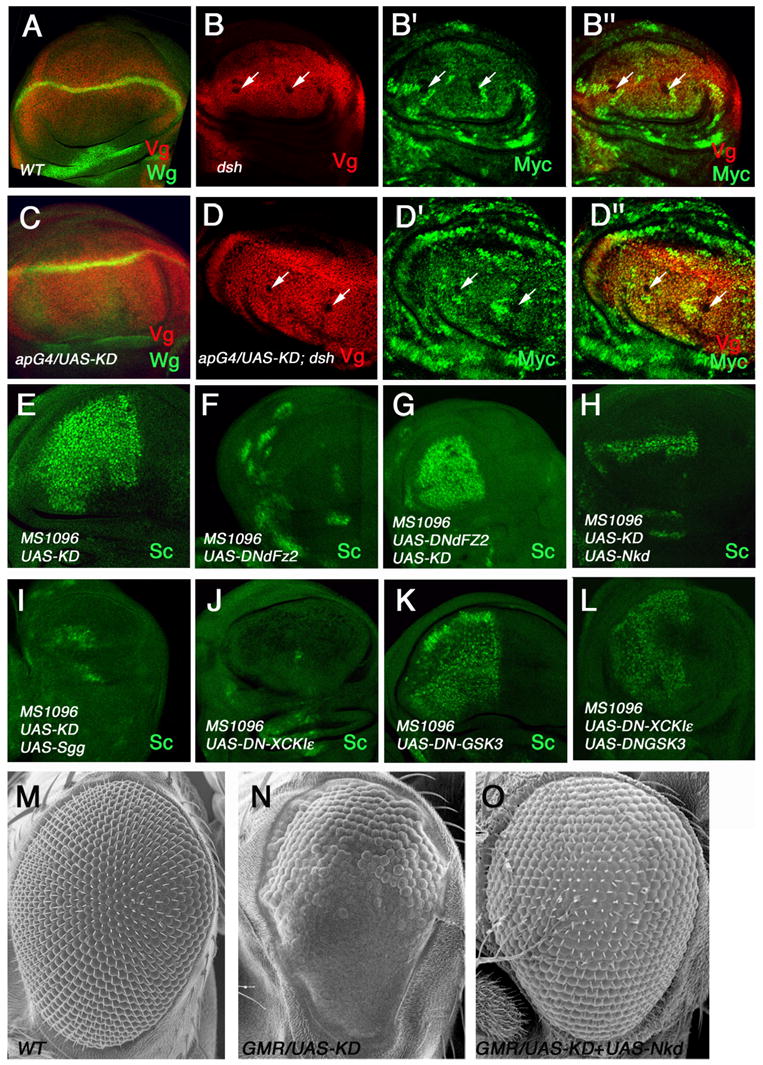
(A, C) Wild type wing disc (A) and wing disc expressing the XCKIε kinase domain (KD) with ap-Gal4 (C) were immunostained with anti-Wg (green) and anti-Vg (red) antibodies. Misexpressing the XCKIε kinase domain via ap-Gal4 resulted in an expansion of the Vg expression domain in the dorsal compartment.
(B-B″, D-D″) Wing disc carrying dshV26 clones without (B-B″) or with (D-D″) XCKIε-KD expressed via ap-Gal4 were immunostained to show the expression of Vg (red) and Myc (green). dshV26 mutant cells are marked by the lack of Myc expression (indicated by arrows). dshV26 mutant cells fail to activate Vg even when they overexpress the XCKIε kinase domain.
(E–L) Late third instar wing discs of the indicated genotypes were immunostained to show Sc expression. Sc expression was blocked by a dominant negative form of dFz2 (DN-dFz2) (F). Coexpressing the XCKIε kinase domain (KD) with DN-dFz2 led to ectopic Sc expression (G), similar to expressing KD alone (E). Coexpressing Nkd or Sgg/GSK3β suppressed the ectopic Sc expression caused by XCKIε-KD (H, I). Coexpression of DN-GSK3β reversed the phenotype caused by DN-XCKIε (compare L with J); however, the levels of ectopic Sc are lower than those in wing discs expressing DN- GSK3β alone (compare L with K).
(M–O) Scanning electron photomicrograph of a wild type adult eye (M), or adult eyes expressing XCKIε-KD (N) or coexpressing Nkd with XCKIε-KD (O) with GMR gal4 driver. Expression of XCKIε-KD blocked the formation of interommatidial bristles (N), which was suppressed by coexpression of Nkd (O).
Mutant clones homozygous for a dsh null allele, dshV26, blocked Wg signaling as evidenced by the loss of vg expression (Fig. 4B-B″). Overexpression of XCKIε-KD in dshV26 clones failed to restore vg expression (Fig. 4D-D″), suggesting that CKIε acts upstream of or parallel to Dsh.
Naked (Nkd) is a Wg-induced pathway inhibitor that binds and inhibits Dsh (Rousset et al., 2001; Zeng et al., 2000). Coexpression of Nkd suppressed ectopic sc expression induced by XCKIε-KD (Fig. 4H), consistent with CKIε targeting Dsh. Similarly, expressing XCKIε-KD in the Drosophila eye blocked the formation of interommatidial bristles (Fig. 4N), a phenotype consistent with ectopic Wg signaling (Cadigan and Nusse, 1996). Coexpression of Nkd completely suppressed the XCKIε-KD-induced eye phenotype (Fig. 4O), strengthening the view that CKIε acts upstream of or parallel to Dsh in activating Wg pathway.
We next addressed the epistatic relationship between Sgg/GSK3β and CKIε. First, we coexpressed Sgg/GSK3β with XCKIε-KD in wing discs using MS1096 and monitored Wg signaling activity by immunostaining with Sc antibody. We found that an excess amount of Sgg/GSK3β inhibits ectopic Wg signaling induced by XCKIε-KD (Fig. 4I), suggesting Sgg/GSK3β acts downstream of CKIε. Second, we coexpressed a dominant negative DN-GSK3β; (Jia et al., 2002) with the dominant negative form of DN-XCKIε. Overexpression of DN-XCKIε alone completely blocked sc expression along the D/V boundary (Fig. 4J). In contrast, coexpression of DN-GSK3β with DN-XCKIε caused ectopic sc expression (Fig. 4L), a phenotype similar to that caused by overexpressing DN-GSK3β alone (Fig. 4K), further arguing that CKIε acts upstream of Sgg/GSK3β. However, We found that the levels of ectopic sc expression in wing discs coexpressing DN-GSK3β and DN-XCKIε were consistently lower than those in wing discs expressing DN-GSK3β alone (compare Fig. 4K with 4L), suggesting that CKIε may have an additional positive role downstream of Sgg/GSK3β.
