

Abstract
PACT is a stress-modulated, cellular activator of interferon (IFN)-induced double-stranded (ds) RNA-activated protein kinase (PKR) and is an important regulator of PKR-dependent signaling pathways. The research presented here is aimed at understanding the regulation of PACT expression in mammalian cells. PACT is expressed ubiquitously in different cell types at varying abundance. We have characterized the sequence elements in PACT promoter region that are required for its expression. Using deletion analysis of the promoter we have identified the minimal basal promoter of PACT to be within 101 nucleotides upstream of its transcription start site. Further mutational analyses within this region, followed by electrophoretic mobility shift analyses (EMSAs) and chromatin immunoprecipitation (ChiP) analysis have shown that Specificity protein 1 (Sp1) is the major transcription factor responsible for PACT promoter activity.
Keywords: interferon, protein kinase, dsRNA, promoter, stress signaling
1. Introduction
Interferons (IFNs) mediate their antiviral and antiproliferative actions by inducing a number of genes at the transcriptional level (Sen and Ransohoff, 1993; de Veer et al., 2001). PKR is an IFN-induced, dsRNA-activated serine/threonine protein kinase involved in mediating the inhibition of protein synthesis (Meurs et al., 1990; Clemens and Elia, 1997). Among the known cellular substrates of PKR activity are the α subunit of eIF2 (Samuel, 1993) and B56α, the regulatory subunit of protein phosphatase 2A (PP2A) (Xu and Williams, 2000). Phosphorylation of these substrates leads to a global inhibition of protein synthesis (Samuel, 1993), thought to eventually lead to apoptosis. Although the impact of PKR activation has been best studied on antiviral pathways, PKR has also been shown to be involved in several cytokine and dsRNA-induced signaling pathways in the inflammatory response (Kumar et al., 1997; Ramana et al., 2000). PKR has also been known to play a major role in apoptosis in response to stress (Jagus et al., 1999; Williams, 1999), and in growth inhibition in several organisms (Chong et al., 1992; Koromilas et al., 1992; Meurs et al., 1993; Dever et al., 1998). It is present in all cell types and IFN induces the levels of cellular PKR, however the protein remains latent until bound by an activator. In virally infected cells dsRNA serves as the activator of PKR (Katze, 1995). PKR has been shown to bind to dsRNAs as short as 11 bp (Bevilacqua and Cech, 1996), however, a perfectly duplexed stretch of 50 bp is required for its activation (Manche et al., 1992).
The dsRNA activation of PKR has been well-studied and characterized. The dsRNA binding domain (DRBD) of PKR resides at the amino terminus (Green and Mathews, 1992; Patel and Sen, 1992) (amino acids 1-170) and is composed of two copies of a dsRNA binding motif (dsRBM), a sequence motif conserved in many RNA binding proteins (St Johnston et al., 1992). Binding of dsRNA to PKR through these motifs causes a conformational change in the protein that leads to unmasking of the ATP-binding site in the kinase domain and results in autophosphorylation of PKR on several sites (Galabru and Hovanessian, 1987). The domains involved in dsRNA binding are also involved in mediating dimerization of PKR, which is essential for its kinase activity in the presence of dsRNA (Patel et al., 1995; Patel and Sen, 1998b; Tan et al., 1998). Although the same domain mediates both PKR’s dsRNA binding and its dimerization, distinct residues have been identified that contribute to one or both these properties (Patel et al., 1996; Patel and Sen, 1998b).
Studies with wild-type and PKR null mouse embryonic fibroblasts (MEFs) indicate that PKR plays a major role in initiating apoptosis in response to stresses such as tumor necrosis factor-α (TNF-α) and lipopolysaccharide (Der et al., 1997; Balachandran et al., 1998; Gil et al., 1999; Gil and Esteban, 2000). Because dsRNA is not present in cells in the absence of a viral infection, a protein activator is thought to initiate this activation of PKR resulting in the induction of apoptotic pathways. This function is provided by PACT (PKR activating protein) which heterodimerizes with PKR and activates it in the absence of dsRNA (Patel and Sen, 1998a). It has been shown that cellular stress signals activate PKR through PACT, ultimately leading to eIF2α phosphorylation and apoptosis. PACT itself undergoes phosphorylation in response to stress signals, and the phosphorylated PACT becomes increasingly associated with PKR (Ito et al., 1999; Patel et al., 2000).
PACT itself binds dsRNA and contains three conserved dsRBMs (Patel and Sen, 1998a; Huang et al., 2002). PACT has the ability to activate PKR in the absence of dsRNA and dsRBMs 1 and 2 (M1 and M2) of PACT are essential for its ability to bind to PKR. The third dsRBM (M3) is not necessary for PACT’s ability to bind PKR, but is shown to be essential in activating PKR’s kinase activity once the PACT/PKR heterodimer has formed (Peters et al., 2001; Huang et al., 2002). Thus, PACT is a stress-modulated, cellular activator of PKR and is an important regulator of PKR dependent signaling pathways. The research presented here is aimed at understanding the regulation of PACT expression. We have analyzed the PACT promoter using mutational analysis and attempted to reveal the factor(s) involved in the transcriptional regulation of PACT. Our results demonstrate that two Sp1 binding sites are essential for PACT expression from its TATA-less promoter.
2. Materials and Methods
2.1: Plasmids
The transcription start site of PACT was previously mapped using primer extension (unpublished observations). PCR was used to amplify a region of the BAC cosmid clone RP11-65L3 (Accession no. AC009948, BacPac Resources Center) that included a region of PACT corresponding to nucleotides -2002 to +45 relative to its transcription start site. The primers used for amplification were:
HPACT-2002: 5′-CGCTCGSGCGTAAAAATATGACCAATGTC-3′, and
HPACT-DS: 5′-CGCTCGAGCGCTGGTCCCCGGGAGGAGC-3′.
