

Abstract
Previous studies suggest that inflammatory cell adhesion molecules may modulate endothelial cell migration and angiogenesis through unknown mechanisms. Using a combination of in vitro and in vivo approaches, herein we reveal a novel redox sensitive mechanism by which ICAM-1 modulates endothelial GSH that controls VEGF-A induced eNOS activity, endothelial chemotaxis, and angiogenesis. In vivo disk angiogenesis assays showed attenuated VEGF-A mediated angiogenesis in ICAM-1−/− mice. Moreover, VEGF-A dependent chemotaxis, eNOS phosphorylation, and nitric oxide (NO) production were impaired in ICAM-1−/− MAEC compared to WT MAEC. Decreasing intracellular GSH in ICAM-1−/− MAEC to levels observed in WT MAEC with 150 μM buthionine sulfoximine (BSO) restored VEGF-A responses. Conversely, GSH supplementation of WT MAEC with 5 mM glutathione ethyl ester (GEE) mimicked defects observed in ICAM-1−/− cells. Deficient angiogenic responses in ICAM-1−/− cells were associated with increased expression of the lipid phosphatase, PTEN, consistent with antagonism of signaling pathways leading to eNOS activation. PTEN expression was also sensitive to GSH status, decreasing or increasing in proportion to intracellular GSH concentrations. These data suggest a novel role for ICAM-1 in modulating VEGF-A induced angiogenesis and eNOS activity through regulation of PTEN expression via modulation of intracellular GSH status.
Keywords: nitric oxide, glutathione, nitric oxide synthase, adhesion molecules, endothelial migration
Introduction
Angiogenesis, the formation of new blood vessels from preexisting capillaries, is integral in physiological processes such as wound healing, menstrus, and embryonic development (1). This phenomenon also contributes to the pathogenesis of a variety of diseases including atherosclerosis, diabetes, cancer, rheumatoid arthritis, and a host of other chronic inflammatory disorders (2; 3). The mechanisms regulating pathological angiogenesis remain unclear however, and their elucidation will provide key insights for therapeutic targeting of these diseases.
Most, if not all, of the pathologies listed above involve both inflammation and angiogenesis. However, the mechanisms of cross-talk between inflammatory and angiogenic pathways are poorly understood. Interestingly, recent evidence implicates inflammatory adhesion molecules of the immunoglobulin superfamily in the regulation of angiogenesis. DeLisser et al demonstrated that antibodies against PECAM-1 inhibited vascular growth into subcutaneous bFGF treated gels implanted in mice (4). Moreover, Huang and colleagues demonstrated impaired vascular growth into matrigel plugs implanted in ICAM-2 null mice along with attenuated tube formation by cardiac endothelial cells isolated from these mice (5). ICAM-1 is a key adhesion molecule implicated in the development of inflammatory vascular disease, and evidence suggests it may also play an important role in angiogenesis as well. Radisavljevic et al have reported that ICAM-1 immunoneutralization attenuates vascular endothelial growth factor A (VEGF-A) mediated chemotaxis of brain endothelial cells (6). Moreover, we and others have shown that VEGF-A stimulation of endothelial cells significantly increases ICAM-1 expression suggesting that this molecule plays an important role in modulating angiogenic activity (7; 8). However, little is known about the molecular mechanisms through which ICAM-1 or other inflammatory adhesion molecules influence the angiogenic process.
VEGF-A is a pluripotent vasoactive cytokine that stimulates endothelial cell proliferation and chemotaxis, and increases endothelial solute permeability (9–13). The chemotactic effects of VEGF-A are mediated through binding of VEGF receptor 2 (VEGFR2) and stimulation of a signaling pathway involving phosphatidyl inositol-3-kinase (PI3K), Akt, and activation of eNOS and nitric oxide (NO) formation (14; 15). Moreover, VEGF-A also stimulates inflammatory responses as indicated by increased leukocyte rolling and adhesion in postcapillary skin venules of VEGF-A transgenic mice and VEGF-A stimulated endothelium (8; 16). Thus, the signaling through which VEGF-A stimulates vascular growth and inflammation could very well overlap.
Recent work from our laboratory has revealed a novel role of ICAM-1 in regulating both endothelial redox status and basal cell motility (17; 18). Considering these findings, along with previous reports suggesting that VEGF-A may exert its pathological effects via inflammatory pathways, we examined the molecular mechanisms by which ICAM-1 regulates angiogenesis and endothelial cell chemotaxis. Here we present data identifying a novel pathway through which ICAM-1 governs VEGF-A mediated eNOS activity and endothelial chemotaxis that is dependent on intracellular GSH. These effects are concomitant with redox dependent up regulation of PTEN, a tumor suppressor and endogenous negative regulator of PI3K signaling that has been shown to play an important role in VEGF-A mediated endothelial function. These data provide unique insight into the relationship between inflammation and angiogenesis highlighting a novel means for endothelial regulation of these processes.
Methods
Animals and Reagents
C57BL/6J and C57BL/6J-ICAM-1tm1Bay mice were bred and housed at the Association for Assessment and Accreditation of Laboratory Animal Care, International-accredited LSUHSC-Shreveport animal resource facility and maintained according to the National Research Council’s Guide for Care and Use of Laboratory Animals. Disks, membranes, and cement for in vivo angiogenesis disk assays were purchased from Millipore (Bedford, MA). Six-well tissue culture inserts with 8 μm pores were purchased from BD Falcon (Franklin Lakes, NJ). VEGFR2 antibody, PTEN antibody, and phospho-eNOS Ser1177 antibodies were purchased from Cell Signaling Technology (Beverly, MA), and total eNOS antibody was purchased from BD Transduction Labs (San Diego, CA). Mouse recombinant VEGF164 and okadaic acid were purchased from Calbiochem (San Diego, CA), and M199, MCDB 131, PEI, GEE, BSO, PMSF, leupeptin, and aprotinin were purchased from Sigma (St. Louis, MO). Protogel, 4x resolving buffer, 4x stacking buffer, and 10x tris/glycine/SDS were purchased from National Diagnostics (Atlanta, GA). PVDF membrane was purchased from BioRad (Hercules, CA). The pVitro2GFP dual expression vector was purchased from InvivoGen (San Diego, CA), and PTEN cDNA was a kind gift from Dr. Charles Sawyers at UCLA.