RNAi knockdown of CKIα induces ectopic Wg signaling
CKIα has been implicated as a negative regulator of the Wnt/Wg pathway that serves as a priming kinase for GSK3β mediated phosphorylation of β-catenin/Arm (Amit et al., 2002; Liu et al., 2002; Yanagawa et al., 2002). The role of CKIα in the Wnt/Wg pathway has been deduced from experiments with cultured cells. To assess the In vivo function of CKIα, we used heritable RNAi technique to inactivate CKIα (Kalidas and Smith, 2002; Kennerdell and Carthew, 2000). We employed three CKIα RNAi constructs, CRL, CRS, and CRS2, which produce hairpin-loop double stranded RNA (dsRNA) targeting CKIα coding sequence for aa 11–337, aa 153–337, aa 192–337, respectively (Fig. 1C; (Jia et al., 2005). CRL knocked down CKIα more effectively than CRS; however, it also knocked down DBT/CKIε to certain extent, likely due to the presence of a 30-nucleotide contiguous sequence shared between CKIα and CKIε (Fig. 1C; Jia et al., 2005). In contrast, CRS specifically knocked down CKIα but not DBT/CKIε (Jia et al., 2005).
We found that CRS affected Wg signaling in a dose-dependent manner. Expressing one copy of CRS with MS1096 failed to cause detectable change in the expression pattern of sc and sen (Fig. 5A–B). In contrast, expressing two copies of CRS induced patchy ectopic expression of both sc and sen in the wing pouch region (Fig. 5C–D). However, the ectopic sc and sen was restricted to the dorsal compartment cells due to the fact that MS1096 expresses Gal4 at higher levels in dorsal than in ventral compartment cells. The dosage effect of CRS on Wg signaling is also reflected in adult phenotypes. One copy of CRS only induced no or few ectopic margin bristles on the wing blade whereas two copies induced the formation of numerous margin bristles on the wing blade (Fig. 5M–O). Similar results were obtained with CRS2 (data not shown).
Figure 5. CKIα RNAi induces ectopic Wg signaling activity.
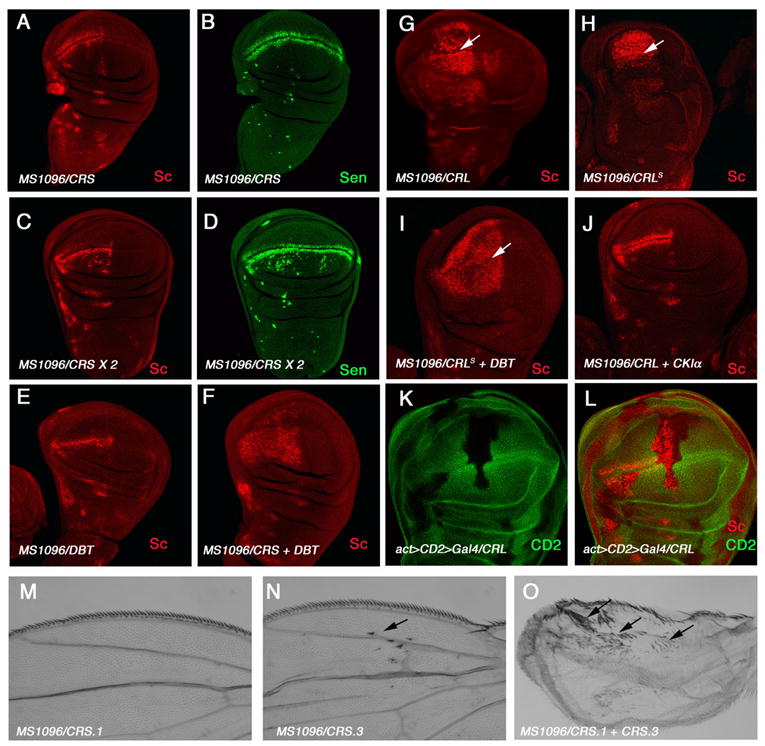
(A–D) Late third instar wing discs expressing one copy (A, B) or two copies of CRS (C, D) with MS1096 were immunostained to show the expression of Sc (red) and Sen (green). Expressing two copies but not one copy of CRS induced ectopic Wg signaling.
(E, F) Sc expression in wing discs expressing DBT/CKIε alone (E) or together with one copy of CRS (F). Expressing DBT/CKIε induced ectopic Wg signaling when CKIα activity was compromised.
(G, H) Sc expression in wing disc expressing low (G) or high (from a strong UAS transgene; H) levels of CRL with MS1096. Of note, MS1096 expresses Gal4 at higher levels in dorsal compartment cells than in ventral compartment cells. CRL induced robust ectopic Wg signaling; however, high levels of CRL reduced the levels of ectopic Sc expression in dorsal cells. Arrows in G–H indicate the D/V boundary.
(I–J) Wing discs coexpressing DBT/CKIε (I) or CKIα (J) with CRL. Coexpression of DBT with high levels of CRL restored the high levels of ectopic Sc in dorsal cells (I) whereas CKIα suppressed ectopic Wg signaling caused by CRL (J).
K–L) Wing discs with flip-out clones expressing CRL with act-Gal4 were immunostained to show CD2 (green) and Sc (red) expression. CRL expressing cells are recognized by the lack of CD2 expression.
(M–O) Wild type adult wing (M) or adult wing expressing one (N) or two (O) copies of CRS with MS1096. Arrows indicate the ectopic wing margin bristles forming on wing blades (N, O).
We also observed a synergistic effect between CKIα knockdown and DBT/CKIε overexpression. For example, expressing either DBT/CKIε or CRS alone did not induce ectopic expression of sc (Fig. 5A, E); however, coexpressing DBT/CKIε with CRS resulted in dramatic expansion of the sc expression domain (Fig. 5F). Hence, reducing CKIα provides a sensitized genetic background that reveals the positive role of DBT/CKIε in the Wg pathway.
