

This review is written as an introduction to those who may be contemplating a foray into the “worm world” but are not familiar with this animal or the toolbox of techniques and reagents that would be useful to get started. The goal is to provide an overview, an idea of what to consider, and where to find additional resources and necessary information. It has been slightly more than 30 years since Sydney Brenner documented the use of Caenorhabditis elegans as a model genetic organism [1]. Since that time more than 7,000 articles have been published investigating all aspects of this organism from its feeding and social behavior to very fine details of neurotransmission. After the sequence of the entire genome of C. elegans became available in 1998 [2], it became apparent that the similarity of genes between this microscopic nematode and that of humans is remarkable with approximately 40% of genes that are associated with human disease having homologs in the C. elegans genome [3]. This similarity validates Brenner’s choice of C. elegans as not only a model for general biological processes but also for understanding the pathogenesis of disease. If the 1990’s is considered the “decade of genome”, where the sequence of organism after organism was reported, the early 2000’s might be considered the “decade of genome mining” in which large scale efforts are aimed at understanding the function of the genomic content. In the C. elegans community, these efforts are directed at understanding of the function, regulation, interaction, and expression of the entire complement of genes in the genome (reviewed in [4]). All of this information is increasing the likelihood that C. elegans will be an attractive system for studying an ever-expanding variety of biological problems. Indeed, a number of investigators have successfully crossed over from another organism to the worm in order to study their homolog of interest in this simple model.
C. elegans has morphological and practical advantages as a model
Model organisms, in general, share a number of common characteristics, and the worm is no exception. The worm is inexpensive to cultivate, easy to physically manipulate, and has a panoply of genetic and molecular tools available. For C. elegans, in particular, there are a number of distinct advantages that will be discussed in greater detail. The animals are simple multicellular organisms: adults contain approximately 1,000 somatic cells yet have a variety of tissue types such as muscles, nerves, and intestinal cells. These animals have a short generation time, which allows for rapid experimentation; they progress from egg to larva to fertile adult in 3 days at room temperature. A single adult can have between 300 and 1,000 progeny. Due to the sexual dimorphism, they are useful for genetics: self-fertilizing hermaphrodites can maintain homozygous mutations without the need for mating and males can be used for genetic crosses. Additionally, the animals are transparent at every stage of their life cycle. The ability to see inside the organism is not only useful for observing cellular events such as mitosis or cytokinesis in real-time but also allows an investigator to use fluorescent reporter genes such as green fluorescent protein (gfp) to mark cells in live animals [5]. A powerful technique called RNA-mediated interference (RNAi) can allow rapid assessment of gene function. In the early days of C. elegans research, a heroic effort led by John Sulston resulted in a map of the entire lineage of every cell during embryogenesis and post-embryonic development [6,7,8]. This information is obviously invaluable for understanding the fine details of cellular events and, in particular, for interpreting the phenotypes of mutant animals at a single-cell level. Finally, a great advantage for C. elegans among its cohort of model organisms is the ability to freeze and recover the animals, thereby allowing long-term storage. Altogether, the distinct advantages of C. elegans make it a useful experimental system to address questions regarding the biology of multicellular organisms.
C. elegans exist as two sexes with interesting anatomical features
There are two sexes of C. elegans: hermaphrodites with 959 somatic cells and males with 1031 somatic cells. Hermaphrodites are essentially females that produce and store sperm at one stage in their life cycle before beginning to produce oocytes, making them self-fertile animals. A single hermaphrodite can produce an entire population of offspring within a few days. Hermaphrodites offer a great advantage from a genetic point of view. For instance, if one is dealing with animals that cannot mate, a population can still be established since mating is unnecessary for a hermaphrodite to produce offspring. Adult hermaphrodites have embryos inside their uterus and a vulval opening in the center of the animal through which the embryos are laid (Fig. 1). These animals also have a tapered tail at their posterior (Fig. 1). Males, which arise infrequently in a population (0.1–0.2%), are smaller and thinner than the hermaphrodites and have a fan-shaped tail that is composed of structures required for mating (Fig. 2). The anatomical differences between the two sexes become apparent at the fourth larval (L4) stage, discussed below. For both sexes, the basic body plan is characterized by two concentric “tubes” that run along the anterior/posterior axis (Fig. 3). These two tubes are separated by a fluid-filled pseudocoelomic space. The inner tube is the digestive system with an anterior pharynx that grinds food, followed by the intestine that extends along the body length to the posterior, and ends at the anal opening where waste is eliminated. The outer tube consists of the cuticle, the hypodermis (“worm skin”), the body-wall muscles and the nervous system. In the adult animal, the gonad is also found in the pseudocoelomic space.
Figure 1.
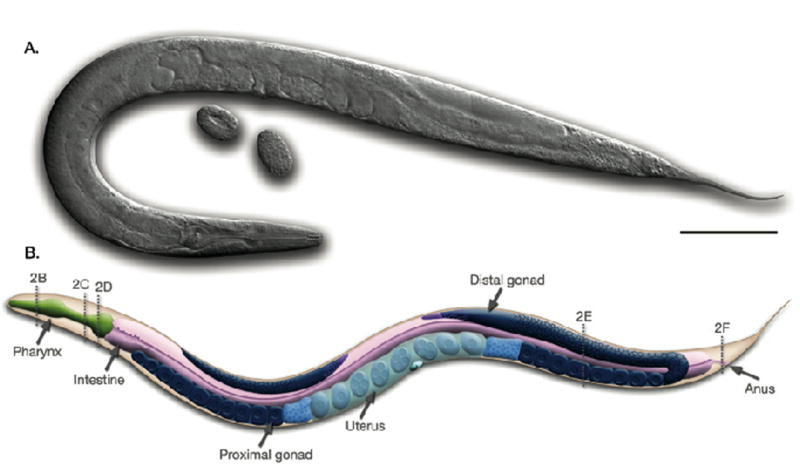
C. elegans hermaphrodite, left lateral view. (A) Micrograph of a hermaphrodite using differential interference optics. Two oval-shaped embryos can be seen near the vulval opening of the animal. (B) Diagram of the major anatomical features of the hermaphrodite. At the anterior (on the left) of the animal the distal gonad bends behind the intestine whereas at the posterior (on the right) the entire U-shaped gonad is visible (shown in shades of blue). The dotted lines labeled 2B–F correspond with the cross sections B–F in Figure 3. Scale bar represents 0.1 mm. Reprinted from Wormatlas (www.wormatlas.org) with permission.
Figure 2.
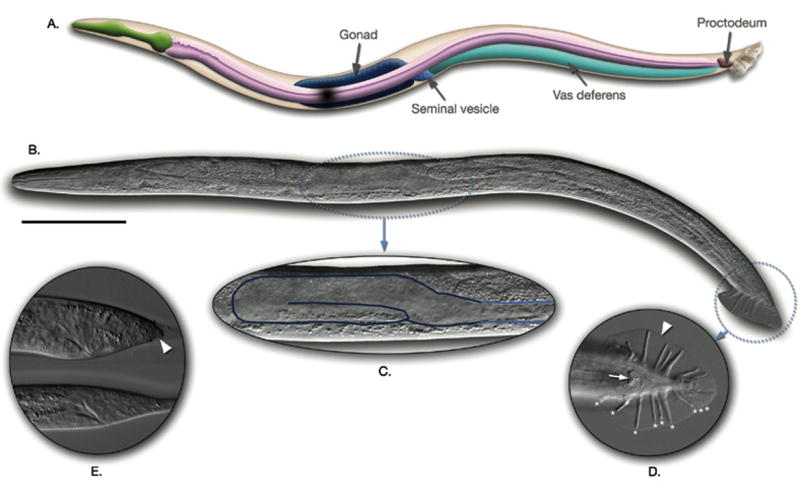
C. elegans male, left lateral view. (A) Diagram of the major anatomical features of the male. (B) Micrograph of a male using differential interference optics. (C–E) Magnified views of the following regions: the one-armed male gonad (C), the tail structures, ventral view (D), and the larval tail prior to differentiation (E). Scale bar represents 0.1 mm. Reprinted from Wormatlas (www.wormatlas.org) with permission.
Figure 3.