Endothelial Cell Isolation and Culture
WT or ICAM-1−/− mice were anesthetized with an IP injection of ketamine (100 mg/kg) and xylazine (8 mg/kg) and aortic endothelial cells were isolated and cultured as previously described (19). Briefly, aortas from WT and ICAM-1−/− mice were excised and collagenase treated to liberate endothelial cells. Cells were treated with FITC tagged BS-1 lectin and sorted. FITC positive cells were plated on 0.2% gelatin in MCDB 131 with 10% FBS, 2 U/mL heparin, 0.1 mg/mL endothelial mitogen (Biomedical Technologies Inc., Stoughton, MA), 0.15% sodium bicarbonate, and antibiotic / antimycotic. Cells used for experiments were between passages 6 and 12.
In Vivo Millipore Disk Angiogenesis Assay
0.45μm nitrocellulose disk filters were glued over the top and bottom of a Millipore disk. Disks were injected with PBS alone or PBS plus 150 ng/mL VEGF. WT (n=6) or ICAM-1−/− (n=6) mice were anesthetized by IP injection with ketamine (100 mg/kg) and xylazine (8 mg/kg) and disks were implanted subcutaneously over the abdomen. After 7 days, the mice were sacrificed and the disks were removed. Pictures were taken of the vasculature in the skin overlying the disk.
Transwell Assay of VEGF Chemotaxis
Six well tissue culture inserts with 8 μm pores were coated with 0.2% gelatin overnight, and WT or ICAM-1−/− MAEC that had been serum starved for at least 12 hours were seeded at 400,000 cells per insert. The cells were allowed to sit for 2 hours, and then mouse VEGF164 at 50 ng/mL was added to the abluminal chamber. After allowing the cells to chemotaxis to the abluminal side for 6 hrs, the inserts were fixed in ice cold 100% methanol for 20 min. The cells were washed in PBS, and the top of each insert was cleared of cells with a PBS soaked cotton swab. Cells on the bottom of each insert were then stained with hematoxylin and washed 3 times in PBS. Cells from five random 20x fields from each insert were counted.
Western Blot Analysis of eNOS Phosphorylation, VEGFR2, and PTEN Expression
Western blots were performed as described previously (17). For eNOS Ser1177 phosphorylation, untreated and 150 μM BSO treated ICAM-1−/− MAEC or untreated and 5 mM GEE treated WT MAEC were stimulated with 50 ng/mL VEGF for 0, 5, 15, and 30 min. 20 μg of total protein per sample were separated on SDS polyacrylamide gels. BSO and GEE treatments were performed overnight. In a separate set of experiments, WT cells were pretreated with 1 μg/ml actinomycin D for 1 hour and then co-incubated with GEE for 12 hours. For PTEN westerns, protein lysates were separated on 10% gels and protein lysates for VEGFR2 or eNOS on 6% gels. Protein was transferred to PVDF overnight at 30 V, blocked in 5% milk TBST at RT for 3 hrs, and then incubated with rabbit-anti-eNOS Ser1177 at 1:1000, rabbit-anti-PTEN at 1:1000, or rabbit-anti-VEGFR2 at 1:1000 in 5% BSA TBST overnight at 4°C. Membranes were then incubated with HRP-linked goat-anti-rabbit secondary at 1:2000 in 5% BSA TBST at room temperature for 3 hrs, incubated with ECL reagents, and then exposed to film. Membranes used to detect eNOS phosphorylation were then stripped and incubated with mouse-anti-eNOS at 1:4000 in 5% BSA TBST at 4°C overnight, incubated with HRP linked goat anti-mouse secondary at 1:2000 in 5% BSA TBST at room temperature for 3 hrs, incubated with ECL reagents, and then exposed to film. Densitometric analysis was performed using Scion imaging software from the NIH.
Measurement of Total Glutathione
Total GSH levels were measured as we have previously reported (18). Endothelium were seeded into 6-well tissue culture plates and incubated 16hr in MCDB 131 with 0, 1, 5, or 10 mM GEE. Cells were washed twice with ice cold PBS with 10 μM DTPA and lysed in 100 μL PBS with 10 μM DTPA and 0.1% Triton X 100. Lysates were centrifuged at 15,000 rpm at 4°C for 15 minutes and supernatant was transferred to a fresh tube. Protein was quantitated using the BioRad Dc Protein Assay. Protein was incubated in PBS with 10 μM DTPA, 1 mM DNTB, 178 μg/mL NADPH, and 9.5x10−6 U/mL Glutathione Reductase for 2.5 minutes. The average change in absorbance at 412 nm over time was used to calculate GSH concentration, which was then normalized to protein concentration.
DAF-FM Fluorescence Measurements
WT and ICAM-1−/− MAEC were seeded into 12 well plates and incubated overnight in serum free MCDB 131 with 5 mM GEE and 150 μM BSO, respectively. The following day, cells were treated with 5 μM DAF-FM for 30 minutes, washed 3 times in PBS, and then treated with 50 ng/mL VEGF in serum and phenol red free M199. DAF-FM fluorescence was measured over time using a TECAN GENios Plus fluorescence plate reader with FITC excitation and emission wavelengths.
PTEN Overexpression and mRNA Measurement
WT human PTEN cDNA was excised from an adenoviral construct kindly provided by Dr. Charles Sawyers, cloned in the pVitro2GFP dual expression vector, and verified by sequencing. For transfections, the PTEN/GFP dual expression vector or the empty vector expressing GFP were mixed with 0.45% polyethylinimine (PEI) at a charge ratio of 1:4, respectively, and allowed to complex at room temperature for 30 min. The complex was then mixed with serum free media and placed on the cells for 2–3 hrs. The cells were then washed and incubated with complete media for 48 hrs. After 48 hrs, the cells were lysed for western blot analysis as described above.
Quantitative real time PCR measurements for mouse PTEN and RPl13a gene expression in response to GEE treatment were performed as we have previously reported with minor modifications (18). 3 micrograms of total RNA was reverse transcribed using Superarray’s RT2 PCR array first strand cDNA synthesis kit (SuperArray, Fredrick, MD). After reverse transcription Real Time PCR was performed on each sample in quadruplicate. Primer sets for mouse PTEN (reference position between nts1155–1176) and RPl13A (reference position between nts 537–556) were purchased from SuperArray (Fredrick, MD). Amplification and detection of PCR amplicons were performed with the iCycler from Bio-Rad and SybR green master mix (SuperArray, Fredrick, MD) with the following PCR reaction profile: 1 cycle of 95°C for 10 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 60s. The delta delta ct method was used for calculating the fold changes in mRNA expression. RPI13a gene expression levels were used as control.
