

Abstract
ClpB is a heat-shock protein that reactivates aggregated proteins in cooperation with the DnaK chaperone system. ClpB belongs to the family of AAA+ ATPases and forms ring-shaped oligomers: heptamers in the absence of nucleotides and hexamers in the presence of nucleotides. We investigated the thermodynamic stability of ClpB in its monomeric and oligomeric forms. ClpB contains 6 distinct structural domains: the N-terminal domain involved in substrate binding, two AAA+ ATP-binding modules, each consisting of two domains, and a coiled-coil domain inserted between the AAA+ modules. We produced 7 variants of ClpB, each containing a single Trp located in each of the ClpB domains and measured the changes in Trp fluorescence during the equilibrium urea-induced unfolding of ClpB. We found that two structural domains: the small domain of the C-terminal AAA+ module and the coiled-coil domain were destabilized in the oligomeric form of ClpB, which indicates that only those domains change their conformation and/or interactions during formation of the ClpB rings.
Keywords: ClpB, AAA+ ATPase, molecular chaperone, protein denaturation, protein stability, protein oligomerization, fluorescence
AAA+ family of ATPases associated with various cellular activities plays an essential role in a wide variety of cellular processes including protein quality control (folding, disaggregation, and degradation), membrane fusion and vesicular trafficking, cytoskeletal regulation and intracellular transport, organelle biogenesis, DNA replication and transcription regulation [1,2,3]. AAA+ proteins are often described as “molecular machines” that use energy from the hydrolysis of ATP to induce structural rearrangements in an impressive gallery of substrates. Approximately 80 genes encoding canonical AAA+ ATPases are found in the human genome [2]. Mutations in many AAA+ genes are linked to severe cellular pathologies and human diseases. Two neurological disorders, hereditary spastic paraplegia and torsion dystonia, correlate with mutations in spastin and torsinA, respectively [3]. AAA+ ATPases TIP and SPAF are linked to oncogenic transformation [2]. Mutations in p97/VCP cause a form of dementia and Paget’s bone disease [3]. Importantly, AAA+ subunits are an essential part of proteasome, the central protein degradation machine of eukaryotic cells [4].
The functional diversity of AAA+ ATPases appears to rely on common protein architecture and principles of molecular mechanism. All AAA+ ATPases contain one or two conserved sequence modules with Walker A and B ATP-binding motifs. The AAA+ module assumes a conserved three-dimensional two-domain structure consisting of a larger N-terminal Rossman-fold α/β domain and a smaller C-terminal mostly α-helical domain [3]. Importantly, AAA+ ATPases form ring-shaped oligomers with nucleotide binding sites located at the interfaces between the subunits and a narrow pore located at the center of the ring. Nucleotides stabilize the AAA+ rings and, conversely, the ring formation is necessary for binding of nucleotides and ATP hydrolysis [5]. Besides the Walker A and B, AAA+ sequences contain other conserved motifs that may be involved in transmitting the information about the nucleotide occupancy among different sites within the oligomeric ring: sensor-1, arginine finger, and sensor-2 [2]. Thus, formation of the rings appears a characteristic structural feature of AAA+ ATPases. In addition to the AAA+ modules, the ATPases contain N- or C-terminal “attachment domains” that often specify their cellular localization and/or substrate identity.
ClpB is a bacterial AAA+ ATPase that reactivates aggregated proteins in cooperation with the DnaK chaperone system [6,7,8]. The function of ClpB is distinct from that of “classical” molecular chaperones, which prevent but do not reverse protein aggregation. ClpB assists hundreds of aggregation-prone proteins in bacteria [9] and its homologues have been found in yeast [10] and plants [11].
As shown in Fig. 1, ClpB contains six structural domains: a distinct N-terminal domain loosely associated with the rest of the protein [12], two AAA+ modules (D1 and D2), each containing a larger and a smaller domain, and a coiled-coil domain inserted between D1 and D2. In solution, ClpB shows a reversible equilibrium between monomers, heptamers, and an intermediate-size oligomers. High protein concentration or low ionic-strength buffers stabilize heptameric ClpB, which switches to a hexamer upon binding of nucleotides [13]. In the oligomeric ClpB, the coiled-coil domains are located at the outside surface of the ring, whereas parts of the D1 and D2 large domains form the surface of the narrow channel at the center of the ring (left side of the structure in Fig. 1) [14]. The mechanism of protein disaggregation mediated by ClpB involves ATP-dependent extraction of single polypeptides from the aggregates and their forced unfolding by threading through the tight channel in the oligomeric ClpB [15]. The N-terminal domain of ClpB is involved in substrate binding [16], but the role of the coiled-coil middle domain remains unknown.
Figure 1.