Expressing one copy of CRL caused a marked expansion of sc in both dorsal and ventral compartments of the wing pouch region (Fig. 5G, H). The effect of CRL on Wg target gene expression was completely suppressed by coexpressing CKIα (Fig. 5J), suggesting that the ectopic Wg signaling caused by CRL is mainly due to the loss of CKIα. The effect of CRL on Wg signaling is cell autonomous as flip out clones expressing CRL under the control of act-gal4 ectopically activate sc in a cell autonomous fashion (Fig. 5K, L). Intriguingly, wing discs expressing higher levels of CRL (using strong UAS transgenic lines or two copies of UAS transgenes) resulted in more robust ectopic sc expression on the ventral side but lower levels of ectopic sc on the dorsal side of the wing pouch region (Fig. 5H). As CRL also knocks down DBT/CKIε, the reduction of ectopic sc is likely due to diminishing levels of DBT/CKIε when CRL levels increase. Indeed, coexpressing DBT/CKIε with CRL restored high levels of ectopic sc on the dorsal side (Fig. 5I).
A negative role for CKIε in the Wg signaling pathway
Several studies have suggested that CKIε may also act in a partially redundant fashion with CKIα in priming β-catenin/Arm for GSK3β phosphorylation and subsequent degradation and thus might have a negative role in the Wnt/Wg pathway (Amit et al., 2002; Yanagawa et al., 2002). However, it has proved difficult to demonstrate a negative role for CKIε because of its predominantly positive role in the Wg pathway. In addition, endogenous CKIα might suffice to prime β-catenin/Arm for GSK3β mediated phosphorylation. To explore the possibility that DBT/CKIε may have a negative role in the Wg pathway, we took advantage of two observations. 1) DBT/CKIε hypomorphic mutations do not affect the positive role of DBT/CKIε in the Wg pathway (Fig. 6F). 2) Reduction of CKIα by expressing one copy of CRS does not cause ectopic Wg signaling but provides a sensitized genetic background to reveal the consequence of other Wg pathway manipulations (Fig. 5F). Therefore, we examined sen expression in wing discs derived from several dbt transheterozogous combinations, dco3/dco2, dco3/dcoP, and dco3/dcole88, that do or do not express one copy of CRS. We observed normal sen expression in dco3/dco2, dco3/dcoP, or dco3/dcole88 transheterozygous wing discs (Fig. 6F; data not shown), suggesting that these allelic combinations do not perturb Wg signaling. dbt heterozygous wing discs expressing one copy of CRS also exhibited normal sen expression (Fig. 6E). In contrast, dbt transheterozygous wing discs expressing one copy of CRS exhibited ectopic sen expression although wild type wing discs expressing one copy of CRS showed normal sen expression (Fig. 6A–D). Hence, a critical level of DBT/CKIε is required for blocking aberrant Wg signaling when CKIα activity is compromised.
Functional analysis of other CKI family members
There are multiple CKI isoforms in the Drosophila genome and seven of them, including CKIα and CKIε share over 50% amino acid identity in their kinase domains (Fig. 1A; Morrison et al., 2000). To address potential roles of the other five highly conserved CKI isoforms in the Wg pathway, we asked whether they could substitute for CKIα in inhibiting Wg signaling, or could induce ectopic Wg signaling when overexpressed. In the first set of experiments, we coexpressed various CKI isoforms with CRL in wing discs and examined sen expression as readout for Wg signaling activity. All CKI isoforms were Flag-tagged at their N-termini and their expression was firmed by immunostaining with Flag antibody (Supplementary Fig. S1I–N). In addition, we did not observe any significant change in their levels when coexpressed with CRL (data not shown). Unlike CKIα, which completely rescued ectopic sen expression caused by CRL (Supplementary Fig. S1C), all the other CKI isoforms we tested, including CG7094, CG2577, CG12147, Gish/CKIγ, and CG9962, failed to suppress the ectopic sen induced by CRL (Supplementary Fig. S1D–H), suggesting that these CKI isoforms can not functionally replace CKIα for its role in blocking Wg signaling.
Next, we overexpressed each CKI isoform in otherwise wild type wing discs or in wing discs with reduced levels of CKIα due to expressing one copy of CRS. None of the CKI isoforms including DBT could induce ectopic sen expression when expressed alone (Fig. 7C, G, K; data not shown). However, coexpressing DBT with CRS resulted in enormous expansion of sen expression domain, which nearly fills the entire wing pouch region (Fig. 7B). Coexpressing CG12147 with CRS also induced robust ectopic sen expression on the dorsal side of the wing pouch region, suggesting that CG12147 can stimulate Wg signaling activity albeit less potently than DBT (Fig. 7F). CG7094 only induced mild ectopic sen expression whereas CG2577 and Gish failed to induce ectopic sen when coexpressed with CRS (Fig. 7D, D′, E, H). Coexpressing CG9962 with CRS affected Wg signaling in a more complex way. In about half of the discs (14/19), low levels of ectopic sen expression were observed on the dorsal side of the wing pouch region (Fig. 7J). In the other half (15/19), the endogenous sen expression along the D/V boundary (15/29) was diminished, especially on the dorsal side of the wing pouch region (Fig. 7I). Expressing CG9962 alone also resulted in inhibition of endogenous sen (Fig, 7K). These observations suggest that overexpressed CG9962 may have both positive and negative inputs to the Wg pathway and its overexpression in conjunction with CRS may result in near-uniform intermediate levels of Wg signaling activity.
Figure 7.