The PCR amplified fragment was cloned into the pGEMT-Easy plasmid (Promega), and the promoter was subsequently cloned into the pGL2Basic plasmid (Promega) upstream of a firefly luciferase reporter gene.
2.2: Deletion Mutants
Deletion mutants from the 5′ end of the PACT promoter were created using PCR amplification corresponding to the following locations relative to the transcription start site: -2002, -821, -444, -309, -200, -101, -54. The six deletion mutants were then sub-cloned into pGL2Basic to allow for their promoter activity to be tested in a cell culture system using HeLa cells.
2.3: Site-directed mutagenesis
The Gene-Editor kit (Promega) was used to create site-directed mutants of the GC boxes within the PACT promoter at the following locations:
site 1A; -63CCACCC-58 to -63GAATCC-58,
site 1; -69CCGCCC-64 to -69AAGCTT-64,
site 1B; -100GGGGCG-95 to -100GAATCC-95,
site 2; -144GGGCGG-139 to -144GAATCC-139,
site 3; -166GGGCGG-161 to -166GATATC-161,
site 4; -177GGGCGG-172 to -177CATATG-172,
site 5; -187GGGCGG-182 to -187TCTAGA-182,
and site 6; -240GGGCGG-235 to -240AGATCT-235,
These site-directed mutants were also sub-cloned into pGL2Basic. pRL-Null is a plasmid containing a constitutively active promoter linked to Renilla Luciferase. Double mutants were also created using the Gene-Editor kit (Promega). The site 1 mutant-BSIIKS+ construct served as the template and each of the second mutations were subsequently added to it.
2.4: Transfections
HeLa cells maintained in Dulbecco’s Modified Eagle’s Medium supplemented with penicillin/streptomycin were used for transfection. 1μg of DNA per well of a six-well plate was transfected using 5μl Lipofectamine (Invitrogen). Cotransfection with 25ng pRL-Null per well was used to normalize the transfection efficiency. Twenty-four hours post-transfection cells were harvested and the luciferase activity in each cell lysate was measured using a luminometer.
2.5: Electrophoretic Mobility Shift Assays (EMSAs)
The gel shift analyses were performed using following 24 bp oligonucleotides.
site 1: 5′-ACCAAGGCTCCGCCCCCACCCTGC-3′, and
site 2: 5′-AAGGGACAGGGGCGGAGTTGGGCC-3′.
We also used the following mutant 24 bp oligonucleotides identical to the mutations that we previously made to test for the lack of competition for specific binding when used as cold competitors.
site 1: 5′-ACCAAGGCTAAGCTTCCACCCTGC-3′, and
site 2: 5′-AAGGGACAGGAATTCAGTTGGGCC-3′.
As a positive control, we used a consensus oligonucleotide for Sp1, and as a negative control we used a consensus oligonucleotide to an irrelevant transcription factor, TFIID from Promega. The oligonucleotides were annealed and radiolabeled with γ32P-ATP. Promega’s Gel-Shift Mobility kit was used to carry out the reactions. Each reaction included 4.8 μg of HeLa nuclear extracts, 25 ng radiolabeled oligonucleotides (∼150,000 cpm), and binding buffer (4% glycerol, 1mM MgCl2, 0.5mM EDTA, 0.5mM DTT, 50mM NaCl, 10mM Tris-HCl [pH 7.5], poly[dI-dC]•poly[dI-dC]). Competition assays were performed using a 50 fold molar excess of unlabeled oligonucleotides. Supershift assays were performed using purified Sp1 antibody (Santa Cruz, sc-59x). All reactions were assayed by electrophoresis on a 5% acrylamide gel and subsequent autoradiography.
2.6: Chromatin Immunoprecipitation (ChiP) Assay
ChiP analysis was performed using the ChiP Assay kit (Upstate cell signaling solutions, 17-295) according to the manufacturer′s instructions. Briefly, cellular proteins and DNA were cross-linked by adding formaldehyde to the growth media to a final concentration of 1% for 10 minutes. Cells were harvested in ice-cold phosphate-buffered saline and lysed with SDS lysis buffer (50 mM Tris pH 8.1, 10 mM EDTA, and 1% SDS). Lysates were sonicated utilizing a sonicator to shear DNA to an average size of 600 bp and pre-cleared with salmon sperm DNA/protein A-agarose. Lysates were then tumbled overnight at 4 °C with salmon sperm DNA/protein A-agarose with anti-Sp1 (Santa Cruz PEP 2, sc-59X) antibody. Complexes were precipitated and serially washed three times each with low salt (20 mM Tris pH 8.1, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, and 1% Triton X-100); high salt (20 mM Tris pH 8.1, 500 mM NaCl, 2 mM EDTA, 01% SDS, and 1% Triton X-100); LiCl wash (10 mM Tris pH 8.1, 250 mM LiCl, 1 mM EDTA, 1% deoxycholate, and 1% Nonidet P-40); and TE buffer (20 mM Tris pH 8.1 and 2 mM EDTA). Washed complexes were eluted with freshly prepared elution buffer (1% SDS and 100 mM NaHCO3), and the Na+ concentration was adjusted to 200 mM by adding NaCl followed by incubation at 65°C for 4 h to reverse protein/DNA cross-links. DNA was purified utilizing a PCR purification kit (Qiagen). Purified DNA was then amplified across the PACT promoter region utilizing the primers 5′-CTGCCCCCTCGCTGGAGCAACG-3′ and 5′-CTTTGTCCTTAGCAGGAGGCCG-3′. 275 bp long PCR products were then resolved on a 1.2% agarose gel.