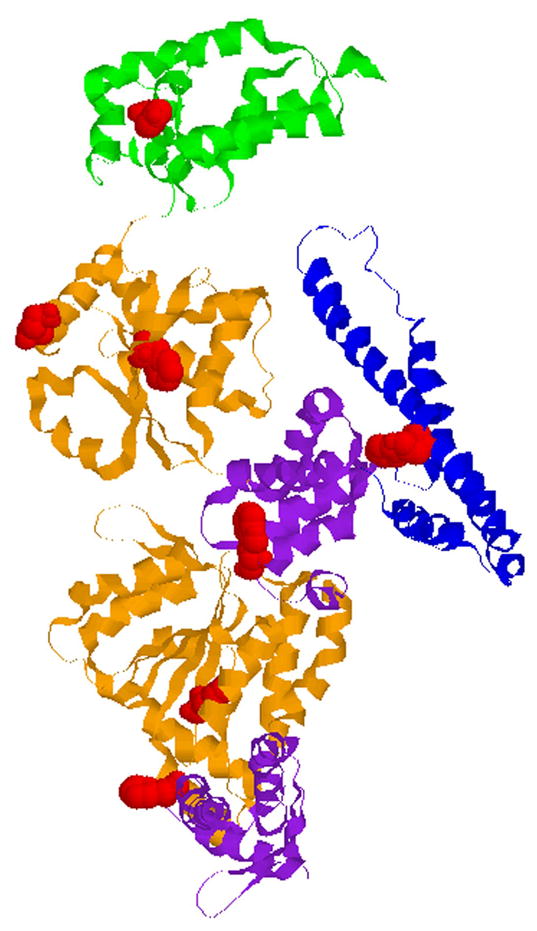
Location of Trp fluorescence probes in ClpB. Shown is the monomer structure of ClpB from Thermus thermophilus [14]. The N-terminal domain (green) is on top of the figure, followed by the D1 AAA+ large (orange) and small domain (purple), the middle coiled-coil domain (blue), and the D2 AAA+ large (orange) and small (purple) domains. Positions of Trp fluorescence probes in E. coli ClpB are shown by those of the corresponding residues in T. thermophilus ClpB (red). From top: Tyr105 (corresponding to Phe105 in E. coli ClpB), Glu246 (approximating the position of Tyr251 at the channel entrance in E. coli ClpB), Phe268 (Phe276 in E. coli ClpB), Trp453 (Trp462 in E. coli ClpB), Trp533 (Trp543 in E. coli ClpB), Phe593 (Phe603 in E. coli ClpB), and Phe803 (Tyr812 in E. coli ClpB).
It can be proposed that the mechanism of other AAA+ ATPases might parallel that of ClpB and could involve insertion or enclosure of substrates in the central pore of the oligomeric ring followed by an ATP-dependent work resulting in a change of substrate conformation. To understand that mechanism, it is essential to characterize the conformational effects that accompany formation of the AAA+ ring. In particular, it is important to understand the role of distinct protein domains within and outside of the AAA+ module in maintaining the ring structure. To reach this goal, we investigated the thermodynamic stability of ClpB in the monomeric and oligomeric form. This work is the first protein stability study of a AAA+ ATPase.
Materials and methods
Proteins
Previously published procedures were used to produce and purify Escherichia coli ClpB and its mutants [5]. Site directed mutagenesis of ClpB was performed using the QuickChange method (Stratagene). Proteins were dialyzed against 50 mM Tris/HCl pH 7.5, 0.2 M KCl, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT, with 10% glycerol. Protein concentration of ClpB was determined spectrophotometrically.
Differential scanning calorimetry (DSC)
DSC experiments were performed with a VP-DSC calorimeter (MicroCal Inc., Northampton, MA) at a scan rate of 1 K/min. The instrument baseline was obtained first by measuring the heat-capacity profile of the dialysis buffer.
ClpB ATPase assay
ClpB samples were incubated for 15 min at 37 °C in 100 mM Tris-HCl pH 8, 10 mM MgCl2, 5 mM ATP, 1 mM EDTA, 1 mM DTT without or with 0.1 mg/ml κ-casein (Sigma). Inorganic phosphate production was determined using the malachite green method [17,18].
Urea-induced unfolding of ClpB
Solutions containing urea at the concentration 0 – 8 M were prepared with buffer A (50 mM Tris/HCl pH 7.5, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT). To investigate monomeric ClpB, single-Trp ClpB variants were diluted to 0.1 mg/ml in the buffer A urea solutions containing 0.3 M KCl. To investigate heptameric ClpB, the ClpB variants were diluted to 0.5 mg/ml in the buffer A urea solutions. ClpB samples were incubated overnight (~15 h) at room temperature before measuring fluorescence spectra.
Fluorescence spectroscopy
Fluorescence spectra of the single-Trp ClpB variants were measured with a Cary Eclipse spectrofluorimeter (Varian) using a 1-cm quartz fluorescence cuvette. In experiments with 0.1 mg/ml ClpB, excitation and emission slits were 5 and 10 nm, respectively. In experiments with 0.5 mg/ml ClpB, the slits were 5 nm. The spectra of urea-containing buffers (without ClpB) were subtracted from the protein spectra.
Results and discussion
Fig. 2 shows the thermal unfolding transitions of ClpB as revealed by differential scanning calorimetry. The experiment was performed at low ionic strength to populate the oligomeric form of ClpB [13]. The thermogram shows two distinct thermal transitions (at 53.5 and 63.7 °C) and a shoulder (~57 °C), which indicates that ClpB contains multiple folding domains that likely correspond to the structural domains identified in Fig. 1. As it is often observed for unfolding of multi-domain proteins, the thermal unfolding of ClpB is irreversible, as documented by the lack of the transitions in the re-scan of the protein sample (data not shown). The irreversibility of thermal unfolding is due to aggregation of ClpB, which occurs when partially folded conformations are populated at high temperature. The irreversibility of protein aggregation distorts the thermograms and prevents their equilibrium thermodynamic analysis.