Statistical Analysis
One way analysis of variance with Bonferroni’s post test was used to analyze chemotaxis transwell data. Students T-test was used to analyze differences in DAF-FM signal at various time points. p<0.05 was considered significant. All experiments were performed in triplicate and reproduced on separate occasions.
Results
ICAM-1 Facilitates VEGF-A Mediated Angiogenesis
Recent studies strongly link inflammation and angiogenic responses during disease, yet minimal information exists regarding the importance of specific inflammatory adhesion molecules for angiogenic activity (20–23). Based on previous observations regarding ICAM-1 expression, inflammation, and angiogenic activity, we hypothesized that ICAM-1 regulates VEGF-A mediated angiogenesis. To test this, disk angiogenesis assays were performed in WT and ICAM-1−/− mice. A clear increase in dermal capillary density was observed in response to VEGF-A in WT mice (Figure 1, Panel B), compared to the dermal vasculature overlying PBS treated disks (Figure 1, panel A). However, ICAM-1−/− mice displayed no increase in capillary density in response to VEGF-A (Figure 1, panel D) versus PBS (Figure 1, panel C) demonstrating a role for this adhesion molecule in facilitating VEGF-A mediated angiogenesis. WT and ICAM-1−/− MAEC were cultured to further examine the manner in which ICAM-1 governs VEGF-A angiogenesis. 50 ng/ml VEGF-A was used for chemotaxis assays as we have previously shown this concentration to be optimal in stimulating ICAM-1 expression and is consistent with pathological levels of VEGF-A in diseased tissue (8). Transwell chemotaxis assays using VEGF-A revealed a significant attenuation of the chemotactic response in ICAM-1−/− cells compared to WT cells (Figure 1, panel E). VEGFR2 is the dominant receptor governing VEGF-A induced endothelial chemotaxis, suggesting that changes in receptor expression could account for differential chemotactic responses (14). Importantly, western blot analysis of VEGFR2 showed comparable expression levels between the two cell types, suggesting that differences in VEGF chemotaxis were not due to changes in receptor expression (Figure 1, panel F).
Figure 1. ICAM-1 governs VEGF mediated angiogenesis and endothelial chemotaxis.
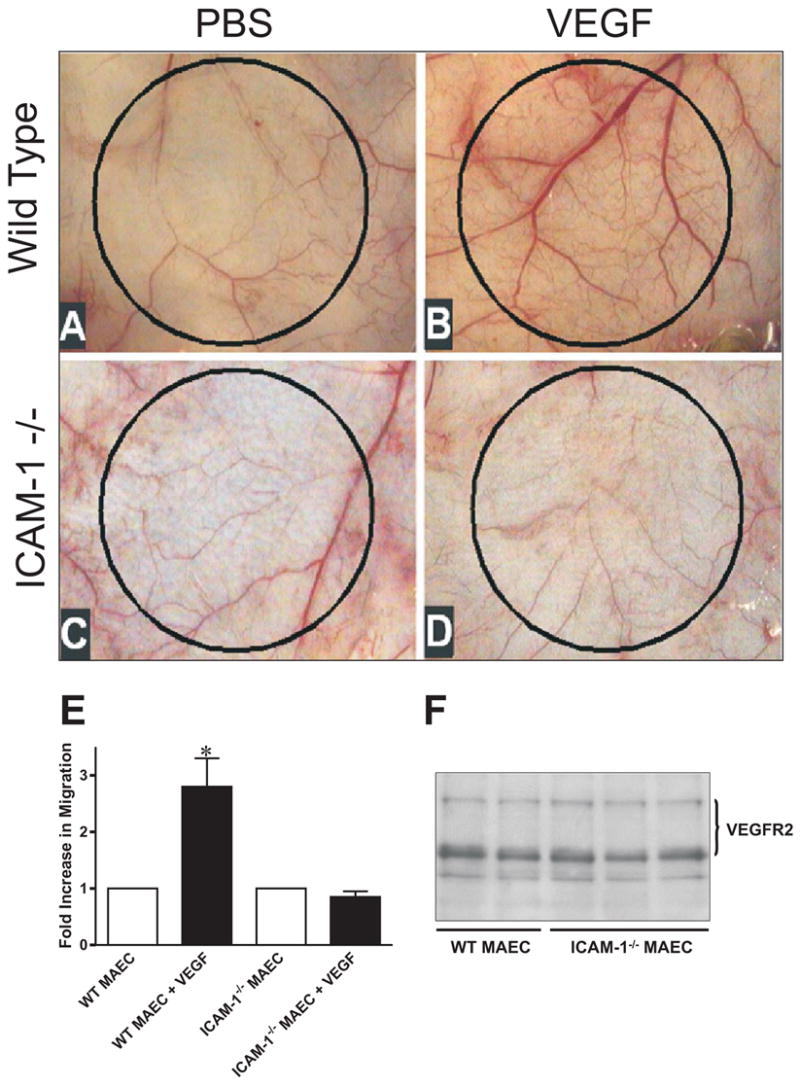
Panel A and B present representative photomicrographs showing the dermal vasculature overlying PBS or VEGF164 150ng/mL treated disks in WT mice respectively. Panel C and D show the vasculature overlying PBS or VEGF treated disks in ICAM-1 knockout mice respectively. Panel E reports a transwell assay of VEGF164 50 ng/mL chemotaxis in WT and ICAM-1−/− MAEC. * p < 0.01. Panel F shows a western blot comparing expression of VEGFR2 in WT and ICAM-1−/− cells.
ICAM-1 Facilitates VEGF-A Mediated eNOS Activity
Work from several laboratories has shown an important role for eNOS derived NO in regulating angiogenesis and VEGF-A dependent signaling (15; 24; 25). To test if altered eNOS activation was contributing to the loss of VEGF-A induced migration in ICAM-1−/− cells, eNOS Ser1177 phosphorylation and nitric oxide production were examined between the two cell types in response to VEGF-A stimulation. In WT endothelial cells, VEGF-A stimulated a maximum increase in eNOS phosphorylation at 5 minutes that returned to baseline by 30 minutes. In ICAM-1−/− cells, phosphorylation at Ser1177 was largely blunted compared to WT at all time points (Figure 2, panel A). NO levels were measured via intracellular DAF-FM fluorescence to confirm decreased NOS activity. Figure 2, panel B shows that VEGF-A dependent NO formation was significantly blunted in ICAM-1−/− cells compared to WT MAEC. Importantly, VEGF dependent as well as basal NO production were substantially attenuated. This observation is consistent with our previous finding that basal NO production is essentially lost in ICAM-1−/−MAEC (17). Indeed, the amount of NO produced in ICAM-1−/− MAEC is on the low range of detection via chemiluminescent techniques, thus no change in basal NO fluorescent detection over time in ICAM-1−/− MAEC is not surprising. Together, these data demonstrate that ICAM-1 expression is required for optimal VEGF-A mediated eNOS phosphorylation and NO production.