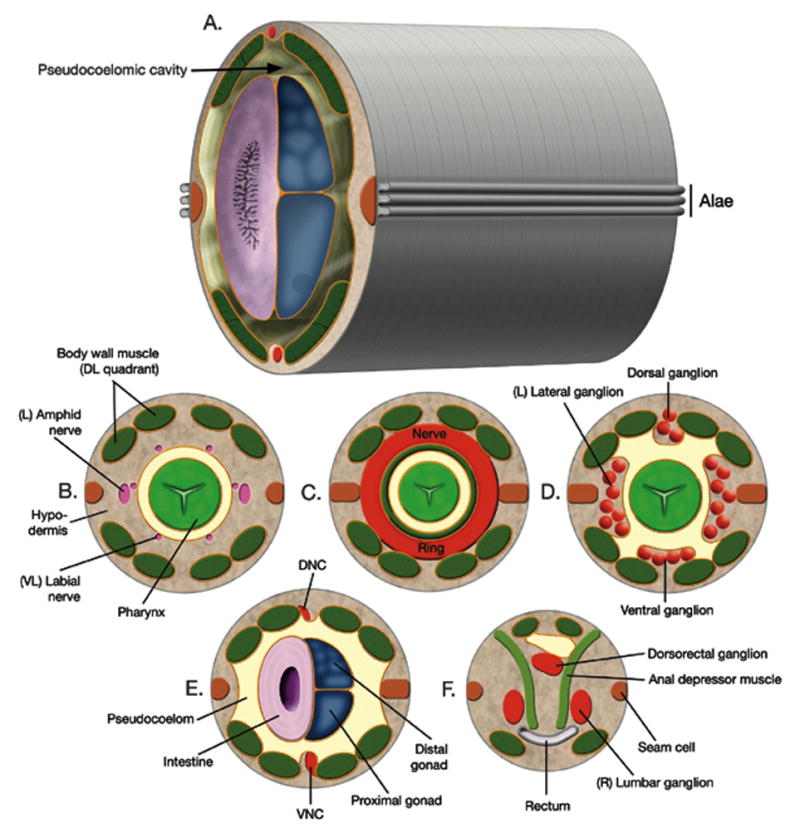
Anatomical features in the body cavity of C. elegans. (A) This diagram shows the major body plan of the animal with two concentric tubes and the intervening pseudocoelomic cavity. Alae are exterior structures derived from the hypodermis. (B–F) Cross-sections that correspond with the approximate position of the dotted lines on the hermaphrodite in Figure 1B. DNC, dorsal nerve cord. VNC, ventral nerve cord. Reprinted from Wormatlas (www.wormatlas.org) with permission.
C. elegans has a simple nervous system
These animals have a nervous system composed of 302 neurons. This simplicity was a key feature in Sydney Brenner’s choice of C. elegans as a new model organism [1]. The neurons come together at the anterior of the animal in a primitive brain called the nerve ring; all of the connections that these neurons make have been mapped in a complete wiring diagram [9, 10]. Their major function is to innervate the body wall muscles that run along the length of the animal in four quadrants resulting in a sinusoidal type of movement (Fig. 3). The nervous system contributes to a variety of behaviors that can be observed in the laboratory including response to soft and harsh touch, movement towards food and other attractive odorants, and repulsion from undesirable chemicals (reviewed in [11]).
C. elegans has a rapid life cycle
The life cycle of C. elegans consists of several stages (Fig. 4). First, regardless of whether the sperm that fertilizes the oocyte comes from the spermatheca of the hermaphrodite or from mating with a male, the embryo begins development inside the hermaphrodite. Once the embryo has approximately 28–30 cells, it is typically laid into the environment through the vulval opening. Embryogenesis is then completed outside the hermaphrodite during an approximately 13-hour period. Just prior to hatching, the egg contains a fully formed larva of 558 cells that can be seen tumbling inside the eggshell in a 3-fold pretzel configuration. At hatching, this L1 larva will arrest in development if there is no food available. In the presence of food, the larva proceeds through four stages that are punctuated by intervening molts when the animal sheds its cuticle to accommodate increasing body growth. During the larval period, a number of blast cells divide and the germline and sexually dimorphic features such as the hermaphrodite vulva and male tail develop so the animal will be able to reproduce when it reaches young adulthood. An alternative pathway in the life cycle is taken when the animal encounters poor conditions such as a limited food supply at the L1/L2 larval molt (Fig. 4). In this scenario rather than developing into an L3 larva, the animal enters the dauer or diapause pathway. Dauer larvae are highly resistant to stress and will reenter the normal life cycle at the L4 stage when conditions improve such as when food becomes available. The genes that control the decision to enter the dauer pathway are interesting because they are homologs of the vertebrate insulin-signaling pathway and many of them play an independent role in the longevity of C. elegans (reviewed in [12].
Figure 4.
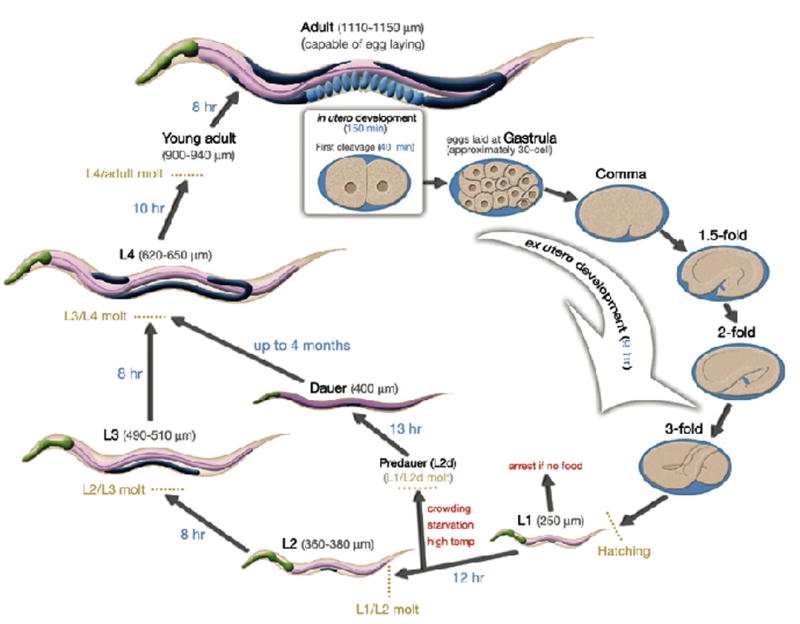
Life cycle of the C. elegans hermaphrodite at 22°C. An adult hermaphrodite with developing oval-shaped embryos inside is shown at the top of the life cycle. Once the embryos reach the 28–30-cell stage, they are laid into the environment and continue to develop. When embryogenesis is complete, L1 larvae hatch out of the eggshell and begin post-embryonic development in the presence of adequate nutritional sources. The animal continues larval development through the L2–L4 stages until reaching adulthood. Note the dark-colored gonad that is expanding during this time period. Prior to the L1/L2 molt, if there is a lack of food or there are too many animals or the temperature is too high then the animals will enter the alternative life cycle and become dauer larvae until these unfavorable conditions improve. The size of the animals at each stage of development is indicated in parentheses, and the amount of time spent at each stage in development is indicated along the arrows.
Studying an enzyme or gene product in C. elegans
When working with an enzyme, an enzymatic pathway, or a specific cellular process, one may be interested in whether the findings translate into a C. elegans study. For simplicity, assume the question is “how does a particular enzyme function in the worm?” Fortunately, there are many useful resources available that one can search before a single worm enters the laboratory. Information about the enzyme of interest can be found by searching WormBase (http://www.WormBase.org), an essential C. elegans website [13, 14]. This site contains a tremendous amount of C. elegans data that has been curated and is available in a user-friendly, searchable format. Examples of information found on WormBase are data on specific genes, mutants, RNAi phenotypes, expression patterns, and whole genome screens among others. Genomic information from other nematodes, from Caenorhabditis briggsae for example, is also becoming available at this website. WormBase has a helpful link to Textpresso, a search engine for worm literature and unpublished abstracts [15]. The abstracts can indicate the type of data related to the enzyme of interest that was presented at the latest C. elegans meeting. WormBase contains links that will help one to obtain C. elegans strains and mutant alleles from the Caenorhabditis Genetics Center (CGC) (www.cbs.umn.edu/CGC/) and the National Bioresource Project (www.shigen.nig.ac.jp/c.elegans/ChangeLocale.do?url=home&lang=en). For general information about the worm, a great place to start is Leon Avery’s C. elegans WWW server (elegans.swmed.edu). This site is the gateway to information, reagents, and investigators in the field. Free online reviews are available on a myriad of C. elegans topics at the WormBook website (www.wormbook.org). The chapters in WormBook have been written by leaders in the field, are available as soon as they are written, and will be updated directly on the web. Prior to WormBook, there has been two review compilations: The nematode Caenorhabditis elegans (1988) and C. elegans II (1997) that provide detailed information on all aspects of C. elegans [16,17]. The entire contents of the C. elegans II book is available for searching on the National Center for Biotechnology Information (NCBI) Bookshelf ([18]; www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Books&itool=toolbar). Two additional references are C. elegans: A practical approach [19], written for those who are outside the field and Caenorhabditis elegans: Modern biological analysis [20], providing detailed protocols that are standards in the field. Finally, there are Wormatlas (www.wormatlas.org/index.htm) and Wormimage (www.wormimage.org). These sites include many EM micrographs that are paired with diagrams detailing the C. elegans anatomy (Wormatlas) and immunofluorescence and reporter gene expression images (Wormimage). A flow chart of how one may approach studying a specific enzyme in the worm is presented in Fig. 5.