3. Results
3.1: Minimal promoter for PACT
In order to characterize PACT expression in different tissues, we performed northern blot analysis with multiple tissue northern (MTN) blots (Clontech). This analysis revealed that PACT mRNA is expressed ubiquitously, with highest levels in placenta and testis (Figure 1). The transcription start site for PACT promoter was mapped by primer extension (data not shown) and PCR was used to amplify a region of PACT -2002 nucleotides upstream of the transcription start site from a genomic cosmid clone. This region could drive a reporter gene expression in multiple human cancer cell lines including HeLa, HT1080, SW480, and murine NIH3T3 cells in an orientation-specific manner. The same region in reverse orientation showed drastically reduced expression of the reporter gene indicating the presence of promoter sequences within this region (data not shown). Upon analyzing PACT’s promoter region we observed that the promoter does not contain a TATA box. The only obvious regulatory elements recognized were GC boxes, which are known binding sites for the transcription factor Sp1 (Figure 2). PACT promoter deletion mutants were created by PCR amplification and analyzed for their activity in HeLa cells. Figure 3 shows that the -54 PACT-pGL2B construct did not yield any more luciferase activity than pGL2B vector alone. The -101 PACT-pGL2B construct yielded 50-fold more luciferase activity than the -54 PACT-pGL2B construct or pGL2B alone. Sequence analysis reveals no GC boxes within the first 54 bp of PACT’s transcription start site. This indicates that PACT’s minimal, basal promoter lies within 101 bp upstream of its transcription start site, and that GC boxes may play a role in regulating the promoter. The promoter activity was increased about 2-fold compared to -101 PACT-pGL2B when the constructs included either 200 bp or 309 bp sequences. The reason for this increase is unclear at present. The -414 PACT-pGL2B construct showed about 5-fold enhanced activity as compared to the -101 PACT-pGL2B construct. This significant increase in promoter activity may be due to the presence of a CCAAT box at position -404 to -400. Further mutational analysis will be required to understand any functional role and significance of this CCAAT box. Inclusion of sequences upstream of -414 did not show much effect on the promoter activity. These results indicate that sequences within the first 101 bp may contain sequence elements that are required for transcription of PACT promoter.
Figure 1.
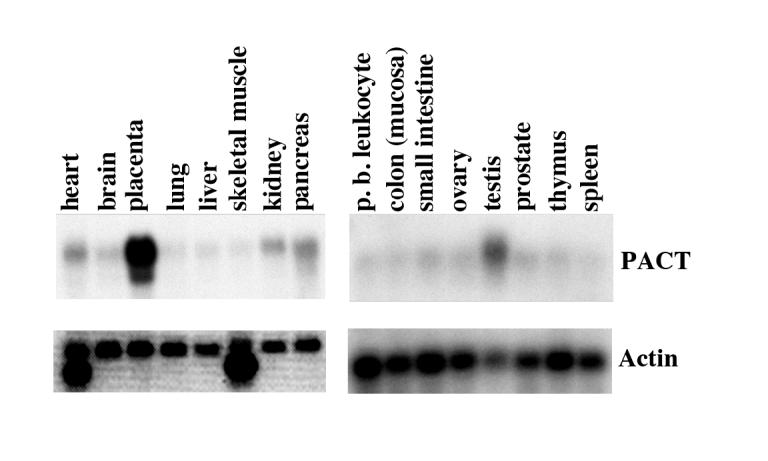
Multiple Tissue Northern blot. Human multiple tissue northern blots I and II (Clontech) were hybridized to a random-primer labeled PACT cDNA probe. Each lane contains 2 μg of poly(A)+ RNA isolated from the indicated tissues. The same blot was stripped and re-probed with a cDNA probe for actin as shown in the bottom panels to ascertain equal loading in all lanes.
Figure 2.

PACT promoter sequence indicating the location of GC boxes within the promoter. PACT promoter sequence (Accession no. AC009948) to the position -821 is shown. The GC boxes are shown in bold underlined print as site 1-6. The transcription start site mapped using primer extension is indicated by an arrow. The six GC boxes/sites were mutated using Promega’s Gene Editor kit.
Figure 3.

PACT’s minimal promoter resides within the first 101 bp upstream of the transcription start site. PACT promoter deletion mutants in pGL2B were transiently transfected into HeLa cells as described in Materials and Methods. Values given are relative Firefly luciferase units standardized to relative Renilla luciferase units obtained by co-transfection of pRL-null plasmid for normalization of transfection efficiency. Each transfection was performed in triplicate and repeated at least once. Values shown are an average of both experiments. The error bars indicate standard error.
3.2: GC boxes are necessary for transcription from PACT promoter
We then tested the effect of mutating each of the six GC boxes individually within the -414 PACT-pGL2B construct. None of these mutations significantly affected the promoter activity compared to wild-type -414 PACT-pGL2B (Figure 4A). We reasoned that this could be due to the functional redundancy offered by the multiple GC boxes within this region. In order to test this idea, we examined the effect of mutating a single GC box within a region in which there were no other GC boxes that could confer redundancy. The -101 PACT-pGL2B construct does not contain GC boxes 2 through 6, however it does contain site 1 (Figure 2). We therefore tested the effect of mutating this site within the -101 PACT-pGL2B construct. A mutation of site 1 abolished PACT’s promoter activity when compared to wild-type -101 PACT-pGL2B (Figure 4B). These data suggest that when site 1 is mutated in the longer -414 PACT-pGL2B construct, one or more of the other GC boxes may be able to replace its function. However, when site 1 is mutated within a context that does not include the other GC boxes, this mutation abolishes PACT’s promoter activity, thereby indicating that site 1 plays an important role in mediating transcription from this promoter. To test the effect of mutating more than one GC box within the same construct we created double mutations. To accomplish this, we created the double mutations within the -309 PACT-pGL2B construct which contains 6 identifiable GC boxes (Figure 1, and 4 A). The double mutation of sites 1 and 2 completely abolished PACT’s promoter activity compared to wild-type - 309 PACT-pGL2B (Figure 4C). These data suggest that sites 1 and 2 contribute significantly to PACT’s promoter activity in HeLa cells. In addition, when one of these two sites is mutated (Fig. 4 A), the other site could drive the expression from this promoter. However, for full activity of the promoter, either site 1 or site 2 is essential, thus when both sites were mutated, the promoter lost its activity. When sites 3-6 were mutated one at a time in combination with site1, there was no deleterious effect on the promoter activity indicating that in the absence of site 1, site 2 can fully substitute for its activity. Thus, for full activity of PACT promoter only one of either site 1 or site 2 GC boxes is sufficient.