Figure 2. ICAM-1 governs VEGF mediated eNOS activity.
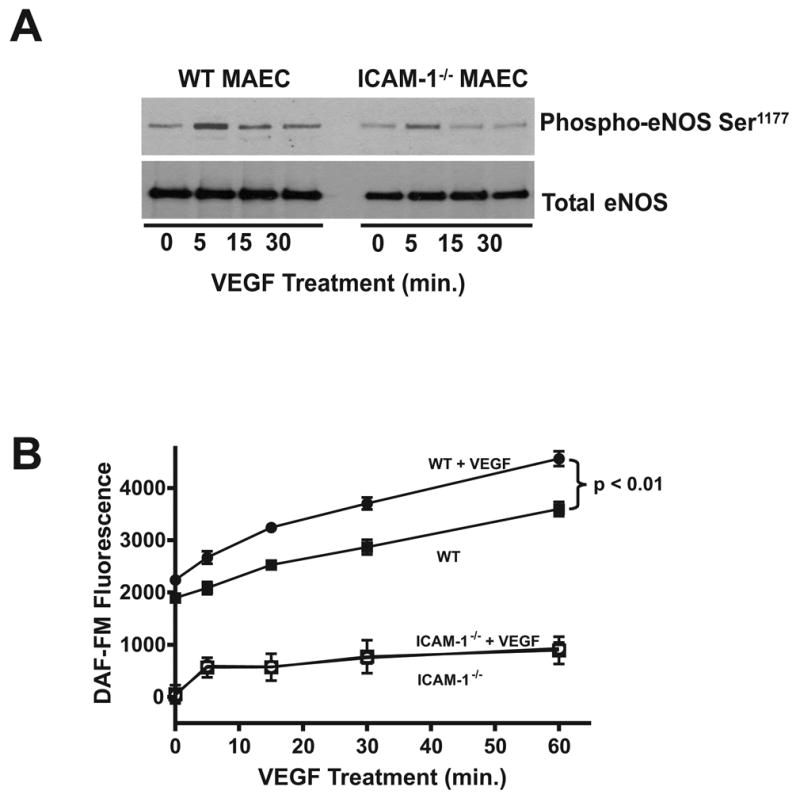
Panel A shows is a western blot comparing eNOS phosphorylation at Ser1177 in response to 50 ng/mL VEGF164 in WT and ICAM-1−/− MAEC over 30 min. Panel B illustrates DAF-FM fluorescence in response to VEGF164 in WT and ICAM-1−/− MAEC over 1 hour. Closed squares indicate WT MAEC, closed circles indicate WT MAEC + 50 ng/mL VEGF164, open squares indicate ICAM-1−/− MAEC, and open circles indicate ICAM-1−/− MAEC + 50 ng/mL VEGF164.
Intracellular Glutathione Modulates VEGF Chemotaxis
We have recently reported that genetic deletion of ICAM-1 results in a γGCL dependent increase in intracellular endothelial GSH levels (18). Therefore, we tested whether increased GSH concentrations in ICAM-1−/− MAEC were responsible for the absence of VEGF-A chemotaxis in these cells. To test this hypothesis, ICAM-1−/− cells were treated with either 50 μM or 150 μM BSO to block GCL activity and decrease GSH levels, and then assayed for chemotactic responses to VEGF-A. We have previously reported that such BSO treatments decrease intracellular GSH levels in ICAM-1−/− MAEC to that of WT cells (18). Transwell chemotaxis assays revealed that treatment with 50 μM BSO significantly increased VEGF-A chemotaxis in ICAM-1−/− cells and that 150 μM BSO treatment further enhanced VEGF-A chemotaxis (Figure 3, panel A). Importantly, BSO treatment of WT cells had no effect on their chemotactic response to VEGF-A (Figure 3, panel B). We next tested if increasing GSH levels in WT cells could attenuate VEGF-A induced chemotaxis. Incubation of WT MAEC with glutathione ethyl ester (GEE), a cell permeable form of GSH, increased intracellular GSH in a dose-dependent manner that was maximal at 10 mM GEE (Figure 3, panel C) resulting in similar intracellular GSH levels found in ICAM-1−/− MAEC (18). Interestingly, 5 or 10 mM GEE treatment of WT MAEC significantly attenuated the chemotactic response to VEGF-A in these cells (Figure 3, panel D). Taken together, these results demonstrate that increased intracellular GSH levels inhibit VEGF-A chemotaxis in endothelial cells.
Figure 3. Glutathione Regulates VEGF Mediated Endothelial Chemotaxis.
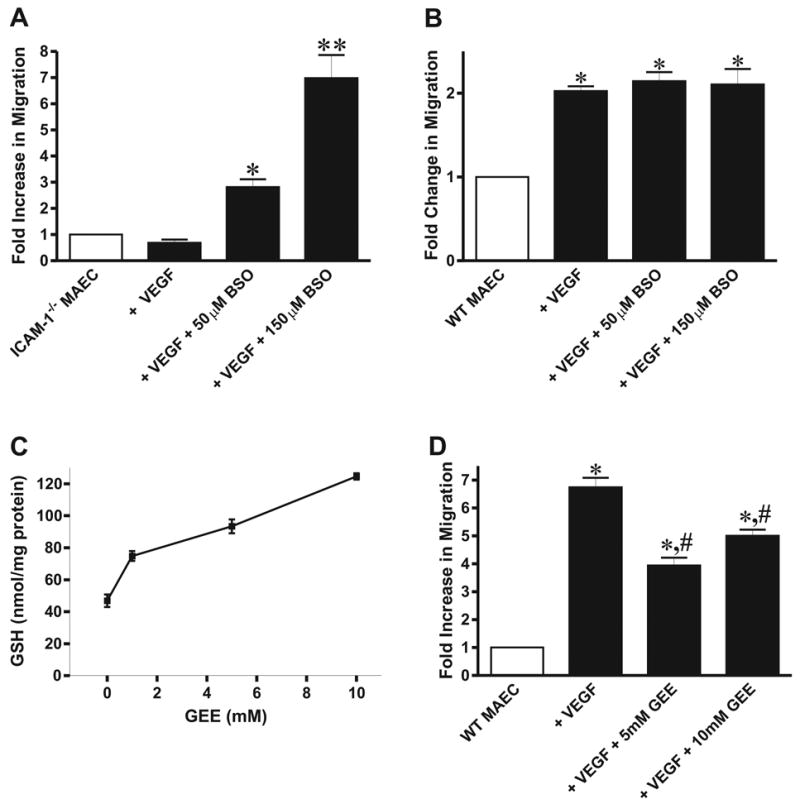
Panel A is a transwell assay of VEGF164 chemotaxis in untreated, 50 μM, and 150 μM BSO treated ICAM-1−/−MAEC. *p<0.05 vs. ICAM-1−/− MAEC + VEGF164. Panel B is a transwell assay of VEGF164 chemotaxis in untreated and BSO treated WT MAEC. *p<0.05 vs. WT MAEC. Panel C illustrates the effect of GEE treatment on total glutathione levels in WT cells, and panel D shows the effect of 5 and 10 mM GEE supplementation on VEGF164 chemotaxis in WT MAEC. *p<0.05 vs. WT MAEC, #p<0.05 vs. WT MAEC + VEGF164.