Figure 5.
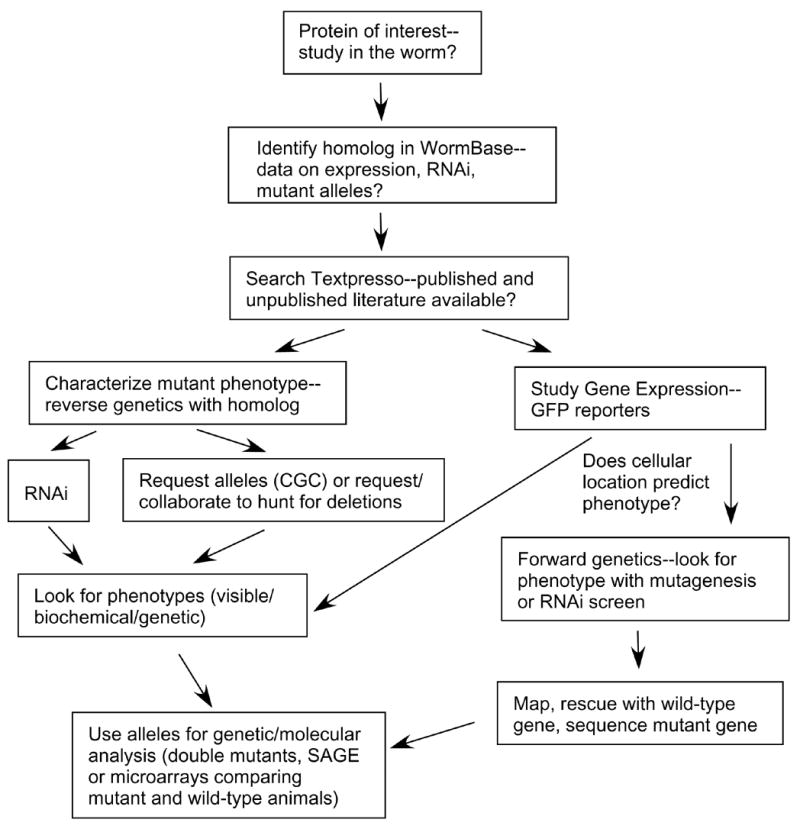
Flowchart for studying a favorite protein in C. elegans.
Growth methods for C. elegans
A relatively small investment is necessary to get started
How does one begin working with the organism? A stereomicroscope that illuminates from below and provides magnification in the 6X–50X ranges will be needed to observe and manipulate the animals. C. elegans are grown on Nematode Growth Medium (NGM) agar plates (for recipe, see [21]). The strain of bacteria that is commonly spread on the NGM plates as a food source for the animals is OP50, a leaky Escherichia coli uracil auxotroph that will grow slowly and provide nutrients for the animals without overgrowing them [1]. This strain can be obtained from the CGC. A worm pick will be needed in order to transfer the animals individually to plates. This pick can be hand-fashioned from a Pasteur pipette into which about an inch of 32-gauge platinum wire, flattened at one end, is attached by melting the glass around the wire. It may also be useful to have access to a compound microscope that is capable of Nomarski or differential interference contrast optics if one will need to examine the anatomy of the animals in detail. Since the animals prefer to grow between 15°C and 25°C, several incubators will be of use as well, although room temperature is convenient for incubation if temperature variation will not affect the experiment. Many useful standard protocols for C. elegans manipulation are found in the review by Theresa Stiernagle, who is currently the main contact at the CGC for strain requests [21].
It is easy to grow and store C. elegans
The commonly used laboratory strain is the Bristol strain called N2. These animals can be obtained from the CGC or any local worm laboratory. They can be cultivated in either liquid culture or on the NGM agar plates in the presence of bacteria [21]. It is also possible to grow the animals in a chemically defined medium without the addition of bacteria [22]. This method is useful because the components of the medium can be altered in order to study the nutrient or other chemical requirements of the animals [23]. For most manipulations of animals, however, they are grown on the agar plates. Once the animals have eaten all of the food on a plate they will burrow into the agar and can be maintained on the “starved” plate for weeks at a time in a 15°C incubator. When one is ready to use the animals again, they can be transferred to an agar plate with fresh bacteria by either cutting and moving a small block of agar from the starved plate with a sterile instrument such as a micropipette tip, or washing the worms off the surface of the plate with sterile water, or by picking one or more individuals onto a fresh plate. Another useful feature of this organism is the resistance of the embryos to harsh treatment such as incubation with a mixture of a strong base and household bleach [21]. Such treatment, followed by hatching in the absence of food allows a synchronous, decontaminated population of animals to be isolated at the L1 larval stage. The L1 animals can then be fed, and they will progress through development at roughly the same rate thereby allowing the isolation of a population of animals roughly synchronized at any stage of development. At any time, the animals can be cryogenically preserved as well. For more details on these and other basic worm techniques there are several good sources [16, 19–21].
C. elegans is a genetically amenable organism
The sexual dimorphism of C. elegans makes this organism excellent for genetic exploitation. If a strain needs to be propagated, single hermaphrodites can be used to self-fertilize and generate a population of offspring. Even if a mutation renders the animal unable to mate, it remains possible for the hermaphrodite to produce progeny. Yet the ability to cross males with hermaphrodites is a vital genetic tool. For example, mating experiments allow genetic markers such as mutations causing visible phenotypes to be placed together in a single organism along with an unknown mutation in order to facilitate mapping of that mutation. Hermaphrodites make only a limited number of sperm and can typically have approximately 300 self-progeny. Mating increases the number of offspring produced by a single hermaphrodite to approximately 1,000 due to the addition of the male-produced sperm. In order to set up a typical mating, non-starved animals at the L4 stage are chosen (Fig. 4). Several hermaphrodites and males are transferred to a new plate that contains only a minimal surface area of bacteria to encourage physical proximity and thus increase the likelihood of mating. It is critical when transferring the males to a mating plate that the transfer of eggs and larva are avoided in order to ensure the distinction between self and cross progeny.
Hermaphrodites have two X chromosomes. Males have a single X chromosome and arise infrequently in hermaphrodite populations due to nondisjunction of the X chromosome. The frequency of nondisjunction can be increased by heat-shock treatment of L4 hermaphrodites (33°C, 4 hours) and males may be observed in the offspring. Once a male is found it can be used to mate to a hermaphrodite of the same genotype so as to generate a population of males since half the offspring of cross progeny (as opposed to self progeny) will be males resulting from half of the sperm carrying the X chromosome and the other sperm lacking a sex chromosome (O genotype). There are also mutations that can be used that increase the frequency of males in the population. High incidence of male (Him) mutations increase the frequency of nondisjunction. A final strategy for generating males for crosses is to mate a strain of interest with N2 wild-type males. If mating is efficient, there will be many males in the offspring, and they will be heterozygous for any specific marker in question. These males can then be used in crosses and the phenotype of interest identified in the second generation. This strategy requires only wild-type males be maintained in the laboratory, since they can be used to generate males from any other strain by simply setting up new crosses when males are needed.
Determining the function of a particular gene
RNAi is a potent and straightforward method of removing a gene product
Once one can comfortably manipulate the animals, it is possible to query the worm for the function of a favorite enzyme or the role of a favorite biochemical process (Fig. 5). If C. elegans has homologs of genes that are of interest, the quickest way to determine gene function is to observe any phenotype generated by RNAi. In RNAi, double-stranded RNA (dsRNA) is delivered to the animal and the endogenous mRNA that is homologous to the dsRNA is destroyed allowing one to mimic a loss-of-function mutation in the corresponding gene. In C. elegans, dsRNA can be delivered by several methods: injecting or soaking the animals with in vitro transcribed dsRNA or, more simply, feeding the animals E. coli that harbor a plasmid engineered to express dsRNA from convergent promoters (Fig. 6) [24, 25]. During injection, adult hermaphrodites typically have dsRNA introduced directly into their developing gonad and the offspring can be observed for phenotypes [26, 27]. This method can be potent since a large amount of dsRNA is presumed to be delivered to the oocytes as they are forming inside the hermaphrodite. Alternatively, soaking and feeding take advantage of the observation that delivery of dsRNA does not need to occur directly in the gonad since the dsRNA can cross the intestinal boundaries and spread into somatic and germ tissues [24]. For a large number of genes, the two methods have been systematically evaluated and found to be comparable [25], although, for any given gene one method may be more potent than the other. For example, feeding and injecting have been reported to be equally penetrant for embryonic lethal phenotypes whereas feeding was more potent for post-embryonic phenotypes [25]. An advantage of feeding and soaking is that one can start with animals at any stage as opposed to the need for adult hermaphrodites that are easiest to manipulate for injection. Therefore, if a gene product is required for embryogenesis it is possible to soak or feed at the L1 stage after completion of embryogenesis and then investigate potential post-embryonic phenotypes.