Figure 4.
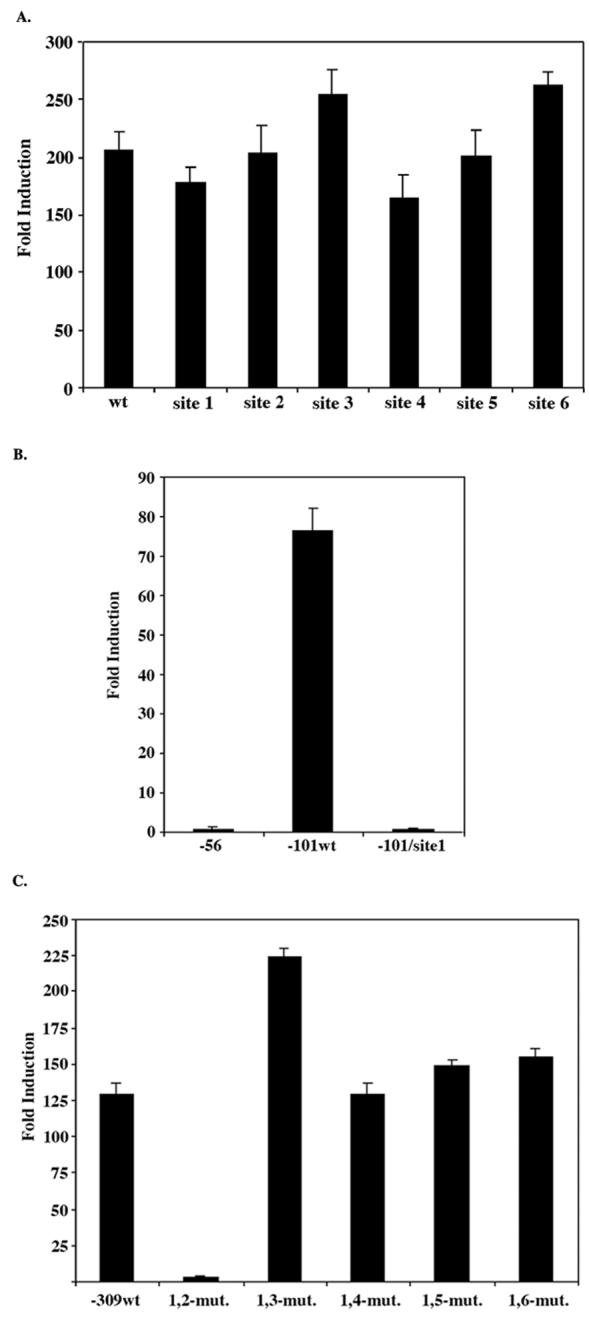
Effect of GC box mutations on PACT promoter activity. All mutant constructs were transiently transfected in HeLa cells as described in Materials and Methods. Values given are relative Firefly luciferase units standardized to relative Renilla luciferase units obtained by cotransfection of pRL-null plasmid for normalization of transfection efficiency. Each transfection was performed in triplicate and repeated at least once. Values shown are an average of both experiments. The error bars indicate standard error. (A) Individual mutations at sites 1-6 created within the -414PACT-pGL2B construct. (B) Mutation of site 1created within the -101 PACT-pGL2B construct. (C) Double mutations created within the -309PACT-pGL2B construct.
3.3: Sp1 transcription factor binds to GC boxes in PACT promoter
To further investigate site 1 and site 2′s ability to bind the transcription factor Sp1 we performed electrophoretic mobility shift assays (EMSAs) using HeLa nuclear extracts. We observed a mobility shift using radiolabeled oligonucleotides corresponding to site 1 and site 2 of the PACT promoter (Figure 5A, lanes 1 and 6). Cold, unlabeled Sp1 consensus oligonucleotide efficiently competed for binding with the radiolabeled site 1 and site 2 oligonucleotides (Figure 5A, lanes 2 and 7). Cold, unlabeled site 1 and site 2 oligonucleotides also efficiently competed for binding (Figure 5A, lanes 3 and 8). However, a cold unlabeled TFIID consensus oligonucleotide did not compete for binding (Figure 5A, lanes 5 and 10). These data suggest that Sp1 binds specifically to site 1 and site 2 of PACT’s promoter. Furthermore, labeled oligonucleotides corresponding to the mutations created at site 1 and site 2 were not able to compete with wild-type site 1 and 2 oligonucleotides for Sp1 binding (Figure 5A, lanes 4 and 9), indicating that the mutations we created did abolish the ability for those sites to bind Sp1. This is significant in that it suggests that the effect of these mutations that we observe in HeLa cells is due to an inability to bind Sp1. The addition of an Sp1 antibody to the reaction resulted in a supershift of one of the radiolabeled bands (Figure 5B). This further indicates that Sp1 is the factor binding to the GC boxes at site 1 and site 2 of the PACT promoter, and that Sp1 is the transcription factor necessary for PACT’s basal transcriptional activity.
Figure 5.