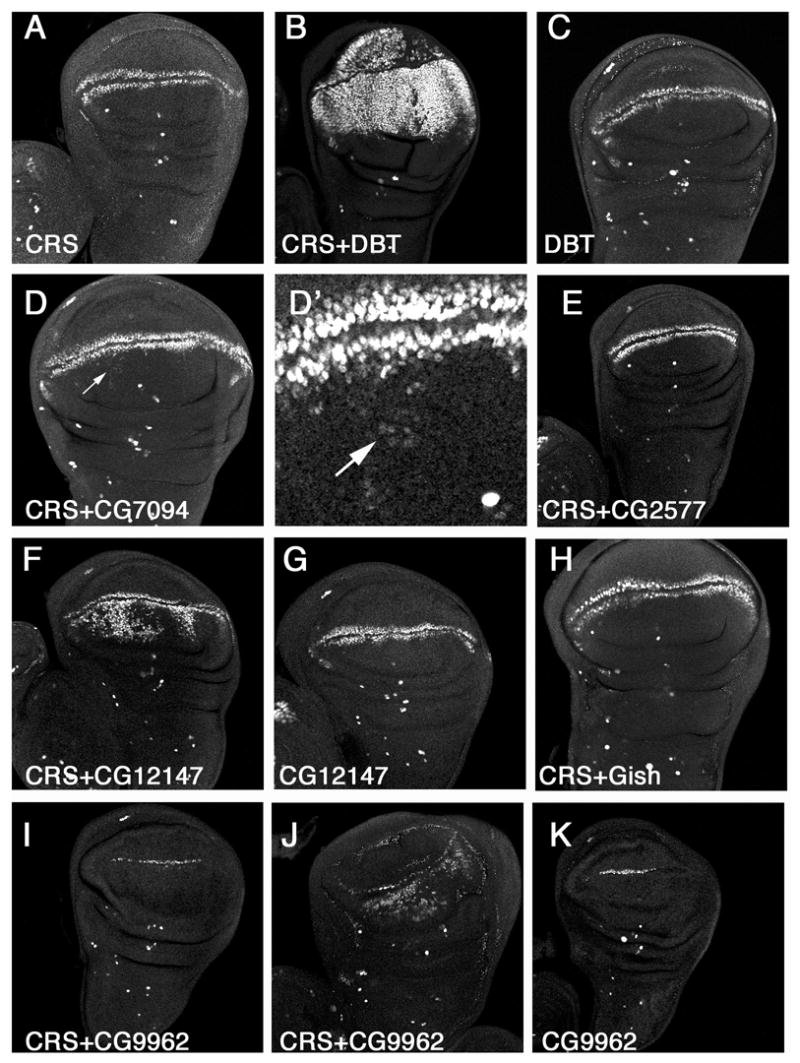
Overexpression of various CKI isoforms in wing discs with CRS sen expression in wing disc expressing CRS alone (A), CRS plus DBT (B), DBT alone (C), CRS plus CG7094 (D, D′), CRS plus CG2577 (E), CRS plus CG12147 (F), CG12147 alone (G), CRS plus Gish (H), CRS plus CG9962 (I, J), or CG9962 alone (K) with MS1096. DBT alone did not induce ectopic sen expression (C) but together with CRS induced robust ectopic sen expression (B). CG12147 also induced ectopic sen expression when coexpressed with CRS (F) whereas CG7094 induced weak ectopic sen expression (arrows in D and D′). Expressing CG9962 alone suppressed sen expression at the D/V boundary (K). When coexpressed with CRS, CG9962 suppressed sen expression in some discs (I) but induced ectopic sen expression in others (J).
A positive role for Gish/CKIγ and CKIα in the Wg pathway
We further explored the role of Gish/CKIγ in Wg signaling by generating gish mutant clones in wing discs and examining the expression of Wg-responsive genes including sen and vg. As shown in Fig. 8, wing discs carrying gishe01759 mutant clones exhibited reduced levels of sen and vg expression (Fig. 8B), suggesting a positive role for Gish/CKIγ in the Wg pathway. In further support of this notion, we found that loss-of-gish enhanced dbt mutant phenotypes. In the majority of wing discs carrying dbtle88 mutant clones, residual expression of sen and vg persisted (Fig. 8C). In contrast, in all the discs (n=25) carrying dco gish double mutant clones, sen and vg expression in the wing pouch region was completely lost (Fig. 8D).
Similarly, reduction of CKIα also enhanced the loss-of-Wg phenotypes caused by dbtle88 mutation. For example, expressing CRS2 in wing discs carrying dbtle88 clones resulted in a complete blockage of sen and vg expression (Fig. 8E), suggesting that CKIα may also have an unappreciated positive role in the Wg pathway in addition to its well documented negative role.
Phosphorylation of Arr by multiple CKI isoforms
Two recent studies showed that CKI phosphorylated multiple sites in the intracellular domain the Wnt co-receptor LRP6, and that phosphorylation of LRP6 recruited Axin to the cell surface, leading to the activation of the Wnt-β-catenin pathway (Davidson et al., 2005; Zeng et al., 2005). However, which CKI isoform(s) are responsible for LRP6 phosphorylation remains controversial. One study suggested that CKIγ is the exclusive CKI isoform for LRP6 phosphorylation (Davidson et al., 2005), whereas the other suggested that multiple CKI isoforms including CKIα and CKIδ/ε could participate (Davidson et al., 2005; Zeng et al., 2005). We sought to examine which CKI isoforms can phosphorylate Arr, the Drosophila orthologue of LRP6 (Pinson et al., 2000; Tamai et al., 2000; Wehrli et al., 2000). We examined two forms of Arr: a full-length Arr (ArrFL-HA) and a membrane tethered Arr intracellular domain (Myr-ArrC-HA), both of which have three copies of HA-tag at their C-termini (Culi and Mann, 2003). ArrFL-HA or Myr-ArrC-HA was coexpressed with each of the seven Drosophila CKI isoform in S2 cells, followed by western blot analysis. As shown in Fig. 9A, CG12147, Gish/CKIγ, and CG9962 phosphorylate both ArrFL and ArrC robustly, as indicated by the mobility shift of Arr proteins on the SDS PAGE, with Gish/CKIγ exhibiting the highest activity. CKIα and DBT/CKIε also phosphorylate Arr albeit less effectively whereas CG7094 and CG2577 failed to phosphorylate Arr under our experimental conditions. The modest shift of ArrC induced by CKIα or DBT/CKIε is due to phosphorylation as it was eliminated by CIP treatment (Fig. 9B). The expression of individual CKI isoforms was confirmed by western blot with a Flag antibody (Fig. 9A, bottom panel).
Figure 9. Phosphorylation of Arr cytoplasmic domain by multiple CKI isoforms.