Figure 6.
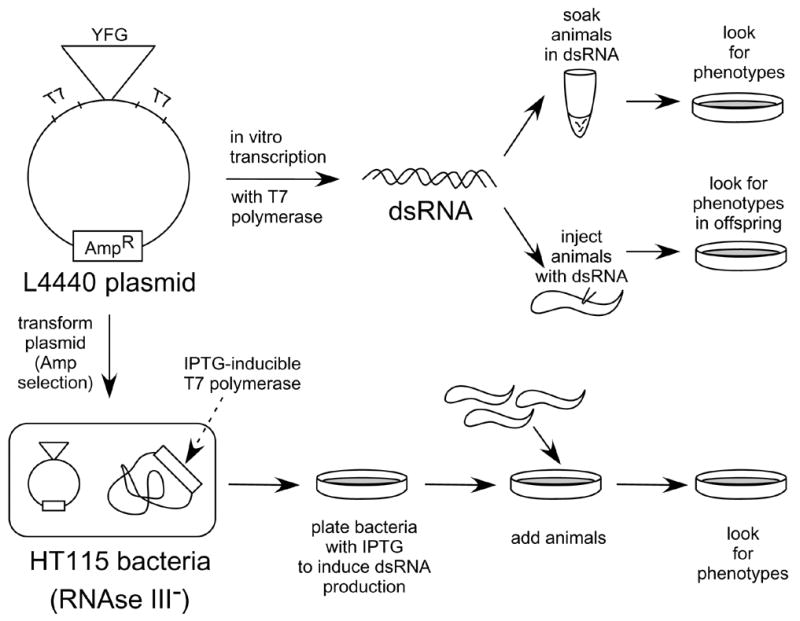
Methods used for RNA interference (RNAi) in C. elegans. A plasmid with opposing T7 promoter sequences containing “your favorite gene (YFG)” is constructed. The plasmid can be used in two ways. The top of the figure shows dsRNA made by in vitro transcription that is used for either soaking or injecting the animals. The bottom of the figure shows how animals can be fed bacteria that are producing dsRNA in vivo.
An appropriate plasmid will need to be constructed for RNAi
For all RNAi delivery methods, a plasmid needs to be constructed containing an insert with 500 to 3,000 base pairs of the target gene (Fig. 6) [25]. This insert can be obtained by PCR amplification of genomic DNA or cDNA. The insert can then be cloned into the commonly used L4440 feeding vector [24] or cloned into a vector containing either two convergent T7 promoters or T3/T7 promoters that surround a multiple cloning site. Then, the insert can be transcribed in vitro using T7 polymerase or T3 and T7 polymerase to make dsRNA for injection or soaking. Alternatively, if the plasmid contains only T7 promoter sites, it can be transformed into an RNase III-deficient strain of E. coli (HT115) that can produce T7 polymerase from an IPTG-inducible promoter [24, 25]. These bacteria are grown in the presence of IPTG to induce dsRNA expression and are fed to C. elegans (Fig. 6).
As a result of genome-wide RNAi studies, many clones of C. elegans genes in the L4440 feeding vector exist in RNAi libraries that are available for purchase [28, 29]; www.geneservice.co.uk/products/rnai/. Phenotypes from these global RNAi studies are listed in WormBase. It should be noted that these experiments are done on a very large scale and only certain classes of noticeable phenotypes are examined and reported: embryonic lethality, sterility, movement problems, slow growth and other obvious phenotypes. Therefore, RNAi-treated animals in which the gene or genes of interest was targeted, are unlikely to have been examined carefully for subtle phenotypes. Therefore, the RNAi phenotype on WormBase may be listed as wild-type even though under certain conditions one may be able to find a phenotype. It is important to be cautious when obtaining RNAi clones from the genome-wide libraries. The RNAi library clones were constructed on a large scale and have not been individually sequenced. A number of colleagues have found that clones from this library are incorrect or missing inserts so the plasmids need to be examined carefully before use on a small number of genes. In many cases, it may be easiest to construct one’s own RNAi plasmid.
Strain selection for RNAi can be useful for interpreting phenotypes
Before working with RNAi, the choice of C. elegans strain to be used for RNAi treatment requires careful consideration. In testing the effect of the dsRNA on a specific cell type, many reporter strains are available at the CGC or are reported in the literature that can be used as the starting strain (see discussion of reporter gene expression below). For example, in assessing the effect of removing a particular muscle actin-associated protein during post-embryonic development, it would be effective to use a myo-3::gfp strain that marks body wall muscles in order to evaluate the integrity of the muscle cells even if the RNAi does not result in any outward movement phenotypes. The myo-3::gfp strain expresses gfp with a nuclear localization signal from a myosin heavy chain promoter and, since the GFP in this strain is located in the nucleus, each muscle cell in the four quadrants can be counted and examined easily [30]. This gfp strain, however, is less useful for examining cytoplasmic actin phenotypes in the early embryo or the gut brush border [30].
There are also strains available that are more sensitive to RNAi treatment such as those defective for the RNA helicase RRF-3, the 3′ exonuclease ERI-1, and Rb pathway components [31–34]. If such strains are used for RNAi treatment, care must be taken to examine the negative controls (using a plasmid without insert DNA in the RNAi treatment, for instance) because these strains can also have defects that include the possibly interfering feature of temperature-sensitive sterility [33]. Gene products that function post-embryonically and in the nervous system have been found to be resistant by some investigators to RNAi [33]. Although, the problem of resistance may be overcome by expression of the dsRNA directly in the animal. Direct expression is accomplished by introducing a plasmid into the animal that will express a hairpin RNA (i.e. dsRNA in one linear nucleic acid molecule) from a tissue-specific promoter [35]. With few exceptions, RNAi is a very potent method of removing mRNA from the animal to quickly evaluate the contribution of the corresponding protein to normal function.
Deletion alleles are useful genetic tools
For the most part, RNAi is not heritable. And, as already noted, certain tissues are more resistant than others to RNAi treatment. It may be useful, therefore, to isolate mutations in genes of interest. Certainly, for any extensive genetic studies, mutants are essential reagents. Unfortunately, it is not yet possible to efficiently target particular loci in C. elegans by homologous recombination in order to replace wild-type genes with in vitro engineered mutated copies. There are, however, large-scale efforts in the C. elegans community to systematically isolate deletions in genes. Several websites allow one to check the availability of deletion strains and to place requests for making specific deletions. The National Bioresource Project will send deletion strains from their collection, and the CGC will send the strains made by the C. elegans Gene Knockout Consortium (www.celeganskoconsortium.omrf.org/). Deletions obtained directly through these sources need to be outcrossed several times to wild-type males before using the strain since mutagenesis can modify other genes in addition to the one of interest. It is also possible, of course, to isolate one’s own deletion alleles. Following mutagenesis of a population, one can screen for animals that harbor a specific deletion (Fig. 7A) [36, 37, for detailed protocols, see [38]]. This method uses a PCR-based strategy where primers are designed to amplify an entire open reading frame or the predicted important domains from a particular gene. The DNA from pools of mutagenized animals is amplified with the gene-specific primers. The PCR fragments from the entire population will be predominantly wild type. However, a pool containing the rare individuals with a deletion in a particular gene can be identified since a smaller PCR fragment will be amplified more rapidly than the larger wild-type fragment (Fig. 7A). Then, the pool of animals can be furthered screened using PCR until a hermaphrodites that are heterozygous or homozygous for the deletion are found [38].
Figure 7.
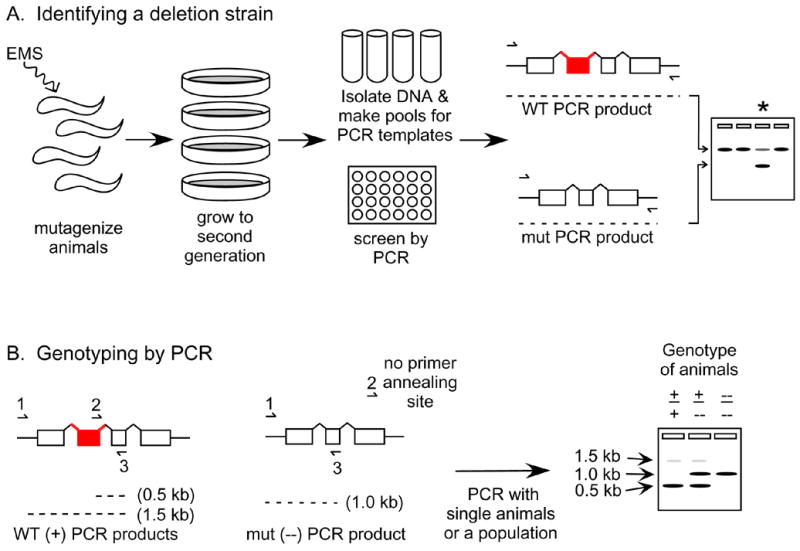
Isolating and following C. elegans genomic deletions. (A) An abbreviated schematic for finding a strain that contains a deletion of interest is shown. Animals from hundreds of plates are harvested for DNA isolation and the templates are pooled together for PCR in a 96-well format. PCR primers (lines with half arrowheads) are designed to flank a gene or gene region of interest. A pool of templates that contains animals with a deletion will be apparent by the smaller PCR fragment shown on the gel on the right (lane of interest labeled with an asterisk). (B) Animals containing a deletion can be distinguished from wild-type animals by designing 3 PCR primers that amplify different sizes of PCR products based on the genotype of the animal. Hypothetical PCR products from wild-type (+/+), heterozygous (+/−), and homozygous mutant (−/−) animals are shown on the gel on the right. WT, wild-type animals. mut, mutant animals.