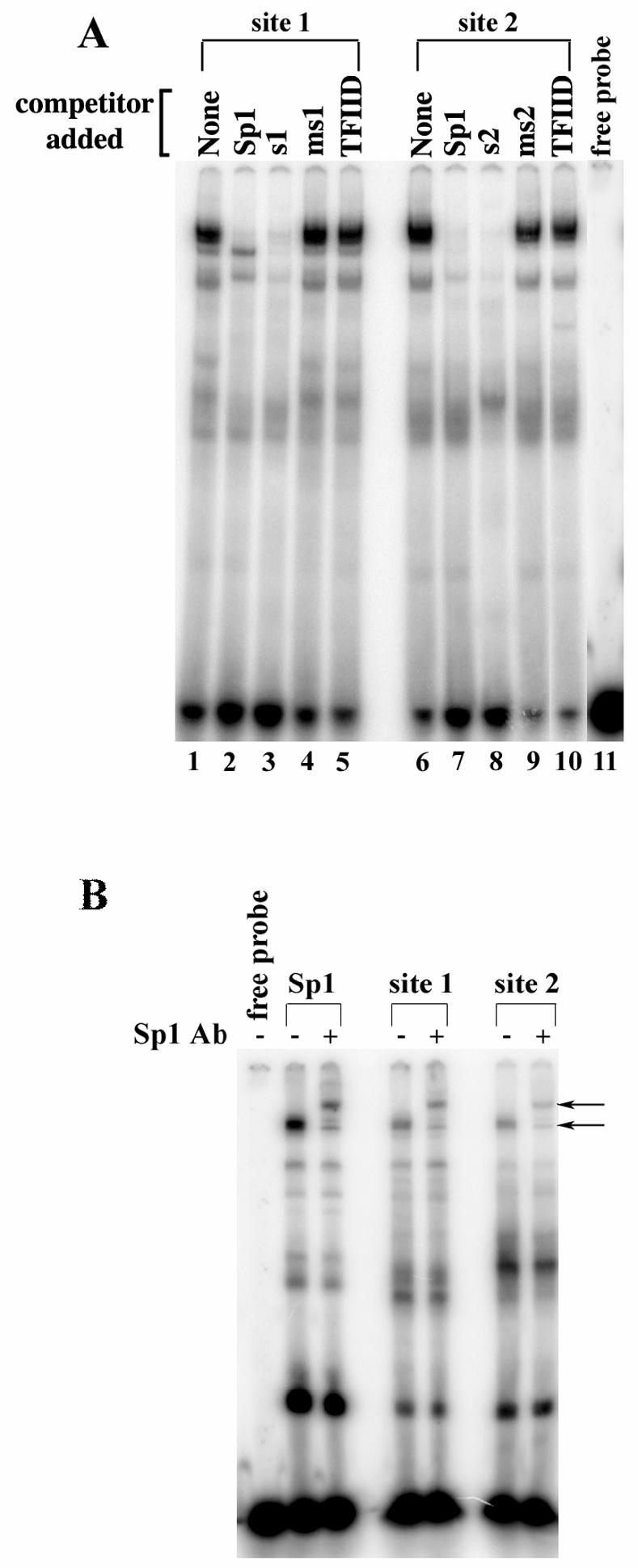
Electrophoretic Mobility Shift Assays (EMSAs) showing the site 1 and site 2 specifically bind Sp1. EMSAs were performed using 24 bp oligonucleotides identical to site 1 and site 2 with HeLa nuclear extracts. (A.) Lanes 1 and 6 are HeLa nuclear extract alone. Lanes 2 and 7 show specific competition for binding with an Sp1 consensus oligonucleotide. Lanes 3 and 8 show competition with unlabeled site 1 and site 2 oligonucleotides (s1 and s2 respectively), respectively. Lanes 4 and 9 show competition with site 1 and site 2 mutant oligonucleotides (ms1 and ms2 respectively). Lanes 5 and 10 show non-specific competition with an TFIID consensus oligonucleotide. The unlabelled competitor oligonucleotide added to the binding reactions are indicated on top on each lane. (B.) An Sp1 antibody causes a specific supershift of the bound proteins in site 1 and site 2. Lane 1 is free probe. Lanes 2 and 3 were probed with the Sp1 consensus oligonucleotide. Lanes 4 and 5 were probed with the site 1 oligo. Lanes 6 and 7 were probed with the site 2 oligonucleotide.
3.4: Sp1 is bound to PACT promoter in native chromatin
In order to verify that Sp1 is bound to the GC boxes within PACT promoter, we tested its presence in native chromatin by performing chromatin immunoprecipitation (ChiP) assay. ChiP assay is a technique that allows quantification of protein-DNA interactions within the context of native chromatin and involves three steps: chemical cross-linking of protein-DNA complexes in intact cells; recovery of specific proteins by immunoprecipitation; and detection of co-precipitating DNA sequences by PCR (Das et al., 2004). As seen in Fig. 6, Sp1 antibody could successfully immunoprecipitate PACT promoter DNA with it from HeLa cells. The negative control with no antibody added during the immunoprecipitation step showed no PCR product. The input DNA control with PCR performed on cross-link reversed total DNA showed a good PCR reaction as expected. These results establish that Sp1 does bind to PACT promoter and regulate its transcriptional activity in HeLa cells, thereby confirming the role of Sp1 as suggested by our mutational analysis of the GC boxes within this promoter.
Figure 6.