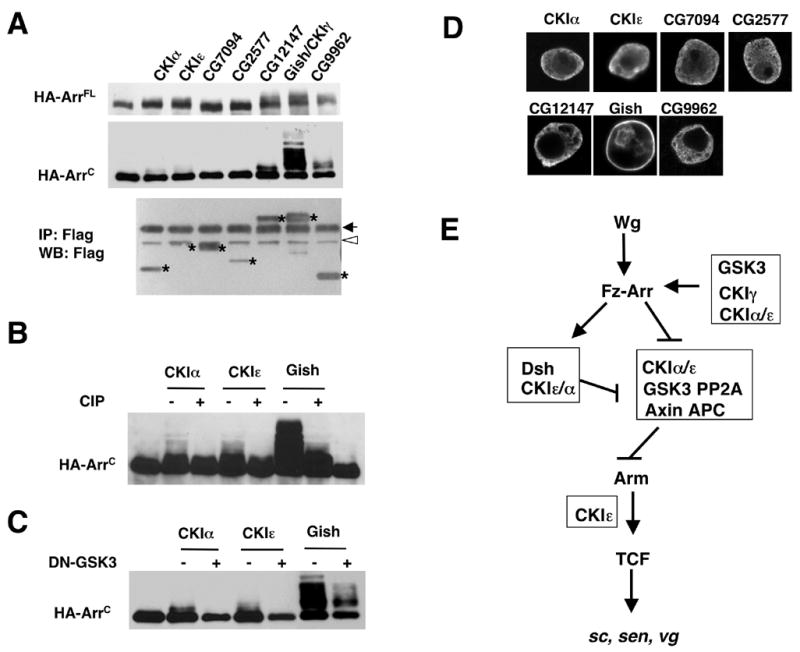
(A) HA-tagged full-length Arr (HA-ArrFL) and membrane-tethered Arr cytoplasmic domain (HA-ArrC) were cotransfected into S2 cells with indicated Flag-tagged CKI family members. Cell lysates were subjected to western blot analysis with anti-HA antibody (top and middle panels) or immunoprecipitated with anti-Flag antibody, followed by western blot anti-Flag antibody (bottom panel). Asterisks indicate the corresponding CKI bands. Arrow indicates IgG and open arrowhead indicates a nonspecific band. Of note, both CKIε and CG7094 run very closely to the nonspecific band.
(B) Cell lysates were prepared from S2 cells cotransfected with HA-ArrC and indicated CKI isoforms with (+) or without (−) CIP treatment and subjected to western blot analysis with anti-HA antibody.
(C) Cell lysates were prepared from S2 cells cotransfected with HA-ArrC and indicated CKI isoforms with (+) or without (−) dominant negative GSK3β (DN-GSK3) and subjected to western blot analysis with anti-HA antibody.
(D) S2 cells were transfected with constructs expressing various Flag-tagged CKI isoforms and immunostained with anti-Flag antibody.
(E) A genetic pathway indicating the multiple inputs of CKI family members in Wg signaling.
GSK3β phosphorylates multiple PPPSP motifs in the cytoplasmic domain of LRP6 and primes CKI phosphorylation at adjacent sites (Zeng et al., 2005). To determine if GSK3β is also required to prime CKI phosphorylation of Arr, we coexpressed DN-GSK3β with CKIα, DBT/CKIε or Gish/CKIγ. As shown in Fig. 9C, phosphorylation of ArrC by CKIα, DBT/CKIε, or Gish/CKIγ was largely inhibited by DN-GSK3β.
We also examined the subcellular localization of various Flag-tagged CKI isoforms expressed in S2 cells. As shown in Fig. 9D, Gish/CKIγ is predominantly associated with the plasma membrane. Similar subcellular distribution for the vertebrate CKIγ has also been observed (Davidson et al., 2005). All other CKI isoforms are uniformly distributed in the cytoplasm with CKIα, DBT/CKIε, and CG2577 also exhibiting nuclear staining.
Discussion
CKI family members have been implicated as positive and negative regulators of the Wnt/Wg pathway that act at multiple levels in the pathway (Price, 2006). However, most of the previous studies were carried out in cell culture systems and relied heavily on overexpression strategies. As such, conflicting results were obtained with regard to which CKI family members participate in a specific process in the Wnt/Wg signaling pathway. Here we employed a combination of genetic mutations, RNAi, dominant negative kinases, and overexpression to explore the function of the CKI family in Wg signaling during Drosophila limb development. We provide the first genetic evidence that DBT/CKIε plays a pivotal positive role in the Wg pathway and provide evidence that DBT/CKIε exerts its positive influence both upstream and downstream of GSK3β. Moreover, we provide the first genetic evidence that DBT/CKIε has a negative role in addition to its predominantly positive role in the Wg pathway. Using RNAi, we provide evidence that CKIα is the major CKI isoform that negatively regulates Wg signaling in Drosophila wing development. In addition, we provide evidence that CKIα may also have an unappreciated positive role and this could be achieved, at least in part, at the level of Arr phosphorylation. Finally, we provide genetic evidence that Gish/CKIγ has a positive role in the Wg pathway. Consistent with our finding, a recent study showed that RNAi knockdown of Gish in cultured cells reduced Wg-stimulated luciferase reporter gene expression (Davidson et al., 2005). In addition, we find Gish/CKIγ, like its vertebrate counterpart, is mainly localized on the cell surface, and can effectively phosphorylate Arr, which may account for its positive role in the Wg pathway. The roles of various CKI isoforms in the Wg pathway are outlined in Fig. 9E.
Yin and Yang of CKIε in the Wg pathway
CKIε was initially identified as a positive regulator in the Wnt pathway based on overexpression studies (Peters et al., 1999; Sakanaka et al., 1999). Indeed, overexpression of XCKIε in Drosophila limb caused cell autonomous accumulation of Arm and activation of Wg responsive genes, leading to pattern abnormality consistent with ectopic Wg signaling (Fig. 2). Although DBT/CKIε shares over 85% amino acid sequence identity with XCKIε in the kinase domain, overexpression of DBT or its kinase domain didn’t induce ectopic Wg signaling (Fig. 5E, data not shown). Nevertheless, overexpression of DBT induced ectopic Wg signaling in a sensitized genetic background (Fig. 5F, 7B).