Once a deletion allele is obtained, it will be necessary to continue using PCR to identify animals containing the mutation until the mutation has either been recombined with a visible marker mutation or the phenotype has been characterized well enough to be certain that the characteristics are exclusively due to the deleted gene in question. First, the breakpoint of the deletion can be identified by sequencing the PCR product that was initially followed to isolate the strain. Then, an additional primer can be designed with homology to a stretch of DNA that resides within the deletion (Fig 7B) [38]. Genomic DNA from animals may then be amplified using PCR primers that reside outside the deletion in the same test tube as the primer that is within the deletion. These primers will generate two PCR products that distinguish the wild type from the mutated allele (Fig. 7B). Genotyping by PCR can be used in initial crosses to follow the mutated allele without assuming anything about the phenotype of the animals.
Assessment of the phenotype of animals missing a gene product
Once a mutation in a particular gene has been obtained and genetically characterized, the specific phenotypes must be established. A careful comparison between the mutated strain and the wild-type N2 strain under standard laboratory conditions will be necessary. First, the outward appearance of the animals can be noted. Are they short and fat (or dumpy, Dpy)? Are they pigmented or clear (Clr)? Are they fertile or sterile (Ste)? Can they move or are they uncoordinated (Unc)? Do they have a normal brood size? Are their progeny dead (Emb) or are there defects in their grandchildren from maternal-effect problems? More subtle defects in embryogenesis and larval development include defects in programmed cell death or in developmental timing, as well as in alterations in aging, lifespan, and entry into the dauer pathway. Looking for the mutant phenotypes that were revealed in RNAi experiments may be a good starting place. Additionally, knowing the expression pattern of the wild-type gene (see below) will be helpful in predicting the possible phenotype that will result from the absence of a functional gene product. For example, a gene that is expressed in some or all of the neurons in the nervous system would be predicted to play a role in one or more behaviors and may alter fine or gross motor movement, chemotaxis, or egg-laying. The animals can be examined for response to harsh or light touch (with the end of a pick or an eyelash, respectively), tested for their ability to respond to attractive or repulsive chemicals, or inspected to see if a normal number of offspring are produced.
An interesting example of the phenotypic characterization of animals that lack an enzyme comes from a study of a deletion allele of ogt-1. ogt-1 codes for an enzyme predicted to transfer O-linked N-acetylglucosamine (O-GlcNAc) to a variety of protein substrates in the cell [39]. C. elegans that are missing this enzyme do not exhibit any obvious developmental defects or visible phenotypes in contrast to mammalian mutants which are embryonic lethal [40]. However, the animals are missing the O-GlcNAc modification on nuclear pore proteins, and biochemical assays to test the presence of certain molecules such as lipids droplets indicate that macronutrient storage is altered in the mutant animals [40]. Furthermore, since entry into the dauer pathway of the C. elegans life cycle is regulated by nutrient availability, double mutants were constructed with ogt-1 and dauer defective mutants that revealed a requirement for ogt-1 in the dauer pathway [40]. Since gene products involved in dauer formation are homologs of an insulin-like signaling pathway, these results have led to a proposed model for studying nutrient-driven insulin resistance in C. elegans [40]. This example illustrates how simple, direct observation of mutant animals may not reveal the underlying biochemical defects but further testing and genetic studies may be required. Clearly, some knowledge of the gene function either in the worm or in other organisms will be useful in directing a phenotype hunt.
Finding genes that mutate to a certain phenotype
Forward genetic screens can lead to new mutations
The previous discussion of assessing mutant phenotypes is an example of reverse genetics, or going from mutant gene to phenotype when the gene in question is known. A powerful technique in C. elegans is forward genetics, or going from predicted phenotype to mutated gene, since this approach is unbiased in terms of knowing which genes are involved in a process and can lead to new genetic discoveries. The large brood of offspring that each hermaphrodite can produce facilitates forward genetics. For a thorough review on the variety of genetic screens that can be performed using C. elegans, see Jorgensen and Mango [41]. Traditionally, animals are treated with a chemical mutagen such as ethylmethane sulfonate (EMS) to induce point mutations or small deletions. The first-generation offspring from the mutagenized hermaphrodites are examined for rare dominant mutations, and the second-generation offspring are examined for the more common recessive mutations. The approach presumes an accurate prediction of the phenotype. Many mutations can lead to a single phenotype. For example, uncoordination can result from nervous system or muscle structural defects. If one were interested in studying the innervation rather than the structure of muscles, it would be useful to use a starting strain that could help to discriminate the two types of mutations. For example, animals harboring a pan neuronal gfp reporter such as snb-1::gfp [42] could be mutagenized to look for uncoordinated offspring. The gfp pattern of any Unc animals could then be examined to see if any nervous system defects are apparent. gfp reporters have also been used in screening for altered morphology as a means for identifying genes that are involved in the development of a tissue. One example of such a screen used the expression of myo-3::gfp, a reporter of body wall muscles and non-striated muscles; after mutagenization, offspring were examined for altered muscle patterning leading to the identification of a gene, mls-1, that encodes a predicted transcription factor involved in controlling a cell-fate switch between two classes of muscles [30]. If the same screen had been performed without the gfp reporter, this mutant would not have been identified since the phenotype is only revealed when the muscle cells are examined directly. Animals can also be subjected to chemicals after mutagenesis to look for mutants that are more resistant to the effects of that chemical. Genes that are involved in acetylcholine-mediated neurotransmission, for example, have been identified by finding animals that, when mutated, are resistant to the anthelmintic drug levamisole [1, 43, 44]. This screen and other chemistry-to-gene screens are reviewed by Jones and colleagues [45].
Useful tools exist for mapping C. elegans genes
Once mutants have been identified and outcrossed, and the phenotype is sufficiently compelling enough to convince one that the gene will be interesting, the process of mapping can begin. This task has become easier with the availability of genome information. A popular method for mapping takes advantage of small nucleotide polymorphisms (SNPs) that exist between the common laboratory strain from Bristol, England (N2) and a Hawaiian strain (CB4856) [46]. Many of the SNPs lead to differential restriction sites, or snip-SNPs, at the same genomic position in each strain (Fig 8A). The snip-SNP sites are the same as restriction fragment length polymorphisms (RFLPs). These differences can be exploited so that a single mating and analysis reveals a general chromosomal location for a mutation rather quickly (Fig 8B) [46–48]. Briefly, Bristol mutant animals are crossed to the CB4856 strain and heterozygous wild-type cross progeny are selected. The second-generation offspring are separated into two populations: wild-type and mutant animals. PCR amplification is performed using lysates of each population to amplify single SNP regions followed by restriction enzyme digestion. Most snip-SNPs will be identical in the two populations because of independent assortment of the chromosomes; each population will have equivalent amounts of the Bristol and Hawaiian strain DNA at each chromosomal location. However, snip-SNPs that are in close proximity, or linked, to the mutation will predominantly have the Bristol pattern in the mutant population and will identify the chromosomal region of the mutation (Fig. 8B). Interval mapping may then be performed to place the mutation in a small region of the chromosome. Traditionally, interval mapping has used pairs of visible phenotypic markers to select for recombinant animals in a genetic region. Since SNP markers are phenotypically silent, they cannot be used as markers to select for recombinant animals. Recently, an adaptation of the snip-SNP procedure to a 96-well PCR plate format has made it possible to allow the rapid molecular identification of recombinants which can be followed by the rapid mapping of the mutation to a small manageable interval on a chromosome without the use of visible markers [48].
Figure 8.