Intracellular Glutathione Regulates eNOS Activity
Having observed a role for GSH in ICAM-1 regulation of VEGF-A chemotaxis, we next examined whether intracellular GSH levels modulate VEGF-A mediated eNOS activity. Treatment of ICAM-1−/− cells with 150 μM BSO substantially enhanced VEGF-A mediated eNOS phosphorylation (Figure 4, panel A). BSO treatment also significantly increased NO production both constitutively and in response to VEGF-A (Figure 4, panel B). Conversely, WT MAEC were treated with 5 mM GEE and then stimulated with VEGF-A for 0, 5, 15, and 30 minutes. Figure 4, panel C shows that in WT cells treated with 5 mM GEE, VEGF-A failed to increase eNOS phosphorylation. Moreover, treatment with 5 mM GEE blunted both constitutive and VEGF-A induced NO production in WT MAEC (Figure 4, panel D). These data demonstrate that increased intracellular GSH levels in ICAM-1−/− MAEC inhibit VEGF-A stimulated eNOS activation and NO production and also reveal that intracellular GSH is a critical regulator of VEGF-A dependent eNOS activity in wild type endothelium.
Figure 4. Endothelial Glutathione Regulates eNOS Activity.
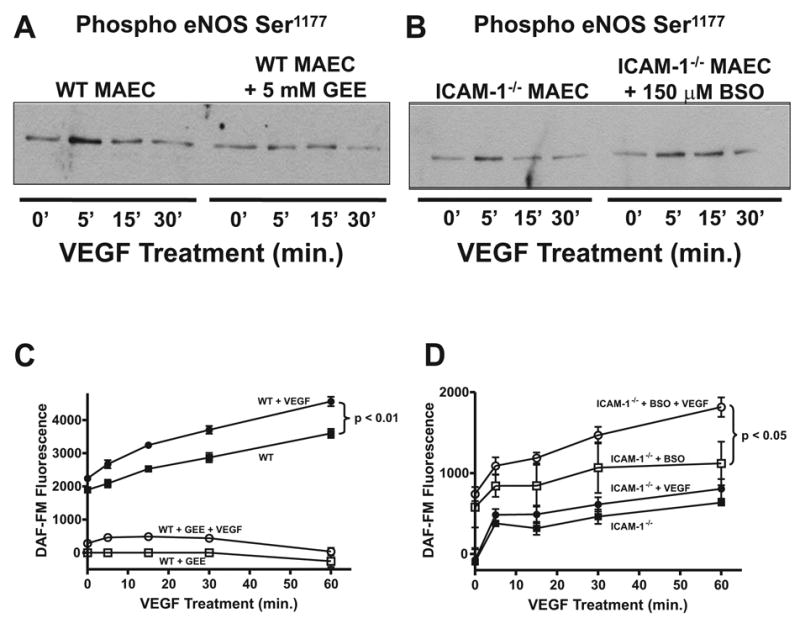
Panel A shows the effect of 150 μM BSO supplementation on VEGF164 mediated eNOS phosphorylation at Ser1177 in ICAM-1−/− MAEC. Panel B illustrates the effect of BSO treatment on DAF-FM fluorescence in response to VEGF164 in ICAM-1−/− MAEC. Closed squares represent ICAM-1−/− MAEC, closed circles represent ICAM-1−/− MAEC + 50 ng/mL VEGF164, open squares represent ICAM-1−/− MAEC + 150 μM BSO, and open circles represent ICAM-1−/− MAEC + 150 μM BSO + 50 ng/mL VEGF164. Panel C shows the effect of 5 mM GEE supplementation on VEGF164 mediated eNOS phosphorylation at Ser1177 in WT MAEC. Panel D illustrates the effect of GEE treatment on DAF-FM fluorescence in response to VEGF164 in WT MAEC over 1 hour. Closed squares represent WT MAEC, closed circles represent WT MAEC + 50 ng/mL VEGF164, open squares represent WT MAEC + 5 mM GEE, and open circles represent WT MAEC + 5 mM GEE + 50 ng/mL VEGF164.