Despite the fact that CKIε has been implicated as a positive regulator of the Wnt/Wg pathway, no genetic evidence for such a role has ever been obtained until now. One reason could be that CKIε participates in multiple cellular processes and null or strong mutations cause cell lethality (Zilian et al., 1999). On the other hand, hypomorphic mutations do not significantly perturb Wg signaling, probably because a low dose of CKIε suffices to transduce the Wg signal and/or because other CKI family members can compensate for the partial loss of CKIε. To facilitate the recovery of mutant clones homozygous for dbt null mutation, we applied a combination of several approaches. First, we generated mitotic clones in the Minute background, which gave mutant cells a growth advantage (Morata and Ripoll, 1975). Second, we coexpressed P35, a cell death inhibitor (Hay et al., 1994), in discs where dco mutant clones were generated to block apoptosis due to loss of CKIε. Finally, we used a wing specific, constitutive source of flipase (MS1096/UAS-flp) to induce FRT-mediated mitotic recombination in the wing pouch region (Jia et al., 2005). Under these conditions, all wing discs of the appropriate genotype contained dco− clones occupying most of the wing pouch region (Fig. 3G–I). These wing discs exhibited diminished levels of Wg target gene expression, demonstrating that DBT/CKIε is a positive regulator of the Wg pathway. The approach described here can be applied to study other cell lethal genes.
Although most of the evidence supports a positive role for CKIε in the Wnt/Wg pathway, several observations implied that CKIε also impinged on β-catenin/Arm phosphorylation and degradation (Price, 2006). For example, it has been shown that CKIε is associated with Axin and DN-CKIε blocks Axin-induced phosphorylation of β-catenin at Ser45 (Amit et al., 2002). In addition, RNAi knockdown of DBT/CKIε resulted in stabilization of Arm in S2 cells, albeit to a lesser extent than CKIα knockdown (Yanagawa et al., 2002), and increased the basal transcription from a Tcf-luciferase reporter gene (Cong et al., 2004). However, one caveat of these studies is that the activities of other CKI isoforms might also be affected by DN-CKIε or DBT/CKIε RNAi. We took a genetic approach to address whether DBT/CKIε has any negative function in the Wg pathway, and found that hypomorphic dbt mutations caused ectopic Wg signaling, but only when CKIα activity was partially blocked (Fig.6). Hence, DBT/CKIε is normally dispensable for Arm degradation due to sufficent CKIα; however, DBT/CKIε levels become critical when CKIα activity is reduced. Our result is not inconsistent with a previous observation that CKIε RNAi did not affect β-catenin phosphorylation and degradation in cultured cells (Liu et al., 2002). In that study, RNAi did not completely block CKIε, and the presence of CKIα in the same cells could have masked any effect CKIε RNAi might have had on β-catenin phosphorylation and degradation. It would be interesting to determine if CKIε RNAi could enhance the effect of CKIα RNAi on β-catenin phosphorylation and degradation, which is predicted by our study.
CKIε regulates the Wg pathway at multiple steps
CKIε binds and phosphorylates Dsh (Gao et al., 2002; Kishida et al., 2001; McKay et al., 2001; Peters et al., 1999; Sakanaka et al., 1999). However, a previous study placed CKIε downstream of Dsh based on the observation that overexpressing XCKIε could rescue Wnt signaling defects caused by a dominant negative form of Dsh (DN-Dsh) (Peters et al., 1999). In contrast, we find that the ability of XCKIε to induce Wg pathway activation depends on Dsh, as dsh null mutant clones overexpressing XCKIε fail to activate Wg target genes (Fig. 4D-D″). Hence our genetic epistasis study places CKIε upstream of or parallel to Dsh. It is possible that DN-Dsh might not completely block endogenous Dsh, and overexpressed XCKIε could transduce the Wnt signal through residual Dsh activity. Consistent with the notion that CKIε acts upstream of or parallel to Dsh, we find that coexpression of Nkd, an inducible Wg pathway inhibitor that acts by binding to Dsh, suppresses the “gain-of-Wg” phenotypes caused by XCKIε (Fig. 4H). In addition, DN-GSK3β can reverse the “loss-of-Wg” phenotypes caused by DN-CKIε (Fig. 4L). Hence a critical role that CKIε plays is to antagonize the activity of the Arm/β-catenin destruction complex, and antagonism of GSK3β alleviates such a requirement. CKIε could bind Dsh and destabilize the Arm/β-catenin destruction complex (Gao et al., 2002). In addition, CKIε could destabilize Axin complex through phosphorylation of Arr (Fig. 9E).
Our epistasis analysis also revealed a role for CKIε downstream of GSK3β phosphorylation. We find that the levels of ectopic sen in wing discs coexpressing DN-CKIε and DN-GSK3β are significantly lower than those in wing discs expressing DN-GSK3β alone, suggesting that DN-CKIε attenuates Wg signaling activity even when phosphorylation and degradation of Arm is blocked by DN-GSK3β. One likely target for CKIε downstream of GSK3β is Tcf as it has been shown that in Xenopus oocyte extracts, CKIε phosphorylated Tcf3 and stabilized its interaction with β-catenin (Lee et al., 2001).
Does CKIα acts solely as a Wnt/Wg pathway inhibitor?
The role of CKIα in the Wnt/Wg pathway has largely been deduced from studies using cell culture systems. Thus, RNAi knockdown of CKIα inhibits β-catenin/Arm phosphorylation and degradation, and induces Tcf/Lef mediated luciferase expression (Liu et al., 2002; Lum et al., 2003; Matsubayashi et al., 2004; Yanagawa et al., 2002). CKIα RNAi in Drosophila embryos resulted in a “naked cuticle” phenotype, consistent with ectopic Wg signaling (Liu et al., 2002; Yanagawa et al., 2002). However, two recent studies revealed that loss-of-CKIα also results in ectopic Hh signaling (Jia et al., 2005; Lum et al., 2003). This finding complicated the interpretation of the “gain-of-Wg” phenotypes resulting from CKIα RNAi as Hh and Wg regulate each other’s expression in Drosophila embryos. To circumvent this problem, we use Drosophila wing development as a model to address the In vivo function of CKIα since Wg and Hh do not regulate each other in this system. We found that overexpressing two shorter forms of CKIα RNAi constructs (CRS and CRS2), which are specific for CKIα, led to ectopic Wg signaling in a dose dependent manner: one copy of CRS or CRS2 barely affected Wg target gene expression whereas two copies resulted in ectopic expression of sc and sen (Fig. 5A–D). A longer form of CKIα RNAi construct (CRL) was more potent than CRS, as expressing one copy resulted in robust ectopic expression of sc and sen. This is likely due to the fact that CRL knocks down CKIα more effectively than CRS (Jia et al., 2005). In addition, CRL may knock down DBT/CKIε to reduce a compensatory effect on loss of CKIα by DBT/CKIε. Intriguingly, expressing CRL at higher levels caused adverse effect on the Wg signaling activity, as manifested by the reduced levels of ectopic sc expression (Fig. 5H). A likely explanation is that high levels of CRL diminish the level of CKIε to the extent that its positive role in the Wg pathway is compromised. In support of this notion, coexpressing DBT/CKIε with CRL restored high levels of ectopic sc expression (Fig. 5I).