Figure 2.
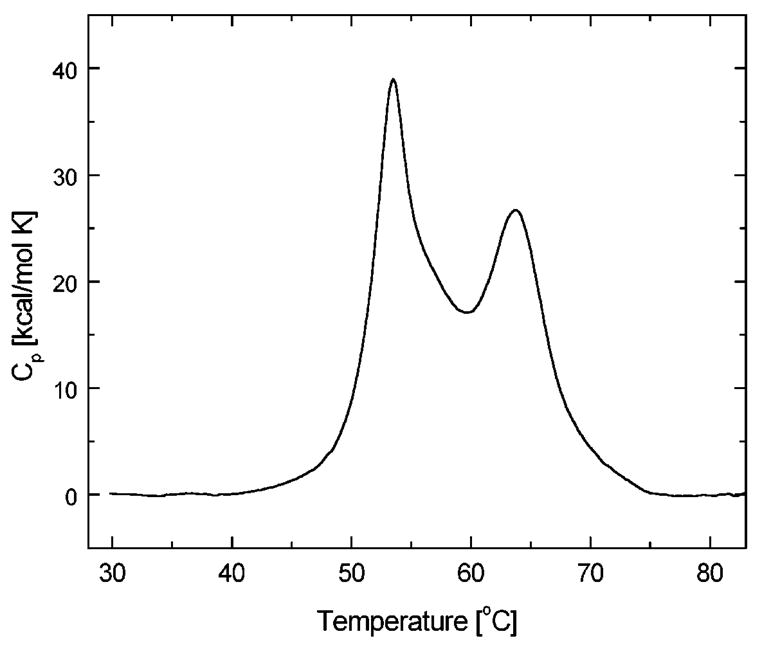
Differential scanning calorimetry of ClpB. The heat capacity profile is shown for 4 mg/ml ClpB in 50 mM Hepes/KOH, pH 7.5. The data have been obtained at a 1 K/min scanning rate, corrected for the buffer baseline and normalized for protein concentration.
To prevent protein aggregation at high temperature, we performed chemical denaturation studies for ClpB at room temperature (25 °C). We used Trp fluorescence to monitor changes in conformation of each domain in ClpB at equilibrium with increasing concentrations of urea. E. coli ClpB contains two Trp residues: Trp462 at the center of the coiled-coil domain (see Fig. 1) and Trp543 in the D1 small domain. First, we produced two single-Trp variants of ClpB, W543F (with the remaining Trp462) and W462F (with Trp543). Next, we produced a Trp-less ClpB with the double mutation W462F/W543F. Subsequently, we produced five new variants of Trp-less ClpB with a single Trp introduced in specific locations containing aromatic residues (see Fig. 1): F105W (within the N-terminal domain, close to the putative substrate binding site [16]), Y251W (at the D1 entrance to the ClpB channel), F276W (within Walker B motif in D1 large domain), F603W (within Walker A motif in D2 large domain), and Y812W (within sensor-2 motif in the D2 small domain). Altogether, we produced 7 variants of ClpB, each with a single Trp located in each of the six protein domains (at positions 105, 251, 276, 462, 543, 603, and 812, see Fig. 1).
We tested the structural integrity and biochemical functionality of the 7 single-Trp variants of ClpB. Similar to wt ClpB, the 7 single-Trp ClpB variants efficiently formed oligomers at high protein concentration, as determined by sedimentation velocity studies (data not shown), which indicates that the site mutations did not disrupt the structure of the ClpB variants. All 7 ClpB variants were active ATPases (Fig. 3A). The basal ATPase activity varied among the single-Trp variants with three proteins showing lower activity than the wt and three proteins showing higher activity. Interestingly, the introduction of a Trp in the vicinity of the Walker A motif in D2 (Trp603) significantly activated the ClpB ATPase. The ATPase of all the variants of ClpB was activated by casein (Fig. 3B, compare the scale with Fig. 3A). The ATPase activity of the ClpB variants measured in the presence of casein paralleled their basal activity. We conclude that a replacement of the selected aromatic residues (either Phe or Tyr) by Trp may modulate the rate of ATP hydrolysis of ClpB. Interestingly, even mutations in the domains that do not contain nucleotide binding sites (W105 in the N-terminal domain, W462 in the coiled-coil domain) affect the ClpB ATPase, which suggests that concerted conformational changes in all domains may contribute to the ATP-dependent functionality of ClpB.
Figure 3.