Endothelial Glutathione Regulates PTEN Expression
PTEN is a lipid/protein phosphatase that has been shown to antagonize PI3K activity by dephosphorylating phosphatidylinositol (3,4,5) trisphosphate (PI(3,4,5)P3) to form phosphatidylinositol (4,5) bisphosphate (PI(4,5)P2) which serves as a negative regulator of VEGF-A signaling (26; 27). Importantly, PTEN function is reversibly controlled by thiol oxidation/reduction suggesting a cross-talk with redox cell signaling processes (28). Since PTEN is a redox sensitive negative regulator of PI3K which governs Akt dependent eNOS phosphorylation, we tested the hypothesis that ICAM-1 modulates VEGF-A mediated eNOS activation by influencing PTEN function. Figure 5A shows that ICAM-1−/− MAEC express 3–4 fold higher levels of PTEN compared to WT MAEC. Importantly, the phosphorylation of PTEN was also increased, resulting in similar ratios of phosphorylated to total PTEN between cell types. These data suggest that loss of ICAM-1 expression increased PTEN activity by up regulating protein expression without affecting the fraction of PTEN that is phosphorylated (Figure 5A). To verify these in vitro findings in vivo, PTEN expression was evaluated in lung homogenates (that contain greater numbers of endothelial cells due to high vascular density) from WT and ICAM-1−/− mice. Consistent with in vitro findings, PTEN protein expression was increased in lungs of ICAM-1−/− mice (Figure 5B). Figures 5C and D suggest that the differences in PTEN expression between cell types resulted from differences in cellular GSH concentrations. Decreasing GSH in ICAM-1−/− MAEC with BSO or increasing GSH in WT MAEC with GEE concomitantly decreased or increased PTEN expression, respectively. Furthermore, co-treatment of WT MAEC with GEE and 1μg/ml actinomycin D, an inhibitor of transcription, had no effect on GSH dependent up-regulation of expression, suggesting that redox regulation of PTEN expression involves post-transcriptional mechanisms (Figure 5E). Consistent with this result, real time PCR analysis of PTEN mRNA expression demonstrated that GEE treatment had no significant effect on PTEN message (Figure 5F). Lastly, we examined if PTEN overexpression in WT cells could decrease eNOS phosphorylation as seen in ICAM-1−/− MAEC. Transfection of human PTEN cDNA increased WT MAEC PTEN protein levels 3-fold (Figure 5G) which was associated with a substantial decrease in eNOS Ser1177 phosphorylation consistent with observations made in ICAM-1−/− MAEC.
Figure 5. Endothelial Glutathione Regulates PTEN Expression.
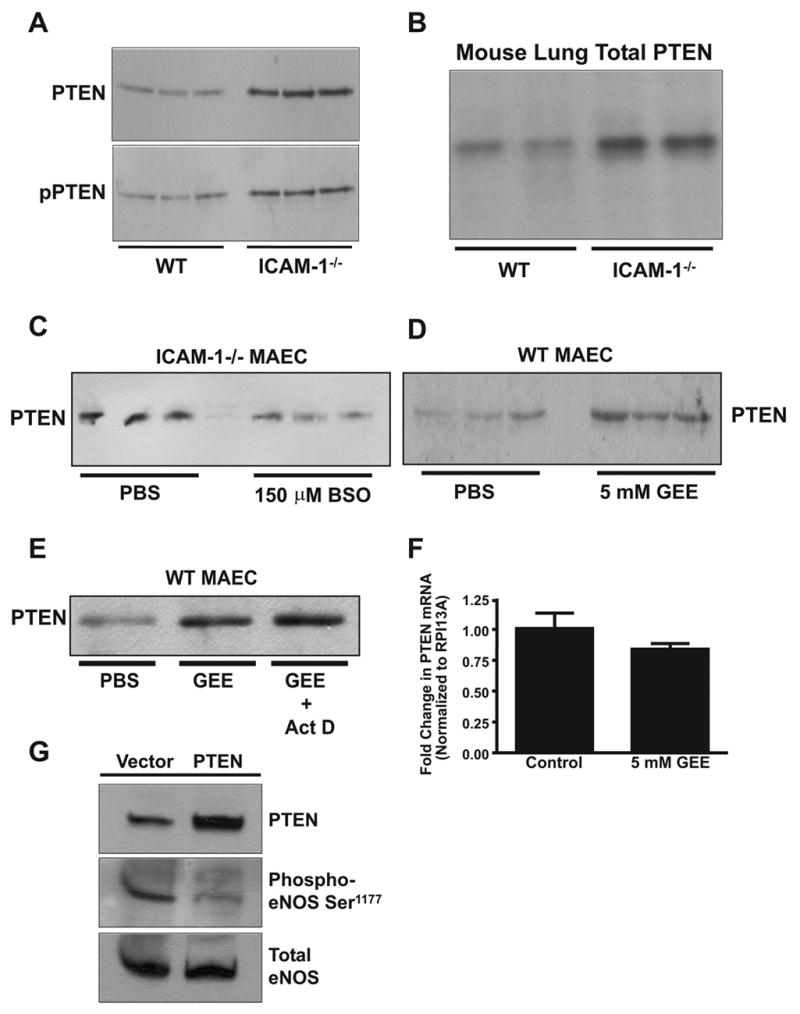
Panel A is a western blot comparing PTEN expression and phosphorylation in WT and ICAM-1−/− MAEC. Panel B is a western blot comparing PTEN expression from the lungs of WT and ICAM-1 knockout mice. Panel C is a western blot comparing PTEN expression in ICAM-1−/− MAEC with and without 150 μM BSO treatment. Panel D is a western blot comparing PTEN expression in WT MAEC with and without 5 mM GEE treatment. Panel E is a western blot comparing PTEN expression in WT MAEC treated with either vehicle, 5 mM GEE, or 5 mM GEE + 1 μg/mL actinomycin D. Panel F shows results of real time PCR analysis comparing PTEN mRNA expression in WT cells with or without overnight treatment with 5 mM GEE. Results are normalized to RPI13A mRNA expression. Panel G shows western blots of PTEN expression, eNOS expression, and eNOS Ser1177 phosphorylation in WT cells after transfection with either a PTEN/GFP dual expression vector or the same vector containing GFP only.
Discussion
Enhanced angiogenesis and increased vascular density are pathological features of chronic diseases such as tumorigenesis, diabetic retinopathy, atherosclerosis, and many others (2). Underscoring the importance of angiogenesis in these disorders is evidence demonstrating that anti-angiogenic therapy helps ameliorate these conditions. Inai et al have shown that a VEGF trap inhibits subcutaneous tumor growth and vascular density, while Joussen et al have shown that anti-VEGF therapy reduces angiogenesis in the ischemic retina (29; 30). The fact that angiogenesis sustains many disease states makes regulation of this phenomenon a key therapeutic goal which requires a detailed understanding of the molecular mechanisms involved.
VEGF-A is a well studied proangiogenic cytokine, shown to mediate angiogenic activity under various physiological circumstances, including embryogenesis, skeletal growth, and ovarian follicular growth (31). However, high levels of VEGF-A have also been observed in many disease states involving pathological angiogenesis (31). The physiological proangiogenic effects of VEGF-A include increased endothelial permeability, proliferation, and chemotaxis, all of which involve the generation of nitric oxide (32; 33). However, higher concentrations of VEGF-A elicit proinflammatory responses, including increases in endothelial permeability and up regulation of endothelial adhesion molecules including ICAM-1(7; 8). Radisavljevic et al have reported that VEGF-A dependent up regulation of ICAM-1 is involved in endothelial cell motility such that antibody blockade of ICAM-1 inhibited VEGF-A mediated chemotaxis independent of immune responses (6). However, the manner in which ICAM-1 modulates VEGF-A dependent chemotaxis remains poorly understood. Here, we reveal a novel mechanism whereby ICAM-1 expression regulates VEGF-A mediated eNOS activity and angiogenesis via regulation of endothelial glutathione levels.