Despite the predominantly negative role of CKIα in the Wg pathway, a positive role has been underscored in our double mutant analysis. We observed that CKIα knockdown enhanced the “loss-of-Wg” phenotypes caused by dbt null mutation, as manifested by more complete loss of sen and vg expression in dbt mutant discs expressing CRS2 (Fig. 8E). CKIα may positively regulate Wg signaling by phosphorylating Dsh, as suggested by previous studies (Matsubayashi et al., 2004; McKay et al., 2001). Alternatively, CKIα could exert a positive influence on the Wg pathway by phosphorylating Arr (Fig. 9E).
Other CKI isoforms in the Wg pathway
We applied overexpression assays to explore the potential role of the other five CKI isoforms that share over 50% amino acid sequence identity in their kinase domains with CKIα (Fig. 1A). First, we asked if any of these CKI isoforms could functionally substitute for CKIα in blocking Wg pathway activation. Unlike CKIα, none of other CKI isoforms including CG7094, CG2577, CG12147, Gish/CKIγ, and CG9962 were able to rescue the “gain-of-Wg” phenotype caused by CRL (Fig. S1), suggesting that these CKI isoforms are unlikely to play any major role in priming GSK3β-mediated phosphorylation and degradation of Arm/β-catenin. On the other hand, we find that CG12147 induced ectopic Wg signaling activity when CKIα was partially blocked, albeit to a lesser extent than DBT (Fig. 7F). Although Gish overexpression failed to induce ectopic Wg signaling activity even when CKIα was partially blocked (Fig. 7H), loss-of-Gish mutation resulted in a reduction in Wg signaling activity and enhanced the “loss-of-Wg” phenotypes caused by the dbt null mutation (Fig. 8B, D), suggesting that Gish/CKIγ positively regulates the Wg pathway.
Phosphorylation of Arr by multiple CKI isoforms
It has recently been shown that CKI family members phosphorylate multiple sites in the cytoplasmic domain of LRP6 (Davidson et al., 2005; Zeng et al., 2005) and a set of these CKI sites are primed by GSK3β phosphorylation of the PPPSP motif (Zeng et al., 2005). Overexpressing CKIγ but not CKIε caused phosphorylation of LRP6, whereas dominant negative CKIγ inhibited Wnt3a-induced LRP6 phosphorylation in HEK293T cells (Davidson et al., 2005), suggesting a specific role for CKIγ in phosphorylating LRP6. In contrast, Zeng et al showed that a combination of dominant negative CKIα and CKIδ but neither CKIα or CKIδ alone blocked Wnt3a-induced LRP6 phosphorylation in CKIε−/− MEF cells, suggesting that CKIα and CKIγ/ε act redundantly in phosphorylating LRP6 in response to Wnt (Zeng et al., 2005). However, dominant negative CKI isoforms may not exhibit absolute specificity, which could account for the discrepancy between these two studies. While it awaits for genetic mutations in individual CKI isoforms to confirm the results obtained with the dominant negative forms of CKI, it is likely that multiple CKI family members could participate in LRP5/6 phosphorylation.
Multiple PPPSP motifs as well as adjacent CKI sites are conserved in the cytoplasmic domain of Drosophila Arr. We found that in Drosophila S2 cells, multiple CKI family members can phosphorylate Arr cytoplasmic domain and this phosphorylation appears to rely on GSK3β primed phosphorylation (Fig. 9A–C). Among all the CKI isoforms that can phosphorylate Arr, Gish/CKIγ exhibited the highest potency whereas CKIα and CKIε show weak activity toward Arr, suggesting that Gish/CKIγ is the major CKI isoform that phosphorylates Arr. Consistent with its high potency toward Arr phosphorylation, Gish/CKIγ is primarily associated with plasma membrane, as is the case for its vertebrate counterpart (Davidson et al., 2005). Phosphorylation of Arr by Gish/CKIγ is likely to account for the positive role that Gish/CKIγ plays in the Wg signaling pathway. We find that gishe01759 attenuates but not completely blocks Wg responsive gene expression. The residual Wg signaling activity in gishe01759 mutant cells could be due to the hypomorphic nature of this mutation. Alternatively, other CKI isoforms could partially substitute for Gish/CKIγ in phosphorylating Arr.
We find that CG12147 and CG9962 phosphorylate Arr more effectively than CKIα or CKIε, although they are less potent than Gish/CKIγ. Consistent with their ability to phosphorylate Arr, overexpressing CG12147 or CG9962 resulted in ectopic Wg signaling in a genetic sensitized background (Fig. 7F and 7J). However, phosphorylation of Arr alone might be insufficient to account for their positive roles as overexpressing Gish/CKIγ did not have the same magnitude of effect on Wg signaling as CG12147 and CG9962. It is possible that CG12147 and CG9962 can phosphorylate other targets in the Wg pathway. Future loss of function study and biochemical analysis should probe the precise roles of these CKI isoforms in the Wg pathway.