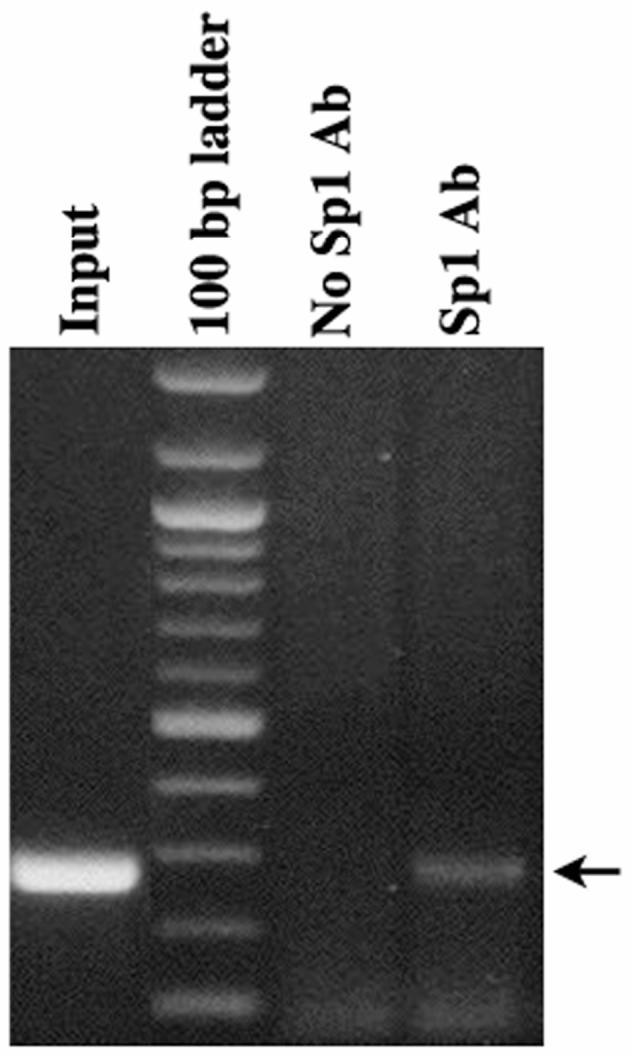
ChiP assay confirms presence of Sp1 on PACT promoter. Chip assay was performed using HeLa cells and Sp1 specific PEP2 antibody (Santa Cruz). The PCR products obtained were analyzed on a 1.2% agarose gel followed by Ethidium Bromide staining. The samples analyzed are as indicated on the top of the lanes. Arrow indicates the position of 275 bp PCR product originating from PACT promoter.
4. Discussion
4.1: Transcription of PACT is regulated by Sp1
PKR is a serine/threonine kinase that is known to be an integral part of the apoptosis pathway in response to cellular stress (Clemens and Elia, 1997; Williams, 2001; Donze et al., 2004). Activation of PKR leads to the phosphorylation of downstream substrates, the best characterized of which is the α subunit of eukaryotic initiation factor 2 (eIF-2) (Samuel, 1993; Clemens, 1996). Phosphorylation of eIF-2α inhibits protein synthesis, and ultimately leads to apoptosis. PACT is the only known cellular activator of PKR that activates PKR by direct protein-protein interaction in response to stress (Patel and Sen, 1998a; Patel et al., 2000; Huang et al., 2002). PACT associates with PKR with an increased affinity in response to stress signals and activates it leading to eIF2α phosphorylation and apoptosis. Here we present our work aimed at characterization of PACT promoter and its regulation by Sp1 transcription factor.
From northern blot analysis it is seen that PACT mRNA is present in a number of tissue types, and is present at an increased abundance in placenta and testis. Sequence analysis revealed that the PACT promoter does not contain a TATA box, however it does have a high GC content. There are six GC boxes within 400 bp upstream of the transcription start site. These GC boxes (GGGCGG) are known binding sequences for the eukaryotic transcription factor Sp1 (Philipsen and Suske, 1999). These features of the PACT promoter resemble the promoters of many housekeeping genes, such as thymidylate synthase (Rudge and Johnson, 2002). Mutational analyses and EMSAs revealed that Sp1 is indeed the transcription factor necessary for PACT basal transcription levels.
From the northern blot analysis shown in Fig. 1, we observed elevated expression of PACT in placenta and testis. To examine if higher levels of Sp1 are responsible for PACT′s elevated expression in these tissues we performed a western blot analysis to examine levels of Sp1 expression in various murine tissues. This analysis did not reveal any higher amount of Sp1 expression in placenta and testis. Although the expression of PACT was higher in placenta on the MTN blot shown in Fig. 1, we did not observe a corresponding higher expression of PACT mRNA or protein in three different human placental cell lines JAR, BeWO, and JEG3 (data not shown). With respect to testis, immunohistochemistry analysis revealed expression of PACT in spermatogonia (unpublished observations) and there is no corresponding tissue culture system to study PACT promoter in spermatogonial cells. Due to this, any promoter elements responsible for higher expression levels of PACT in placenta and testis could not be identified. It is certainly possible that the higher expression of PACT in placenta and testis may be regulated by hormonal stimuli and thus could be mediated by transcription factors other than Sp1, which may regulate the promoter by binding to sequence elements other than GC boxes. It is also possible that the Sp1 activity may be modulated by post-translational modifications such as phosphorylation (Li et al., 2004; Chu and Ferro, 2005) in these tissues to give a higher transcriptional activation. These possibilities remain to be examined.
4.2: PACT activity is regulated post-translationally in response to stress signals
Our research on PACT has demonstrated that PACT plays an important role in mediating apoptosis in response to stress signals (Patel et al., 2000). PACT is phosphorylated rapidly in response to cellular stress such as serum/growth factor deprivation, oxidative stress, arsenite, and actinomycin D (Ito et al., 1999; Patel et al., 2000; Peters et al., 2001). Phosphorylated PACT associates with PKR with an increased affinity and causes activation of PKR leading to eIF2α phosphorylation. The cells overexpressing PACT show enhanced apoptosis in response to these stress agents (Ito et al., 1999; Patel et al., 2000). However, stress signals do not mediate these effects by changing the cellular abundance of PACT mRNA or protein as analyzed by RNase protection or western blot analysis (data not shown). PACT activity is modified by phosphorylation at specific serine residues within the carboxy-terminal M3 motif of PACT in response to stress signals without changing the cellular levels of PACT protein (Peters et al., 2006). Thus, we did not expect PACT promoter to show enhanced activity in response to stress signals and this was confirmed by our promoter analysis. Serum deprivation, peroxide, thapsigargin, or arsenite treatment of transfected cells did not show any enhanced promoter activity in any of the deletion constructs (data not shown).