Endothelial redox status has been shown to modulate cellular proliferation and chemotaxis (34–36). A key element controlling endothelial redox status is the GSH cycling between reduced and oxidized forms. We have previously reported that constitutive ICAM-1 expression acts as a repressor of γGCL activity such that loss of ICAM-1 results in an increase in γGCL-C expression, a two-fold increase in total intracellular glutathione, and an increase in the ratio of reduced to oxidized glutathione (18). In this manner, ICAM-1 may regulate VEGF-A angiogenic activity through endothelial GSH redox pathways. Data in this current study suggest that impaired VEGF-A responses in ICAM-1−/− MAEC are due to increased glutathione levels which prevent eNOS activation and NO production.
Many studies have documented a role for changes in kinase and phosphatase activities in regulating cell migration and chemotaxis. Huang and Kontos have shown that overexpression of PTEN, a lipid/protein phosphatase, inhibits VEGF mediated proliferation and chemotaxis in HUVEC, and that it also inhibits sprouting of aortic explants in matrigel (26). Recent reports also demonstrate that PTEN facilitates directional sensing of chemotactic gradients through its localization at the uropod, facilitating cellular polarization and directional migration (37). Moreover, PTEN function is governed in a redox sensitive manner such that hydrogen peroxide reversibly inactivates the enzyme through the formation of a disulfide bond between cys124 and cys71 (38). This disulfide linkage may be reduced by thioredoxin, thus reactivating the enzyme (28). Our data extend these observations and demonstrate that PTEN expression is also GSH sensitive. Supplementation of WT MAEC with 5 mM GEE significantly increased PTEN expression, while treatment of ICAM-1−/− MAEC with 150 μM BSO significantly decreased PTEN expression. Importantly, PTEN expression in the lungs of WT and ICAM-1 knockout mice paralleled in vitro results suggesting that the observed differences were not due to changes in endothelial cell function during culture and also provides in vivo evidence that ICAM-1 regulates expression of this phosphatase. Furthermore, overexpression of PTEN in WT endothelium is sufficient to decrease constitutive eNOS phosphorylation at Ser1177, which is consistent with our observations in cultured ICAM-1−/− MAEC. Together, these data suggest that ICAM-1 dependent redox regulation of PTEN expression could account for the influence of this adhesion molecule on eNOS activity, endothelial chemotaxis, and angiogenesis.
Previous reports from our lab and others have shown that ICAM-1 is important for VEGF-A mediated immune responses, such as increased recruitment of leukocytes that facilitate the angiogenic process (8; 39; 40). However, in this current study we demonstrate that ICAM-1 influences VEGF-A angiogenesis independent of its role in governing immune cell infiltration. The influence of ICAM-1 on endothelial function shown herein suggests a pivotal role for this adhesion molecule in regulating vascular reactivity, as depicted in figure 6. Under conditions where ICAM-1 expression is low, like in areas of laminar flow, increased γGCL, GSH levels, and PTEN expression could keep the endothelium in a more quiescent state. Conversely, under conditions where ICAM-1 expression is high, as is the case in many chronic inflammatory disorders with a pathological angiogenic component, decreased GSH levels, decreased PTEN expression, and increased eNOS activity could all contribute to increased endothelial activity and perpetuate the proangiogenic state.
Figure 6. Regulation of Endothelial Chemotaxis by ICAM-1.
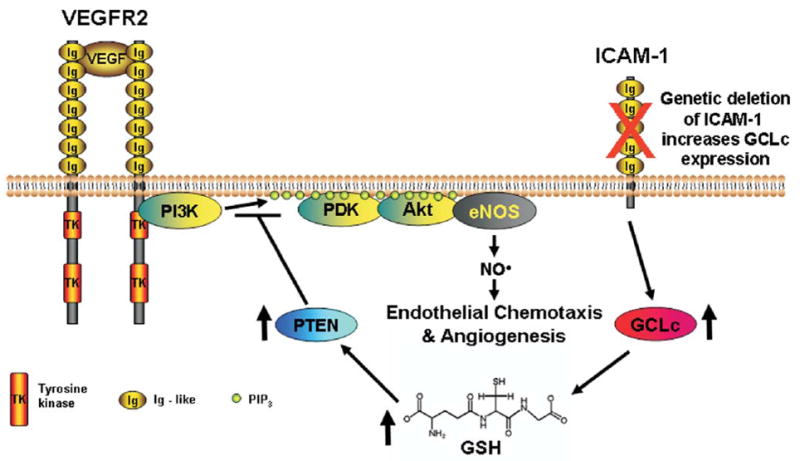
This diagram depicts our current hypothesis on the role of ICAM-1 in VEGF-A stimulated endothelial motility based on present data. ICAM-1 regulates endothelial GSH levels, such that genetic loss of ICAM-1 increases endothelial GSH levels, which in turn decrease eNOS activity and NO bioavailablity. Our data on PTEN expression in ICAM-1−/− cells together with data on GSH regulation of PTEN expression provide a possible mechanistic explanation for the effects of GSH on eNOS activity.