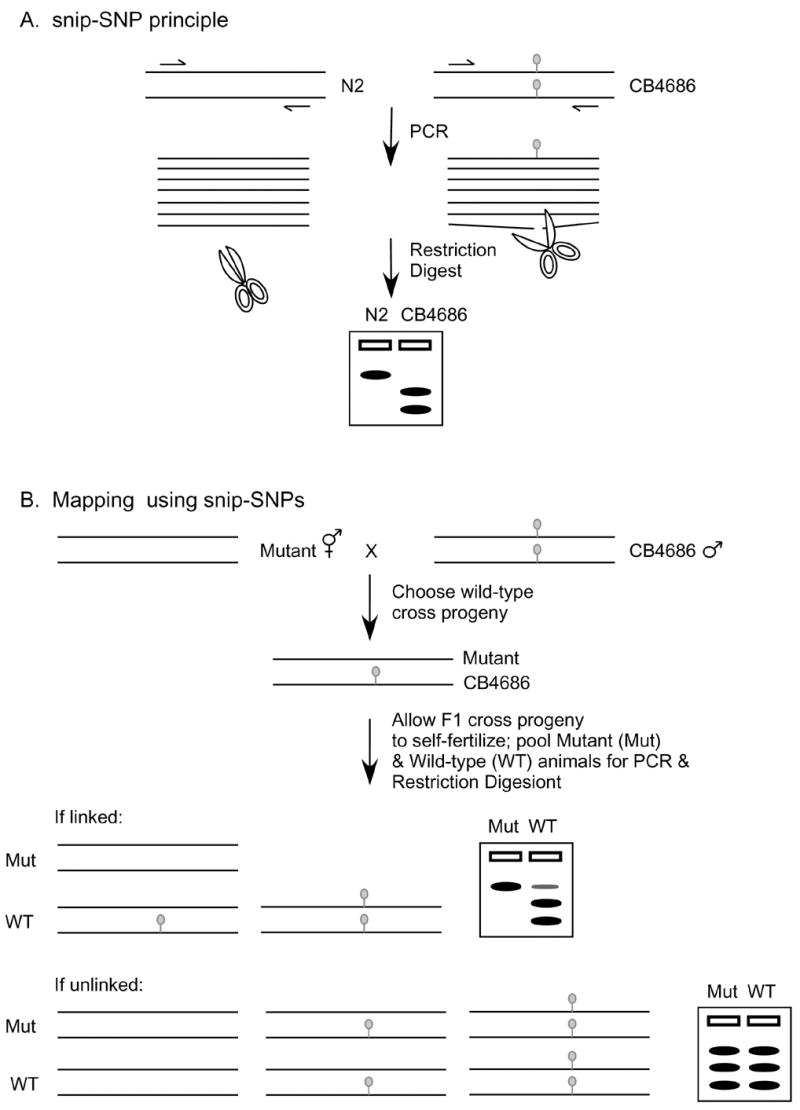
Mapping using single nucleotide polymorphism (SNP) markers. (A) A particular restriction site (grey circle) is not present in the N2 Bristol strain but is present in the CB4686 Hawaiian strain. DNA from each strain can be distinguished by PCR amplification of the genomic region followed by restriction digest (snip) and gel electrophoresis. (B) The method for mapping using snip-SNPs is shown. In this example, Bristol hermaphrodites harboring a recessive mutation are crossed with the Hawaiian males and wild-type heterozygous hermaphrodite cross progeny are selected. The chromosomes segregate during self-fertilization and the second generation offspring are separated based on phenotype into mutant and wild-type populations. DNA from the two populations is amplified at various SNP sites as in (A) with only one site shown here. If the mutation is linked to the SNP site, there will be a bias for Bristol DNA and not CB4686 in the mutant population. If the mutation is elsewhere in the genome or unlinked, the chromosomes with the SNP site with assort randomly and both Bristol and CB4868 DNA will be present and detected by snip-SNP analysis in the mutant and wild-type populations.The techniques and reagents available for studying C. elegans have made it possible to define the role that the HLH-8 protein plays in nematode development, but there is also clinical relevance to the work. In humans, Twist mutations cause birth defects that include craniofacial and digit malformation from defective bone development [90]. Despite the absence of bones in C. elegans, they do have a conserved Twist pathway that is useful for other aspects of mesoderm development. At this stage, multiple genes are known downstream of HLH-8 that have human homologs that cause similar craniofacial syndromes. Therefore, the information learned from the nematode will be relevant for understanding human mesoderm development and birth defects.
After a region with a reasonably low number of genes is established, several options are available for identifying the specific gene that has been mutated. It may be possible to predict a best candidate gene in the region based on sequence homology. The exons and splice junctions of that gene can be directly amplified by PCR from worm lysates and sequenced to see if a mutation can be identified [49]. If several potential candidates exist, or the genes in question are very large and might be difficult to sequence, an alternative approach is to perform RNAi (see above); sequences from a candidate or candidates are used to see if the mutant phenotype can be mimicked in wild-type animals by removing the gene product. If there are many candidates or no obvious candidate is apparent, rescue experiments can be performed using regions of the genome that are contained on cosmids. Cosmids can be obtained from the CGC and are introduced into the animals in successively smaller pools to narrow down rescuing activity to a single cosmid, which can then be subcloned into individual genes. Once a candidate gene has been identified, it will be necessary to perform a rescue experiment with a single gene. Cosmids and plasmids containing single genes are introduced using methods for making transgenic C. elegans. Unlike RNAi, where the physical location for delivery of dsRNA is relatively forgiving, (recall that animals can simply ingest the dsRNA and get phenotypes in non-intestinal tissues), DNA transformation requires delivery directly to the hermaphrodite gonad. The U-shaped gonad contains a syncytium of nuclei that are incorporated into gametes during meiosis (Fig. 1). During gametogenesis, exogenous DNA can be incorporated into nuclei as large cosmid or plasmid-based extrachromosomal arrays [50, 51]. The delivery of DNA into the hermaphrodite gonad can be a challenging technique for non-C. elegans investigators.
Two methods exist for DNA delivery into the hermaphrodite gonad. The first technique involves microinjection of animals mounted on an agarose pad. With the aid of a micromanipulator, the DNA is injected precisely and gently into the gonad using a glass needle [26]. Usually, the mixture of plasmids that is injected includes the experimental DNA plus a dominant marker such as a gfp reporter or a mutant gene such as rol-6 (su6000) (pRF4), which causes the animals to move in a corkscrew pattern rather than the wild-type sine wave motion [51]. The dominant marker can be scored to find the transgenic animals, and the phenotype of those animals can then be assessed for rescue. The success rate of this microinjection technique can be low and is dependent on the skills of the investigator.
A second technique for introducing DNA into the gonad of the hermaphrodite is that of microparticle bombardment. Gold microparticles coated with experimental DNA are delivered at a high velocity using helium pressure to a large population of animals directly on an agar plate [52, 53]. The procedure seems to favor low-copy integration directly into random chromosomal positions and can be advantageous for making stable lines [53]. The success rate of transformation is low, however, so that a large number of animals are required and selection markers are essential for identifying transformants. For example, unc-119 mutations that cause animals to die when starved can be rescued with a plasmid containing a wild-type copy of the unc-119 gene in addition to an experimental gene that is introduced by bombardment [53]. One disadvantage of the bombardment method is that the unc-119 mutation needs to be crossed first into the mutant strain of interest prior to transformation. Aside from the expense of the bombardment equipment, this is a less desirable approach for mutation rescue than injection where any strain can be treated directly. Once rescued transgenic animals are obtained, regardless of the method of transformation, it is necessary to identify the mutation in the gene in question. DNA from the candidate gene can be amplified by PCR using single worm lysates [49] followed by direct sequencing of PCR products. Altogether, finding the lesion in the locus and complementing the mutation by single gene rescue will confirm the identity of the mutated gene in question.
Gene mapping is unnecessary with RNAi screens
A more recent and powerful approach circumvents the need for mapping and cloning genes. The strategy takes advantage of the RNAi libraries that are available for a majority of the genes in the genome [54]. Delivering the RNAi clones to animals as bacterial lawns allows for relatively rapid screening through many genes. Similar to the traditional forward genetics screens described above, the initial strain selection can facilitate identification of phenotypes of interest, e.g. using a gfp reporter strain. This approach was used to identify genes related to unc-34, which plays a redundant role with wsp-1 in morphogenesis that is only revealed when both genes products are removed by RNAi or mutation [55]. In order to identify additional genes that function with unc-34, an RNAi screen was performed using unc-34 mutants that do not have embryonic defects in a search for animals that had decreased embryonic viability in the presence of an RNAi clone [55]. The RNAi screen identified wve-1, a WASP family protein that did not have an embryonic phenotype on its own but did in combination with the unc-34 mutant [55]. With RNAi screens, only loss-of-function and not gain-of-function phenotypes can be examined. Nevertheless, the identification of a missing gene product will be immediately apparent, and the lack of mapping and cloning that will be necessary should compensate for the inability to study gain-of-function mutations by the method.