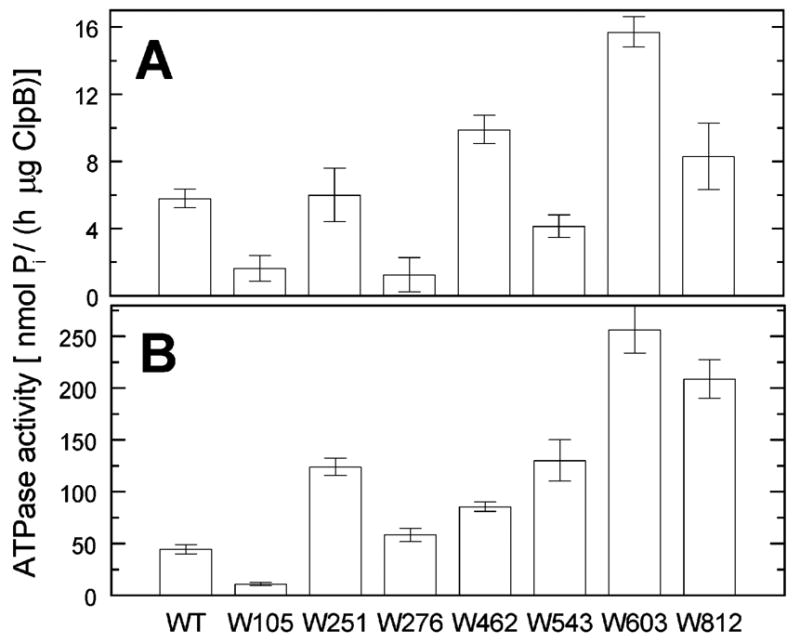
ATPase activity of wild type ClpB and its single-Trp variants. The rate of ATP hydrolysis catalyzed by wt ClpB or its variants, each with a single Trp at the indicated position, was measured at 37 °C without (A) or with 0.1 mg/ml κ-casein (B). Averages from three measurements are shown with the standard deviations.
We have previously shown that ~0.1 mg/ml ClpB is monomeric in high ionic strength buffers (0.2–0.3 M KCl), whereas heptameric ClpB predominates at low ionic strength [13,19]. We compared the Trp fluorescence spectra of the ClpB variants under the conditions where either monomeric (see Fig. 4A) or oligomeric ClpB (see Fig. 4B) predominates. As shown in Fig. 4A, the Trp emission intensity and the wavelength position of the emission band strongly depend on the position of the fluorophore in monomeric ClpB. In particular, the fluorescence of Trp276 (near the D1 ATP binding site) shows the strongest intensity quenching, whereas that of Trp543 (in D1 small domain) shows the lowest quenching. The differences between emission spectra of different ClpB variants indicate that the solvent exposure and the local environment of each of the fluorophores are different for each ClpB variant. Importantly, the intensities and positions of Trp emission spectra for the oligomeric ClpB (Fig. 4B) correlate with those observed for monomeric ClpB (Fig. 4A). This result indicates that major conformational changes are not likely to occur in the ClpB domains during protein oligomerization.
Figure 4.
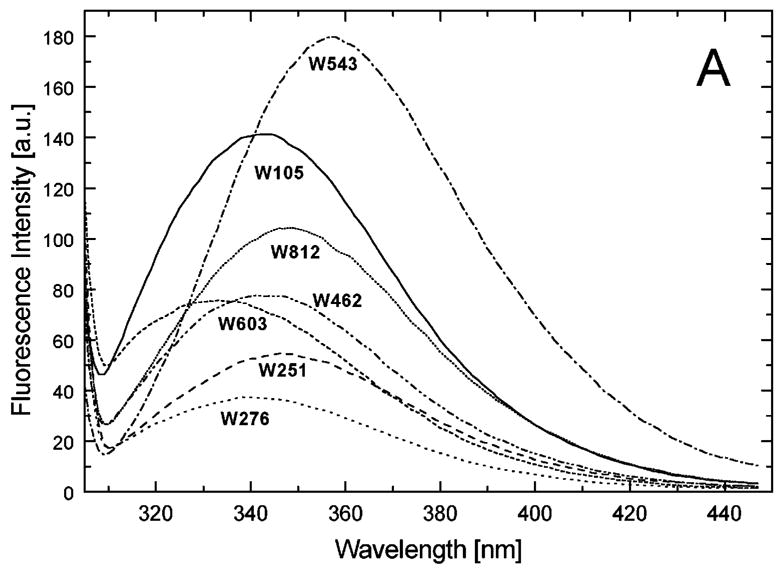
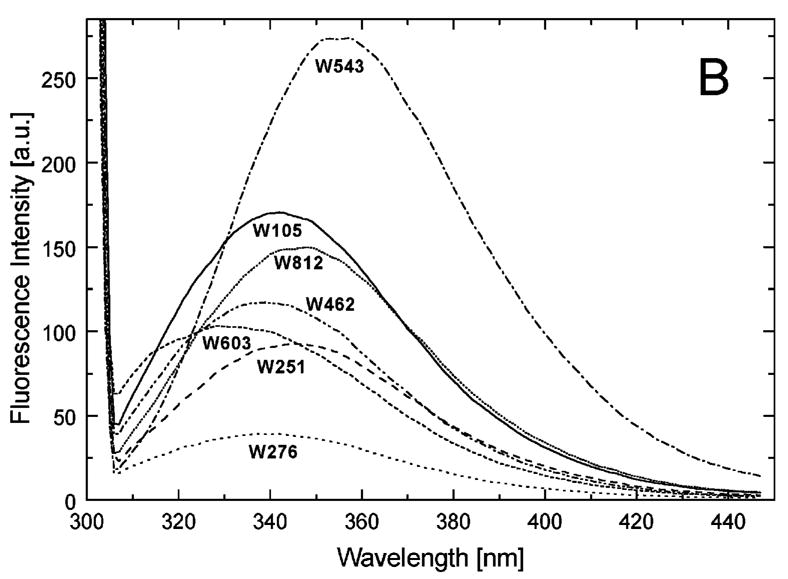
Fluorescence emission spectra of the single-Trp variants of ClpB. The excitation wavelength was 300 nm and the emission spectra were measured for each of the variants of ClpB indicated in the figure at 0.1 mg/ml in buffer A (50 mM Tris/HCl pH 7.5, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT) with 0.3 M KCl (A) or at 0.5 mg/ml in buffer A without KCl (B).