Supplementary Material
Acknowledgments
We thank Liping Luo for excellent technical assistance, Dr. J. Peters for assistance with the XCKIε constructs, Drs. M. Noll, J. Treisman, C. Baht, R. Carthew, R. Mann, S. Carroll, H. Bellen, Bloomington stock center for stocks and reagents. This work was supported by grants from NIH, Welch Foundation, and Leukemia and Lymphoma Society Scholar Program to Jin Jiang (J. J).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–76. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. wingless signaling in the Drosophila eye and embryonic epidermis. Development. 1996;122:2801–12. doi: 10.1242/dev.122.9.2801. [DOI] [PubMed] [Google Scholar]
- Campuzano S, Modolell J. Patterning of the Drosophila nervous system: the achaete-scute gene complex. Trends Genet. 1992;8:202–8. doi: 10.1016/0168-9525(92)90234-u. [DOI] [PubMed] [Google Scholar]
- Cong F, Schweizer L, Varmus H. Casein kinase Iepsilon modulates the signaling specificities of dishevelled. Mol Cell Biol. 2004;24:2000–11. doi: 10.1128/MCB.24.5.2000-2011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culi J, Mann RS. Boca, an endoplasmic reticulum protein required for wingless signaling and trafficking of LDL receptor family members in Drosophila. Cell. 2003;112:343–54. doi: 10.1016/s0092-8674(02)01279-5. [DOI] [PubMed] [Google Scholar]
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–72. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci U S A. 2002;99:1182–7. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–9. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hazelett DJ, Bourouis M, Walldorf U, Treisman JE. decapentaplegic and wingless are regulated by eyes absent and eyegone and interact to direct the pattern of retinal differentiation in the eye disc. Development. 1998;125:3741–51. doi: 10.1242/dev.125.18.3741. [DOI] [PubMed] [Google Scholar]
- Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature. 2002;416:548–52. doi: 10.1038/nature733. [DOI] [PubMed] [Google Scholar]
- Jia J, Tong C, Wang B, Luo L, Jiang J. Hedgehog Signalling Activity of Smoothened Requires Phosphorylation by Protein Kinase A and Casein Kinase I. Nature. 2004;432:1045–1050. doi: 10.1038/nature03179. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell. 2005;9:819–30. doi: 10.1016/j.devcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Protein kinase A and Hedgehog signalling in Drosophila limb development. Cell. 1995;80:563–572. doi: 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Complementary and mutually exclusive activities of decapentaplegic and wingless organize axial patterning during Drosophila leg development. Cell. 1996;86:401–9. doi: 10.1016/s0092-8674(00)80113-0. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature. 1998;391:493–6. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- Kalidas S, Smith DP. Novel genomic cDNA hybrids produce effective RNA interference in adult Drosophila. Neuron. 2002;33:177–84. doi: 10.1016/s0896-6273(02)00560-3. [DOI] [PubMed] [Google Scholar]
- Kennerdell JR, Carthew RW. Heritable gene silencing in Drosophila using double-stranded RNA. Nat Biotechnol. 2000;18:896–8. doi: 10.1038/78531. [DOI] [PubMed] [Google Scholar]
- Kishida M, Hino S, Michiue T, Yamamoto H, Kishida S, Fukui A, Asashima M, Kikuchi A. Synergistic activation of the Wnt signaling pathway by Dvl and casein kinase Iepsilon. J Biol Chem. 2001;276:33147–55. doi: 10.1074/jbc.M103555200. [DOI] [PubMed] [Google Scholar]
- Lee E, Salic A, Kirschner MW. Physiological regulation of [beta]-catenin stability by Tcf3 and CK1epsilon. J Cell Biol. 2001;154:983–93. doi: 10.1083/jcb.200102074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Lum L, Yao S, Mozer B, Rovescalli A, Von Kessler D, Nirenberg M, Beachy PA. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science. 2003;299:2039–45. doi: 10.1126/science.1081403. [DOI] [PubMed] [Google Scholar]
- Matsubayashi H, Sese S, Lee JS, Shirakawa T, Iwatsubo T, Tomita T, Yanagawa S. Biochemical characterization of the Drosophila wingless signaling pathway based on RNA interference. Mol Cell Biol. 2004;24:2012–24. doi: 10.1128/MCB.24.5.2012-2024.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay RM, Peters JM, Graff JM. The casein kinase I family in Wnt signaling. Dev Biol. 2001;235:388–96. doi: 10.1006/dbio.2001.0308. [DOI] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975 Feb;42:211–21. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Nolo R, Abbott LA, Bellen HJ. Drosophila Lyra mutations are gain-of-function mutations of senseless. Genetics. 2001;157:307–15. doi: 10.1093/genetics/157.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, McKay RM, McKay JP, Graff JM. Casein kinase I transduces Wnt signals. Nature. 1999;401:345–50. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, Zipursky SL. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development [published erratum appears in Cell 1998 Feb 20;92(4):following 585] Cell. 1997;91:881–91. doi: 10.1016/s0092-8674(00)80480-8. [DOI] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–8. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, Young MW. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell. 1998;94:83–95. doi: 10.1016/s0092-8674(00)81224-6. [DOI] [PubMed] [Google Scholar]
- Price MA. CKI, there’s more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- Rousset R, Mack JA, Wharton KA, Jr, Axelrod JD, Cadigan KM, Fish MP, Nusse R, Scott MP. Naked cuticle targets dishevelled to antagonize Wnt signal transduction. Genes Dev. 2001;15:658–71. doi: 10.1101/gad.869201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Tice DA, Polakis P. Axin-dependent phosphorylation of the adenomatous polyposis coli protein mediated by casein kinase 1epsilon. J Biol Chem. 2001;276:39037–45. doi: 10.1074/jbc.M105148200. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Casein kinase iepsilon in the wnt pathway: regulation of beta-catenin function. Proc Natl Acad Sci U S A. 1999;96:12548–52. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–5. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- Wang G, Wang B, Jiang J. Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes & Dev. 1999;13:2828–37. doi: 10.1101/gad.13.21.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O’Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–30. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Yanagawa S, Matsuda Y, Lee JS, Matsubayashi H, Sese S, Kadowaki T, Ishimoto A. Casein kinase I phosphorylates the Armadillo protein and induces its degradation in Drosophila. Embo J. 2002;21:1733–42. doi: 10.1093/emboj/21.7.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca M, Basler K, Struhl G. Direct and long-range action of a Wingless morphogen gradient. Cell. 1996;87:833–844. doi: 10.1016/s0092-8674(00)81991-1. [DOI] [PubMed] [Google Scholar]
- Zeng W, Wharton KA, Jr, Mack JA, Wang K, Gadbaw M, Suyama K, Klein PS, Scott MP. naked cuticle encodes an inducible antagonist of Wnt signalling. Nature. 2000;403:789–95. doi: 10.1038/35001615. [DOI] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–7. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Carthew RW. Interactions between Wingless and DFz2 during Drosophila wing development. Development. 1998;125:3075–85. doi: 10.1242/dev.125.16.3075. [DOI] [PubMed] [Google Scholar]
- Zilian O, Frei E, Burke R, Brentrup D, Gutjahr T, Bryant PJ, Noll M. double-time is identical to discs overgrown, which is required for cell survival, proliferation and growth arrest in Drosophila imaginal discs. Development. 1999;126:5409–20. doi: 10.1242/dev.126.23.5409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.