4.3: Sp1, a general transcription factor
Sp1 was the first transcription factor to be identified and cloned (Dynan and Tjian, 1983; Kadonaga et al., 1987). It was shown to be a sequence-specific DNA binding protein that was required for transcription of several mammalian and viral genes (Suske, 1999). Sp1 binds to GC/GT boxes via three C2H2-type zinc fingers present in the carboxy-terminal part of the protein (Kadonaga et al., 1988). Several different Sp1like proteins have been identified in mammalian cells including the Kruppel-like factors (KLFs) that have similar modular structure to Sp1 factors and are important components of the eukaryotic cellular transcriptional machinery (Kaczynski et al., 2003). By regulating the expression of a large number of genes that have GC-rich promoters, Sp1-like/KLF transcription regulators may take part in many important facets of cellular function, including cell proliferation, apoptosis, differentiation, and neoplastic transformation (Black et al., 2001). Individual members of the Sp1 family can function either as activators or repressors depending on the promoter they bind and the co-regulators with which they interact (Zhao and Meng, 2005). Of particular interest is the emerging role of Sp1 factors in growth regulatory pathways in cancer (Safe and Abdelrahim, 2005). It will thus be of interest to examine if PACT expression is modulated by modulation of Sp1 levels or activity during malignant transformation.
4.4: Sp1 regulates transcription of both PKR and PACT genes
Although the organization of both human and murine PACT genes has been described earlier, this analysis did not identify the transcription factors involved in its transcription (Rowe and Sen, 2001). The results presented here identify Sp1 as the transcription factor that binds to the GC boxes in PACT promoter and regulates its transcription. The expression of PKR itself is regulated by Sp1 factors in the absence of IFN induction (Das et al., 2006). Similar to PACT promoter, PKR promoter also lacks the TATA box. Study of the promoter region of PKR by deletion and mutational analysis has revealed a consensus 13-bp IFN inducible response element (ISRE), which drives increased expression in response to IFNs (Kuhen and Samuel, 1997; Tanaka and Samuel, 2000). In addition to this element, a novel kinase conserved sequence (KCS) element was described that is required for optimal basal and IFN-induced expression (Ward et al., 2002; Ward and Samuel, 2002). In contrast to many IFN-inducible genes, a considerable amount of expression of PKR is seen in many cell types even in the absence of IFN treatment. This basal level expression of PKR gene has been shown to be dependent on Sp1 and Sp3 factors belonging to the Sp family of transcription factors (Ward and Samuel, 2003). Similar to our PACT promoter data, mutational analysis of the PKR promoter also revealed two different Sp sites to be essential for expression (Das et al., 2006). Regulation of both the PKR and PACT genes by Sp transcription factors emphasizes the important functional role of these transcription factors in regulation of cell proliferation and apoptosis.
Acknowledgements
The authors would like to thank Anna McNeal and Indhira Handy for excellent technical help with generation of constructs. Financial support from National Institutes of Health (HL63359) and American Heart Association (0555503U) to RCP is gratefully acknowledged.
Abbreviations
- IFN
interferon
- ds
double-stranded
- PKR
RNA-activated protein kinase
- Sp1
Specificity protein 1
- DRBD
dsRNA-binding domain
- PP2A
protein phosphatase 2A
- eIF2
eukaryotic initiation factor 2
- EMSA
electrophoretic mobility shift analyses
- dsRBM
dsRNA-binding motif
- MEF
mouse embryonic fibroblast
- TNF-α
tumor necrosis factor-α
- KLFs
Kruppel-like factors
- PCR
polymerase chain reaction
- ChiP
chromatin immunoprecipitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. Embo J. 1998;17:6888–902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–94. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–60. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- Chong KL, Feng L, Schappert K, Meurs E, Donahue TF, Friesen JD, Hovanessian AG, Williams BR. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. Embo J. 1992;11:1553–62. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Ferro TJ. Sp1: regulation of gene expression by phosphorylation. Gene. 2005;348:1–11. doi: 10.1016/j.gene.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Clemens MJ. Protein kinases thet phosphorylate eIF2 and eIF2B, and their role in eukaryotic cell translational control. In: Hershey JWB, Mathews MB, Sonenberg N, editors. Translational Control. Cold Spring Harbor Laboratory Press; 1996. pp. 139–172. [Google Scholar]
- Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–24. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- Das PM, Ramachandran K, vanWert J, Singal R. Chromatin immunoprecipitation assay. Biotechniques. 2004;37:961–9. doi: 10.2144/04376RV01. [DOI] [PubMed] [Google Scholar]
- Das S, Ward SV, Tacke RS, Suske G, Samuel CE. Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. J Biol Chem. 2006;281:3244–53. doi: 10.1074/jbc.M510612200. [DOI] [PubMed] [Google Scholar]
- de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–20. [PubMed] [Google Scholar]
- Der SD, Yang YL, Weissmann C, Williams BR. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:3279–83. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Sripriya R, McLachlin JR, Lu J, Fabian JR, Kimball SR, Miller LK. Disruption of cellular translational control by a viral truncated eukaryotic translation initiation factor 2alpha kinase homolog. Proc Natl Acad Sci U S A. 1998;95:4164–9. doi: 10.1073/pnas.95.8.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze O, Deng J, Curran J, Sladek R, Picard D, Sonenberg N. The protein kinase PKR: a molecular clock that sequentially activates survival and death programs. Embo J. 2004;23:564–71. doi: 10.1038/sj.emboj.7600078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynan WS, Tjian R. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell. 1983;35:79–87. doi: 10.1016/0092-8674(83)90210-6. [DOI] [PubMed] [Google Scholar]
- Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–44. [PubMed] [Google Scholar]
- Gil J, Alcami J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Mol Cell Biol. 1999;19:4653–63. doi: 10.1128/mcb.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5:107–14. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- Green SR, Mathews MB. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev. 1992;6:2478–90. doi: 10.1101/gad.6.12b.2478. [DOI] [PubMed] [Google Scholar]