To further characterize the effects of ClpB oligomerization on the properties of its domains, we investigated the stability of the ClpB variants and measured Trp fluorescence as the function of urea concentration. To ensure the thermodynamic equilibrium during protein denaturation studies, we incubated ClpB in urea-containing buffer overnight and determined that the fluorescence spectra of ClpB samples did not change upon longer incubation. We also investigated the urea-induced unfolding of ClpB in the presence of ATP and ADP. Interestingly, after prolonged incubation with nucleotides, ClpB solutions with low urea concentrations became cloudy, which indicated protein precipitation and made fluorescence measurements unfeasible. This result suggests that nucleotides induce a high-affinity protein-binding conformation in ClpB and promote its non-specific self-association. Indeed, ClpB binds substrates with the highest affinity in its ATP-bound state [20].
Figure 5A-G shows urea-induced changes in the Trp fluorescence intensity of the seven ClpB variants under conditions stabilizing the ClpB monomer and the heptamer. For all chosen locations of the fluorescence probes in ClpB, except Trp251 and Trp276 (Fig. 5B and C), the Trp emission intensities decrease as the concentration of urea increases and show cooperative urea-dependent transitions. The emission intensities of Trp251 and Trp276 are the lowest among all investigated ClpB variants (see Fig. 4), which indicates that both these locations of the fluorophores are significantly exposed to the solvent in the native ClpB. Trp251 is located within a flexible unstructured loop at the entrance to the ClpB channel. The Trp251 loop has not