The study of gene expression in C. elegans
Reporter genes can be used to examine expression patterns
It is obviously necessary to know where an identified gene is expressed. Consulting WormBase will indicate what others have found, but one should be aware that the data may be incomplete. One approach to the site of expression makes use of gfp reporter studies. Recall that C. elegans is transparent at all life cycle stages, making it possible to see green gfp-positive cells in live animals. In order to examine the expression pattern of a gene, one can clone a predicted promoter sequence upstream of gfp cDNA. A variety of gfp vectors have been designed by Andrew Fire’s laboratory that include multiple cloning sites where promoter sequences of interest can be inserted (www.ciwemb.edu/pages/firelab.html; http://www.addgene.org/Andrew_Fire) [56, 57]. These vectors also contain synthetic introns and 3′ UTRs that are important for robust gfp expression [56, 57]. The gfp vectors can be obtained from Addgene (www.addgene.org/pgvec1?f=c&cmd=showcol&colid=1). Most expression elements can be found in the 3–4 kb upstream of the start of translation in genes that are not contained in operons [58]. However, there are examples in which sequences present in the 3′ UTR participate in gene regulation, specifically in cases where a site exists for the binding of micro RNAs [59]. It may be important, therefore, to investigate sequences beyond those present upstream from the start of translation. A rapid method for making gfp expression vectors uses a PCR technique whereby overlapping DNA segments are used to juxtapose two pieces of DNA such as a potential promoter and gfp cDNA [60]. The primers for the fusion PCR are designed so that in three PCR steps a DNA fragment is generated that contains a promoter of interest fused to gfp and a generic 3′ UTR [61]. This PCR product can be injected directly into animals without purification or cloning of the DNA, thereby providing a rapid means for making transgenic animals for examining gene expression [61]. As indicated, a marker of transgenesis such as rol-6 (su6000) (pRF4) can be used to establish a stable line so that the animals can be examined carefully for gfp expression by epifluorescence microscopy.
Genomic approaches can reveal global gene expression patterns
Since the sequence of the entire genome of C. elegans is available, it is possible to examine animals from a “transcriptome” point of view. For example, a genome-wide profile can be examined for changes in gene expression in the presence or absence of an enzymatic activity. Two methods commonly used to investigate this type of question are oligonucleotide microarrays and serial analysis of gene expression (SAGE). For microarray experiments, Affymetrix (www.affymetrix.com/products/arrays/specific-/celegans.affx) provides a Genechip® that contains 22,150 unique C. elegans transcripts and Nimblegen (www.nimblegen.com/products/lit/nimblegen_catalog.pdf ) has one that contains 21,121 unique transcripts. Long oligomer-based microarrays from the C. elegans genome are also available from academic sources (genome.wustl.edu/genome/celegans/microarray/ma_gen_info.cgi; www.mshri.on.ca/microarray). These chips are hybridized with labeled cDNA that can be detected and quantified to measure the level of gene expression at each locus represented on the microarray (for more protocol information, see [62]). The SAGE method is used to identify individual transcripts by sequencing short “tags” from mRNAs in a population of animals or cells [63]. Both methods are used to investigate gene expression in multiple organisms and systems so the method chosen will rely on the resources available to the investigator.
With both methods, the first step is to isolate RNA from the animals. Among many examples, microarrays have been used to examine C. elegans gene expression at various stages of development [64–66], in both sexes to identify male and female germline-specific transcripts [67], in animals that differ in the number of a particular structure such as male tail rays [68], in animals that have been differentially treated with ethanol [69], and in animals that have activated or overexpressed transcription factors to identify downstream target genes [70, 71]. In addition to isolating RNA from a staged population of animals, techniques have been developed for isolating RNA from single tissues or cell types. In one approach, a gfp reporter marked a particular cell type that was then separated from a population of cells [72]. For example, unc-4::gfp, expressed in a subset of motor neurons, was used to isolate neurons for RNA isolation from primary C. elegans cell cultures by fluorescence activated cell sorting (FACS) to identify neuron-specific gene expression [73]. A second approach uses immunoprecipitation of an epitope-tagged poly(A) binding protein (PAB-1) expressed under the control of a tissue-specific promoter [74]. For example, the myo-3 promoter was used to express flag::PAB-1 in muscles in order to precipitate RNA with a flag antibody and obtain a muscle-specific transcription profile [74]. Such gene-expression profiling techniques can generate large amounts of data, but the challenge is in interpreting that data in order to provide biologically meaningful information. Programs have been designed that are intended to be helpful for such analysis (for recent reviews, [75, 76]).
Biochemical approaches being used with C. elegans
There are limited examples of direct biochemical approaches
The power of C. elegans as a model organism has traditionally resided in genetic investigation. The organism has not been used extensively for conventional biochemistry where large amounts of protein are purified from whole organisms or tissue-specific lysates for in vitro study. One limitation is the inability to isolate large quantities of any single tissue from C. elegans that would be suitable for biochemical purification. Established cell lines originating from C. elegans tissues are also not available. However, proteins that either represent a large fraction of the total protein in the animals such as actin or proteins that are a part of a structure that is amenable to purification, as is the cuticle, have been studied at the biochemical level. Actin was purified directly from C. elegans after growth in liquid culture and subsequent extraction of cellular material in the presence of ATP to separate actin filaments from actin binding proteins [77, 78]. The polymerization properties of the C. elegans actin were found to be similar to rabbit muscle actin, although the C. elegans actin interacted specifically with a C. elegans actin binding protein [78].
In a second example, the cuticle, a multi-layered exoskeleton that is composed mainly of cross-linked molecules in the extracellular matrix of hypodermal cells was examined [79, 80]. Because of the extensive cross-linking in the cuticle, it was possible to isolate a mainly intact structure by sonicating synchronized animals followed by boiling in a 1% sodium dodecyl sulfate (SDS) solution [81]. These purified cuticles have been used to examine the morphological features by electron microscopy and the biochemical composition by enzyme digestion and SDS-PAGE analysis [81]. In the biochemical approach, cuticle proteins were found to be predominantly collagen, due the complete digestion with collagenase [81]. These studies of proteins purified directly from C. elegans provide a better understanding of general nematode biology and have the potential to reveal similarities and differences when comparing the biochemical properties of the nematode and mammalian homologous proteins.
C. elegans auxotrophies can be studied biochemically
Because nematodes that are relatives of non-parasitic C. elegans are significant medical and agricultural parasites, unique aspects of nematode biology have been a subject of biochemical investigation. Several biosynthetic pathways have been investigated biochemically by manipulating the medium in which C. elegans is grown because the nematodes are auxotrophs for a number of nutrients. For example, unlike most eukaryotes, C. elegans do not have enzymes necessary for the synthesis of heme [23]. They do, however, require functional hemoproteins for key biological processes and therefore must acquire heme from the diet [23]. Growing animals in a chemically defined liquid medium allows the levels of heme to be manipulated, and this approach can be used to study the transport and utilization of heme in C. elegans [23]. The animals also lack the ability to synthesize their own cholesterol, which they must obtain from the diet as cholesterol itself or as a nonfunctional sterol that can be converted into cholesterol [82]. Cholesterol is required in C. elegans for proper growth and development; hermaphrodites reared in the absence of cholesterol have offspring that arrest in early larval stages (reviewed in [83]). In order to study the cholesterol requirement, animals have been fed both labeled sterols and metabolites in the pathway of cholesterol synthesis [82]. These studies identified enzymes unique to C. elegans cholesterol metabolism as well as compounds such as 25-azacoprostane-HCL (azacoprostane) that are inhibitory to the organisms [84]. Biochemical characterization of nutritional requirements that are unique to nematodes, as are heme and cholesterol, has the potential for identifying potent and specific nematocides.
Proteomic approaches reveal novel aspects of protein function
Proteomics is being applied to problems in C. elegans with increasing frequency. With the complete, annotated genome of C. elegans available, it is possible to identify the source of peptides isolated from particular complexes or from animals subjected to a specific treatment (reviewed in [4, 85]). Thus, extracts of C. elegans grown in the presence of the cholesterol inhibitor azacoprostane, have been examined by 2D gel analysis followed by mass spectrometry. Major changes in unsuspected proteins occurred when the animals were treated and analyzed [84]. Similarly, the role for the RNase III-related enzyme, Dicer (DCR-1) has been examined using a proteomics approach. DCR-1 is an enzyme that plays a critical role in RNAi. Briefly, DCR-1 and its associated proteins were immunopurified and subjected to proteolysis followed by a fractionation/mass spectrometry analysis of the resulting peptides [86]. The identity of the interacting factors revealed several new proteins involved in RNAi as well as suggested a model of competition that exists for the multiple roles that DCR-1 plays in small RNA-mediated gene silencing pathways [86]. As additional proteomics studies are performed with C. elegans, a more complete picture of the biochemistry within the organism will be revealed.