Acknowledgments
This work was supported by NIH grant HL80482 to C.G.K., HL70146 to R.P.P., and HL60849 to D.J.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 4.DeLisser HM, Christofidou-Solomidou M, Strieter RM, Burdick MD, Robinson CS, Wexler RS, Kerr JS, Garlanda C, Merwin JR, Madri JA, Albelda SM. Involvement of endothelial PECAM-1/CD31 in angiogenesis. Am J Pathol. 1997;151:671–677. [PMC free article] [PubMed] [Google Scholar]
- 5.Huang MT, Mason JC, Birdsey GM, Amsellem V, Gerwin N, Haskard DO, Ridley AJ, Randi AM. Endothelial intercellular adhesion molecule (ICAM)-2 regulates angiogenesis. Blood. 2005;106:1636–1643. doi: 10.1182/blood-2004-12-4716. [DOI] [PubMed] [Google Scholar]
- 6.Radisavljevic Z, Avraham H, Avraham S. Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J Biol Chem. 2000;275:20770–20774. doi: 10.1074/jbc.M002448200. [DOI] [PubMed] [Google Scholar]
- 7.Kim I, Moon S-O, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kB activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 8.Goebel S, Huang M, Davis WC, Jennings M, Siahaan TJ, Alexander JS, Kevil CG. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2006;290:G648–654. doi: 10.1152/ajpgi.00466.2005. [DOI] [PubMed] [Google Scholar]
- 9.Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- 10.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 12.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 13.Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986;46:5629–5632. [PubMed] [Google Scholar]
- 14.Zeng H, Dvorak HF, Mukhopadhyay D. Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) peceptor-1 down-modulates VPF/VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent pathways. J Biol Chem. 2001;276:26969–26979. doi: 10.1074/jbc.M103213200. [DOI] [PubMed] [Google Scholar]
- 15.Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477:258–262. doi: 10.1016/s0014-5793(00)01657-4. [DOI] [PubMed] [Google Scholar]
- 16.Detmar M, Brown LF, Schon MP, Elicker BM, Velasco P, Richard L, Fukumura D, Monsky W, Claffey KP, Jain RK. Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J Invest Dermatol. 1998;111:1–6. doi: 10.1046/j.1523-1747.1998.00262.x. [DOI] [PubMed] [Google Scholar]
- 17.Kevil CG, Orr AW, Langston W, Mickett K, Murphy-Ullrich J, Patel RP, Kucik DF, Bullard DC. Intercellular adhesion molecule-1 (ICAM-1) regulates endothelial cell motility through a nitric oxide-dependent pathway. J Biol Chem. 2004;279:19230–19238. doi: 10.1074/jbc.M312025200. [DOI] [PubMed] [Google Scholar]
- 18.Kevil CG, Pruitt H, Kavanagh T, Wilkerson J, Farin F, Moellering D, Darley-Usmar V, Bullard D, Patel R. Regulation of endothelial glutathione by ICAM-1: implications for inflammation. Faseb J. 2004;18:1321–1323. doi: 10.1096/fj.03-1401fje. [DOI] [PubMed] [Google Scholar]
- 19.Kevil CG, Bullard DC. In vitro culture and characterization of gene targeted mouse endothelium. Acta Physiol Scand. 2001;173:151–157. doi: 10.1046/j.1365-201X.2001.00901.x. [DOI] [PubMed] [Google Scholar]
- 20.Mor F, Quintana FJ, Cohen IR. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol. 2004;172:4618–4623. doi: 10.4049/jimmunol.172.7.4618. [DOI] [PubMed] [Google Scholar]
- 21.Bagli E, Xagorari A, Papetropoulos A, Murphy C, Fotsis T. Angiogenesis in inflammation. Autoimmun Rev. 2004;3 (Suppl 1):S26. [PubMed] [Google Scholar]
- 22.Lai WK, Adams DH. Angiogenesis and chronic inflammation; the potential for novel therapeutic approaches in chronic liver disease. J Hepatol. 2005;42:7–11. doi: 10.1016/j.jhep.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Imhof BA, Aurrand-Lions M. Angiogenesis and inflammation face off. Nat Med. 2006;12:171–172. doi: 10.1038/nm0206-171. [DOI] [PubMed] [Google Scholar]
- 24.Amano K, Matsubara H, Iba O, Okigaki M, Fujiyama S, Imada T, Kojima H, Nozawa Y, Kawashima S, Yokoyama M, Iwasaka T. Enhancement of ischemia-induced angiogenesis by eNOS overexpression. Hypertension. 2003;41:156–162. doi: 10.1161/01.hyp.0000053552.86367.12. [DOI] [PubMed] [Google Scholar]
- 25.Lee PC, Salyapongse AN, Bragdon GA, Shears LL, 2nd, Watkins SC, Edington HD, Billiar TR. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol. 1999;277:H1600–1608. doi: 10.1152/ajpheart.1999.277.4.H1600. [DOI] [PubMed] [Google Scholar]
- 26.Huang J, Kontos CD. PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J Biol Chem. 2002;277:10760–10766. doi: 10.1074/jbc.M110219200. [DOI] [PubMed] [Google Scholar]
- 27.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 28.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 29.Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joussen AM, Poulaki V, Tsujikawa A, Qin W, Qaum T, Xu Q, Moromizato Y, Bursell SE, Wiegand SJ, Rudge J, Ioffe E, Yancopoulos GD, Adamis AP. Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol. 2002;160:1683–1693. doi: 10.1016/S0002-9440(10)61115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9 :669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 32.Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol. 1996;270:H411–415. doi: 10.1152/ajpheart.1996.270.1.H411. [DOI] [PubMed] [Google Scholar]
- 33.Wu HM, Huang Q, Yuan Y, Granger HJ. VEGF induces NO-dependent hyperpermeability in coronary venules. Am J Physiol. 1996;271:H2735–2739. doi: 10.1152/ajpheart.1996.271.6.H2735. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz-Gines JA, Lopez-Ongil S, Gonzalez-Rubio M, Gonzalez-Santiago L, Rodriguez-Puyol M, Rodriguez-Puyol D. Reactive oxygen species induce proliferation of bovine aortic endothelial cells. J Cardiovasc Pharmacol. 2000;35:109–113. doi: 10.1097/00005344-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 35.Ushio-Fukai M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 36.Yasuda M, Ohzeki Y, Shimizu S, Naito S, Ohtsuru A, Yamamoto T, Kuroiwa Y. Stimulation of in vitro angiogenesis by hydrogen peroxide and the relation with ETS-1 in endothelial cells. Life Sci. 1999;64:249–258. doi: 10.1016/s0024-3205(98)00560-8. [DOI] [PubMed] [Google Scholar]
- 37.Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J Biol Chem. 2003;278:20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- 38.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasuda M, Shimizu S, Tokuyama S, Watanabe T, Kiuchi Y, Yamamoto T. A novel effect of polymorphonuclear leukocytes in the facilitation of angiogenesis. Life Sci. 2000;66:2113–2121. doi: 10.1016/s0024-3205(00)00537-3. [DOI] [PubMed] [Google Scholar]
- 40.Yasuda M, Shimizu S, Ohhinata K, Naito S, Tokuyama S, Mori Y, Kiuchi Y, Yamamoto T. Differential roles of ICAM-1 and E-selectin in polymorphonuclear leukocyte-induced angiogenesis. Am J Physiol Cell Physiol. 2002;282:C917–925. doi: 10.1152/ajpcell.00223.2001. [DOI] [PubMed] [Google Scholar]