been resolved in the crystal structure [14] and is involved in binding aggregated substrates to ClpB [16]. Urea-induced changes in the fluorescence of Trp251 are small and do not show clear cooperative transitions, which is consistent with the lack of stable structure within the channel loop either for the monomeric or the oligomeric ClpB. Trp276 is located near the conserved acidic residues of the Walker B motif in the D1 large domain of ClpB, which are essential for the ATP hydrolysis [20]. The Walker B motif is located at the tip of a β-strand [14] and its side chains are exposed into the ATP binding site. Unlike for the remaining fluorophore positions, the fluorescence intensity of Trp276 increases with increasing concentration of urea (Fig. 5C), which suggests that urea decreases the emission quenching of the solvent-exposed Trp276. Characteristically, the changes in Trp276 fluorescence show similar patterns for the monomeric and the oligomeric ClpB. We conclude that Trp251 and Trp276 are highly exposed to the solvent in the ClpB structure and their emission intensities may not follow the urea-induced structural rearrangements in their corresponding ClpB domains.
Figure 5.
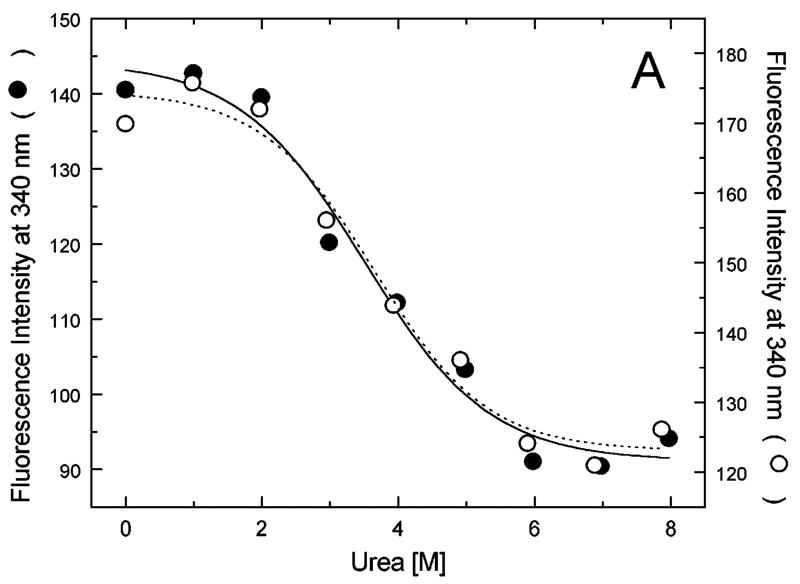
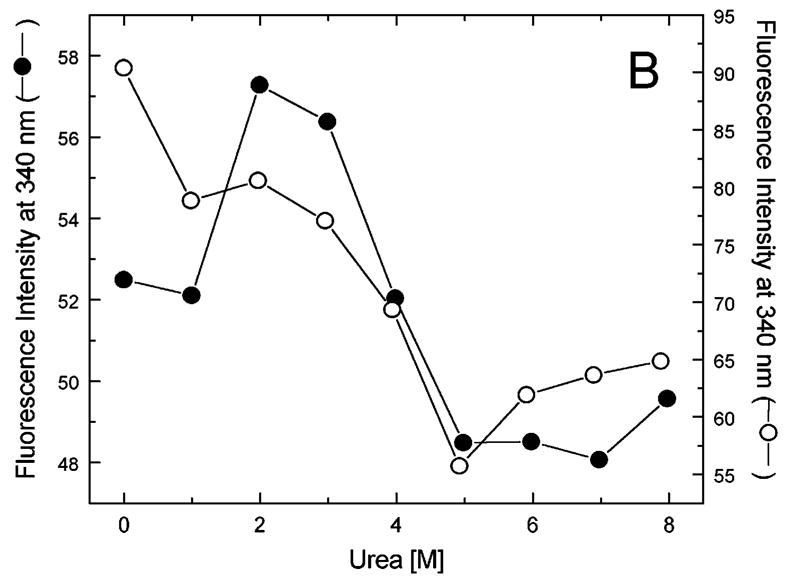
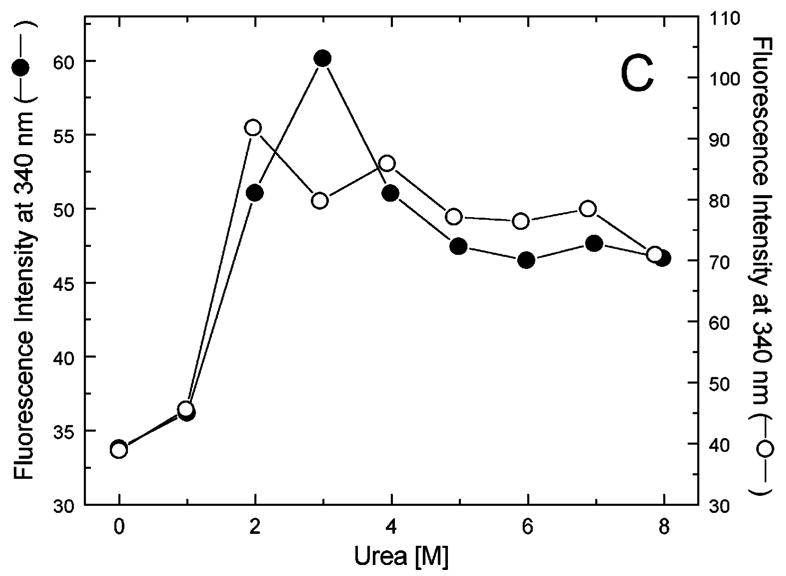
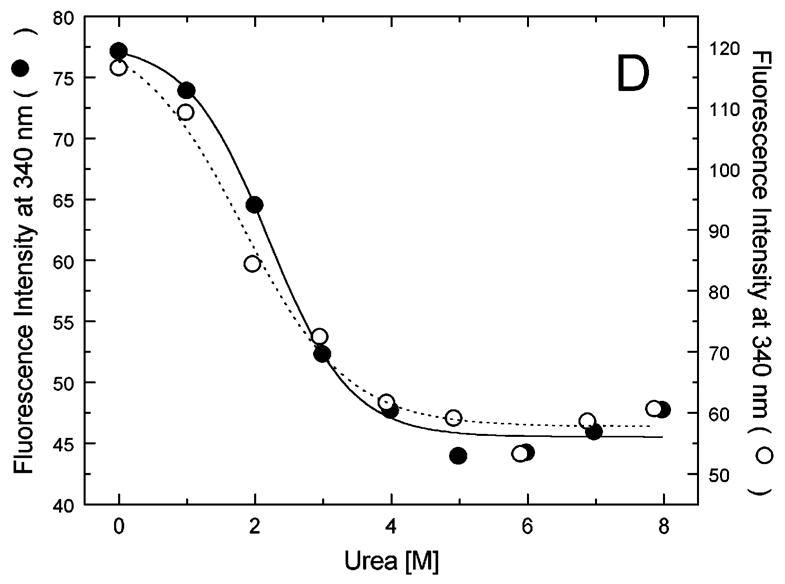
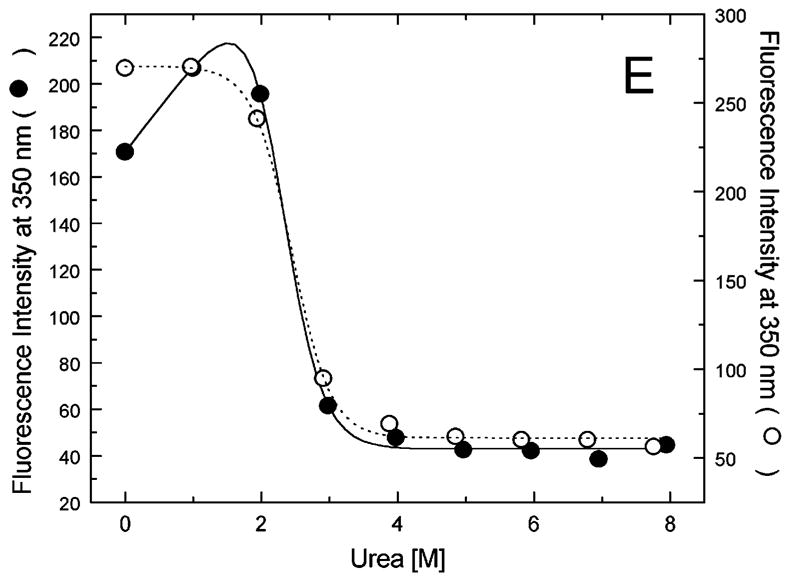
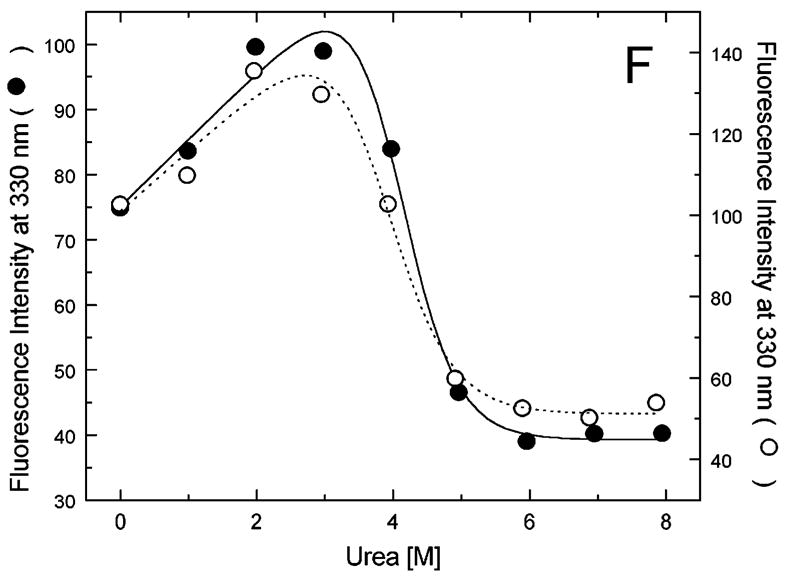
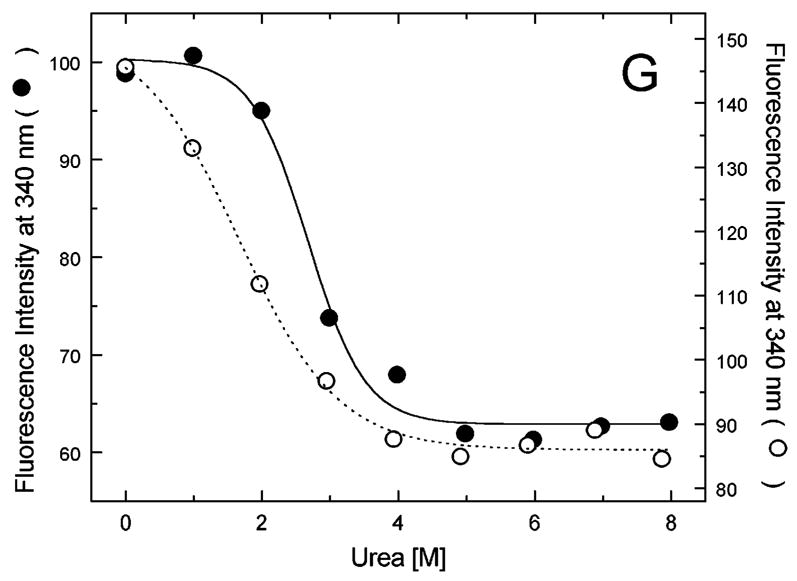