- Huang X, Hutchins B, Patel RC. The C-terminal, third conserved motif of the protein activator PACT plays an essential role in the activation of double-stranded-RNA-dependent protein kinase (PKR) Biochem J. 2002;366:175–86. doi: 10.1042/BJ20020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999;274:15427–32. doi: 10.1074/jbc.274.22.15427. [DOI] [PubMed] [Google Scholar]
- Jagus R, Joshi B, Barber GN. PKR, apoptosis and cancer. Int J Biochem Cell Biol. 1999;31:123–38. doi: 10.1016/s1357-2725(98)00136-8. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell. 1987;51:1079–90. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- Kadonaga JT, Courey AJ, Ladika J, Tjian R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science. 1988;242:1566–70. doi: 10.1126/science.3059495. [DOI] [PubMed] [Google Scholar]
- Katze MG. Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol. 1995;3:75–8. doi: 10.1016/s0966-842x(00)88880-0. [DOI] [PubMed] [Google Scholar]
- Koromilas AE, Roy S, Barber GN, Katze MG, Sonenberg N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257:1685–9. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- Kuhen KL, Samuel CE. Isolation of the interferon-inducible RNA-dependent protein kinase Pkr promoter and identification of a novel DNA element within the 5′-flanking region of human and mouse Pkr genes. Virology. 1997;227:119–30. doi: 10.1006/viro.1996.8306. [DOI] [PubMed] [Google Scholar]
- Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams BR. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. Embo J. 1997;16:406–16. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, He S, Sun JM, Davie JR. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82:460–71. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–48. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–90. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1993;90:232–6. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275:37993–8. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J Biol Chem. 1992;267:7671–6. [PubMed] [Google Scholar]
- Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. Embo J. 1998a;17:4379–90. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RC, Sen GC. Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double-stranded RNA and its antigrowth effects in yeast. Mol Cell Biol. 1998b;18:7009–19. doi: 10.1128/mcb.18.12.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RC, Stanton P, McMillan NM, Williams BR, Sen GC. The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci U S A. 1995;92:8283–7. doi: 10.1073/pnas.92.18.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RC, Stanton P, Sen GC. Specific mutations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J Biol Chem. 1996;271:25657–63. doi: 10.1074/jbc.271.41.25657. [DOI] [PubMed] [Google Scholar]
- Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21:1908–20. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Li S, Sen GC. Phosphorylation of specific serine residues in the PKR-activation domain of PACT is essential for its ability to mediate apoptosis. J Biol Chem. 2006 doi: 10.1074/jbc.M607714200. In Press. [DOI] [PubMed] [Google Scholar]
- Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, Stark GR. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. Embo J. 2000;19:263–72. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe TM, Sen GC. Organizations and promoter analyses of the human and the mouse genes for PACT, the protein-activator of the interferon-induced protein kinase, PKR. Gene. 2001;273:215–25. doi: 10.1016/s0378-1119(01)00588-1. [DOI] [PubMed] [Google Scholar]
- Rudge TL, Johnson LF. Synergistic activation of the TATA-less mouse thymidylate synthase promoter by the Ets transcription factor GABP and Sp1. Exp Cell Res. 2002;274:45–55. doi: 10.1006/excr.2001.5451. [DOI] [PubMed] [Google Scholar]
- Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–48. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Samuel CE. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J Biol Chem. 1993;268:7603–6. [PubMed] [Google Scholar]
- Sen GC, Ransohoff RM. Interferon-induced antiviral actions and their regulation. Adv Virus Res. 1993;42:57–102. doi: 10.1016/s0065-3527(08)60083-4. [DOI] [PubMed] [Google Scholar]
- St Johnston D, Brown NH, Gall JG, Jantsch M. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci U S A. 1992;89:10979–83. doi: 10.1073/pnas.89.22.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- Tan SL, Gale MJ, Jr., Katze MG. Double-stranded RNA-independent dimerization of interferon-induced protein kinase PKR and inhibition of dimerization by the cellular P58IPK inhibitor. Mol Cell Biol. 1998;18:2431–43. doi: 10.1128/mcb.18.5.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Samuel CE. Mouse interferon-inducible RNA-dependent protein kinase pkr gene: cloning and sequence of the 5′-flanking region and functional identification of the minimal inducible promoter [In Process Citation] Gene. 2000;246:373–82. doi: 10.1016/s0378-1119(00)00080-9. [DOI] [PubMed] [Google Scholar]
- Ward SV, Markle D, Das S, Samuel CE. The promoter-proximal KCS element of the PKR kinase gene enhances transcription irrespective of orientation and position relative to the ISRE element and is functionally distinct from the KCS-like element of the ADAR deaminase Promoter. J Interferon Cytokine Res. 2002;22:891–8. doi: 10.1089/107999002760274917. [DOI] [PubMed] [Google Scholar]
- Ward SV, Samuel CE. Regulation of the interferon-inducible PKR kinase gene: the KCS element is a constitutive promoter element that functions in concert with the interferon-stimulated response element. Virology. 2002;296:136–46. doi: 10.1006/viro.2002.1356. [DOI] [PubMed] [Google Scholar]
- Ward SV, Samuel CE. The PKR kinase promoter binds both Sp1 and Sp3, but only Sp3 functions as part of the interferon-inducible complex with ISGF-3 proteins. Virology. 2003;313:553–66. doi: 10.1016/s0042-6822(03)00347-7. [DOI] [PubMed] [Google Scholar]
- Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–20. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- Williams BR. Signal integration via PKR. Sci STKE. 20012001:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- Xu Z, Williams BR. The B56alpha regulatory subunit of protein phosphatase 2A is a target for regulation by double-stranded RNA-dependent protein kinase PKR. Mol Cell Biol. 2000;20:5285–99. doi: 10.1128/mcb.20.14.5285-5299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Meng A. Sp1-like transcription factors are regulators of embryonic development in vertebrates. Dev Growth Differ. 2005;47:201–11. doi: 10.1111/j.1440-169X.2005.00797.x. [DOI] [PubMed] [Google Scholar]