Urea-induced changes of fluorescence intensity in single-Trp ClpB variants. Shown are the data for monomeric ClpB (0.1 mg/ml in 50 mM Tris/HCl pH 7.5, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT with 0.3 M KCl and indicated urea concentrations, filled symbols) and oligomeric ClpB (0.5 mg/ml in 50 mM Tris/HCl pH 7.5, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT with indicated urea concentrations, open symbols). The emission intensity was measured at equilibrium for the ClpB variants containing Trp at the following positions: W105 (A), W251 (B), W276 (C), W462 (D), W543 (E), W603 (F), and W812 (G). In panels A and D through G, shown are the nonlinear least-squares fits of the data using the two-state unfolding model (see text) for monomeric ClpB (solid line) and for oligomeric ClpB (dotted line).
The remaining five single-Trp ClpB variants (W105, W462, W543, W603, and W812) show cooperative urea-induced transitions, which are consistent with two-state unfolding of the corresponding structural domains of ClpB (see Fig. 1). The pre-transition increases in fluorescence intensity are found for W543 under monomeric conditions (Fig. 5E) and for W603 (Fig. 5F). We assume that pre-transition intensity increases are due to urea-induced changes in fluorescence quenching for the fluorophores that are partially solvent-exposed in the native structure of ClpB.
The urea-induced cooperative conformational transitions are often described with the following empirical relationship: ΔG([D]) = ΔG0 − m[D] [21], where ΔG0 is the standard free energy of unfolding of a given domain under urea-free conditions, [D] is molar concentration of urea, and m is an empirical constant. The above equation predicts that the standard free energy of protein unfolding (ΔG) decreases with an increasing urea concentration, which increases the equilibrium population of the unfolded conformation and decreases that of the native state.
We used the above empirical relationship to describe the cooperative urea-induced transitions in the Trp fluorescence intensity (Fig. 5A and D–G). For the data in Fig. 5E and F, we assumed a linear urea-dependent pre-transition fluorescence baseline. The parameters of the equation, ΔG0 and m, are listed in Table I for five single-Trp ClpB variants. The D2 large domain that contains Trp603 is the most stable among the domains of monomeric ClpB (as shown by the values of ΔG0). The coiled-coil domain that contains Trp462 and the N-terminal domain with Trp105 are the least stable. Upon oligomerization of ClpB, no significant changes of domain stability are found for the N-terminal domain (W105), the small D1 domain (W543), and the large D2 domain (W603). However, the ClpB oligomerization significantly destabilizes the small D2 domain (W812) and, to a lower extent, the coiled-coil domain (W462). This result indicates that the two domains may undergo significant structural rearrangements during formation of the ClpB ring. In the oligomeric ClpB, the coiled-coil and D2 small domain may loose some inter-domain contacts that contribute to their stability in the monomeric form. Conversely, in the oligomeric ring, these domains may form new contacts with partner domains of the neighboring subunit. Indeed, our previous studies showed that the contacts mediated by the D2 small domain are necessary for the stability of the ClpB ring. Removal of the D2 small domain completely inhibits the formation of ClpB oligomers in the absence or in the presence of nucleotides [5]. Deletions within the coiled-coil domain also destabilize the oligomers of ClpB [22]. In the high-resolution structure of the ClpB oligomer [14], both the coiled-coil domain and the D2 small domain form the outside “layer” of the ring. The two domains form extensive contacts with the large D1 and D2 domains, respectively, of the neighboring subunit within the ring. The mechanism of stabilization of the ClpB ring by contacts maintained by the coiled-coil and the D2 small domain is consistent with the oligomerization-induced changes in those domains’ stabilities that were detected in the present work. The remaining ClpB domains: the mobile N-terminal domain, the D2 large domain, and the D1 small domain, which serves as a “hinge” for the coiled-coil (see Fig. 1) do not change their stability in the oligomeric ClpB (see Fig. 5). This result indicates that the core of the AAA+ module is a rigid structural unit that can be assembled into an oligomeric ring and “fastened” to its neighbors by contacts maintained by the coiled-coil and the D2 small domain.
Table I.
Parameters of the urea-induced two-state unfolding transitions in single-Trp variants of ClpB (see text and ref. [21])
| ClpB variant | monomer | heptamer | ||
|---|---|---|---|---|
| ΔG0 [kJ/mol] | m [kJ/(mol M)] | ΔG0 [kJ/mol] | m [kJ/(mol M)] | |
| W105 | 9.6 ± 3.2 | 2.7 ± 0.9 | 11.1 ± 3.7 | 3.0 ± 1.0 |
| W462 | 9.3 ± 2.1 | 4.2 ± 0.8 | 6.0 ± 2.1 | 3.2 ± 0.8 |
| W543 | 20.8 ± 1.8 | 9.0 ± 0.8 | 21.8 ± 1.8 | 8.9 ± 0.7 |
| W603 | 26.1 ± 6.8 | 6.4 ± 1.6 | 21.3 ± 7.0 | 5.5 ± 1.7 |
| W812 | 16.2 ± 4.1 | 6.1 ± 1.4 | 5.7 ± 1.2 | 3.4 ± 0.5 |
Acknowledgments
This work was supported by the NIH grant GM58626 and the Kansas Agricultural Experiment Station (contribution 06-234-J). We thank Dr. John Tomich for making the instrumentation in his laboratory available for our studies.
Footnotes
Dedicated to Ann Ginsburg, our mentor in protein stability studies.
References
- 1.Vale RD. J Cell Biol. 2000;150:F13–F19. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogura T, Wilkinson AJ. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 3.Hanson PI, Whiteheart SW. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 4.Pickart CM, Cohen RE. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 5.Barnett ME, Zolkiewska A, Zolkiewski M. J Biol Chem. 2000;275:37565–37571. doi: 10.1074/jbc.M005211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zolkiewski M. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 7.Motohashi K, Watanabe Y, Yohda M, Yoshida M. Proc Natl Acad Sci U S A. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T, Bukau B. Proc Natl Acad Sci U S A. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B. EMBO J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glover JR, Lindquist S. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 11.Queitsch C, Hong SW, Vierling E, Lindquist S. Plant Cell. 2000;12:479–492. doi: 10.1105/tpc.12.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tek V, Zolkiewski M. Protein Sci. 2002;11:1192–1198. doi: 10.1110/ps.4860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akoev V, Gogol EP, Barnett ME, Zolkiewski M. Protein Sci. 2004;13:567–574. doi: 10.1110/ps.03422604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, Tsai FT. Cell. 2003;115:229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 15.Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 16.Barnett ME, Nagy M, Kedzierska S, Zolkiewski M. J Biol Chem. 2005;280:34940–34945. doi: 10.1074/jbc.M505653200. [DOI] [PubMed] [Google Scholar]
- 17.Hess HH, Derr JE. Anal Biochem. 1975;63:607–613. doi: 10.1016/0003-2697(75)90388-7. [DOI] [PubMed] [Google Scholar]
- 18.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 19.Zolkiewski M, Kessel M, Ginsburg A, Maurizi MR. Protein Sci. 1999;8:1899–1903. doi: 10.1110/ps.8.9.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weibezahn J, Schlieker C, Bukau B, Mogk A. J Biol Chem. 2003;278:32608–32617. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 21.Pace CN. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 22.Kedzierska S, Akoev V, Barnett ME, Zolkiewski M. Biochemistry. 2003;42:14242–14248. doi: 10.1021/bi035573d. [DOI] [PMC free article] [PubMed] [Google Scholar]