An example of a gene studied in C. elegans
Many examples are available of interesting proteins identified in mammals whose function in C. elegans has been investigated successfully using some of the methodology described here (Fig. 5). For simplicity, an example is chosen from the author’s laboratory for illustration. The laboratory is interested in the role of transcription factors acting as key regulators in developmental decisions such as cell-fate specification and differentiation. The factor of interest here is Twist, a basic helix-loop-helix (bHLH) protein. In all organisms examined, Twist plays a role in the development of mesoderm, i.e in the middle embryonic germ layer that becomes muscle, bone, and cartilage. A WormBase search reveals that only one Twist homolog, coded for by the hlh-8 gene, is present in C. elegans. When that homolog was first identified in 1998, there were no abstracts, publications, RNAi or other data about hlh-8 that could be found by searching WormBase. RNAi with hlh-8 sequences led to an incompletely penetrant egg-laying phenotype [87]. A gfp reporter constructed using 5′ upstream sequences of hlh-8, was expressed in a subset of mesodermal cells that included precursors of egg-laying muscles [87]. When a gfp reporter was made that included the large 1.5 kb first intron of hlh-8, expression was observed in the egg-laying and defecation muscles (P. Wang and AKC, unpublished). The gfp expression pattern predicted that if HLH-8 was playing an important role in the development of these muscles, then animals that were missing the protein might have problems laying eggs or defecating. Indeed, a screen for egg-laying defective (Egl) and constipated (Con) animals identified such a mutant strain. The allele was partially mapped, and the hlh-8 locus was sequenced to reveal a point mutation in the coding region for the HLH-8 basic DNA binding domain [88]. A deletion allele was isolated in hlh-8 using the PCR-based isolation strategy (Fig. 7A) [37], and these mutant animals were also Egl and Con [89]. Both hlh-8 mutant strains were rescued by using a plasmid containing the genomic hlh-8 DNA [88,89].
The hlh-8 mutant animals have been useful for understanding the role that HLH-8 plays in mesoderm development. gfp reporters that mark various mesodermal cells were crossed into the hlh-8 mutants in order to examine the cellular phenotypes. These reporters allowed the observation of patterning defects in the divisions of the early larval mesoderm in the mutants. Using gfp reporters, it was also possible to observe that the precursors of the muscle cells are born but do not differentiate in the mutants [88,89]. The next challenge was to identify the genes downstream of the HLH-8 transcription factor. Since HLH-8 is found in a limited number of cells, 2% at any given time in development, an overexpression strategy was used to generate samples with and without an excessive amount of HLH-8. The RNA from these samples was hybridized to Affymetrix Genechips® leading to the identification of new target genes [71]. Deletion alleles of several of these genes were obtained from the CGC, and the characterization of the mutant animals is underway.
Conclusions and future frontiers
C. elegans has been providing insights over the years for many basic biological processes that range from cell-cell signaling during embryogenesis and organogenesis to neuronal outgrowth and axonal pathfinding. The molecular tools and techniques have become rather sophisticated in terms of being able to go from envisioning a question to finding out an answer. Nevertheless, methodological challenges remain for investigators who focus on this organism. One challenge is the need for developing the capability of directing extrachromosomal pieces of DNA to specific places in the genome by homologous recombination. Such technology will allow structure/function questions to be examined in vivo since gene replacement will allow point mutations to be engineered directly in the animal. Another problem is the need for a cell culture system of established cell lines in order to study individual C. elegans cell types.
An incredible amount of data is being generated from mining the information in the genome of this multicellular invertebrate. As more data becomes available on a genome-wide scale, it becomes more apparent that it will be necessary to take a step back and tease out the significance for the organism of the individual observations. That is where the expertise of biochemists will need to enter the picture. When those individuals with in-depth knowledge of biochemistry turn their attention to the worm, new possibilities will open for understanding the many biological phenomena in this animal and, by extension, in the human as well.
Acknowledgments
The author is grateful to Zeynep Altun at WormAtlas for permission to reproduce the first four figures, and also wishes to thank Iqbal Hamza, John Hanover, William Jakoby, Michael Krause, and Harold Smith for critical comments on the manuscript. The work in the author’s laboratory is supported by NIH grant K22DE14541.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brenner S. The Genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 3.Culetto E, Sattelle DB. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum Mol Genet. 2000;9:869–77. doi: 10.1093/hmg/9.6.869. [DOI] [PubMed] [Google Scholar]
- 4.Hillier LW, Coulson A, Murray JI, Bao Z, Sulston JE, Waterston RH. Genomics in C. elegans: So many genes, such a little worm. Genome Res. 2005;15:1651–1660. doi: 10.1101/gr.3729105. [DOI] [PubMed] [Google Scholar]
- 5.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–5. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 6.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–56. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 7.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 8.Kimble J, Hirsh D. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev Biol. 1979;70:396–417. doi: 10.1016/0012-1606(79)90035-6. [DOI] [PubMed] [Google Scholar]
- 9.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Phil Trans R Soc Lond. 1986;B314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 10.White J, Southgate E, Durbin R. Neuroanatomy. In: Wood WB, et al., editors. The nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; 1988. pp. 433–455. [Google Scholar]
- 11.Thomas JH, Lockery S. In: Neurobiology. C. elegans a practical approach. Hope IA, editor. Oxford University Press; 1999. pp. 143–179. [Google Scholar]
- 12.Gami MS, Wolkow CA. Studies of Caenorhabditis elegans DAF-2/insulin signaling reveal targets for pharmacological manipulation of lifespan. Aging Cell. 2006;5:31–7. doi: 10.1111/j.1474-9726.2006.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen N, Harris TW, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Bradnam K, Canaran P, Chan J, Chen CK, et al. WormBase: A comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 2005;33:D383–D389. doi: 10.1093/nar/gki066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connell K. There’s no place like WormBase: an indispensable resource for Caenorhabditis elegans researchers. Biol Cell. 2005;97:867–72. doi: 10.1042/BC20040155. [DOI] [PubMed] [Google Scholar]
- 15.Muller HM, Kenny EE, Sternberg PW. Textpresso: an ontology-based information retrieval and extraction system for biological literature. PLoS Biol. 2004;2:e309. doi: 10.1371/journal.pbio.0020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood WB. The nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; 1988. the Community of C. elegans Researchers. [Google Scholar]
- 17.Riddle DL, Blumenthal T, Meyer BJ, Priess JR. C. elegans II. Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 18.Corsi AK. Books for free? How can this be?–a PubMed resource you may be overlooking. Biol Cell. 2006;98:439–43. doi: 10.1042/BC20050093. [DOI] [PubMed] [Google Scholar]
- 19.Hope IA. C. elegans a practical approach. Oxford University Press; 1999. [Google Scholar]
- 20.Epstein HF, Shakes DC. Caenorhabditis elegans: modern biological analysis of an organism. Academic Press; 1995. [Google Scholar]
- 21.Stiernagle T. Maintenance of C. elegans. [February 11,2006];Wormbook, ed. The C. elegans Research Community, WormBook. doi: 10.1895/wormbook.1.101.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- 22.Szewczyk NJ, Kozak E, Conley C. Chemically defined medium and Caenorhabditis elegans, BMC Biotechnology. 2003;3:19. doi: 10.1186/1472-6750-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao AU, Carta LK, Lesuisse E, Hamza I. Lack of heme synthesis in a free-living eukaryote. Proc Natl Acad Sci USA. 2005;102:4270–4275. doi: 10.1073/pnas.0500877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 25.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:research0002.1–0002.10. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mello CC, Fire A. DNA Transformation. In: Epstein HF, Shakes DC, editors. Caenorhabditis elegans: Modern Biological Analysis of an Organism. Academic Press; San Diego, CA: 1995. pp. 452–482. [Google Scholar]
- 27.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;19:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 28.Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–30. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- 29.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 30.Kostas SA, Fire A. The T-box factor MLS-1 acts as a molecular switch during specification of nonstriated muscle in C. elegans. Genes Dev. 2002;16:257–269. doi: 10.1101/gad.923102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–9. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- 32.Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PV, Kamath RS, Fraser AG, Ahringer J, Plasterk RH. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 2003;1:E12. doi: 10.1371/journal.pbio.0000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–9. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Kennedy S, Conte D, Jr, Kim JK, Gabel HW, Kamath RS, Mello CC, Ruvkun G. Somatic misexpression of germline P granules and enhanced RNA interference in retinoblastoma pathway mutants. Nature. 2005;436:593–7. doi: 10.1038/nature04010. [DOI] [PubMed] [Google Scholar]
- 35.Tavernarakis N, Wang SL, Dorovkov M, Ryazanov A, Driscoll M. Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet. 2000;24:180–3. doi: 10.1038/72850. [DOI] [PubMed] [Google Scholar]
- 36.Jansen G, Hazendonk E, Thijssen KL, Plasterk RH. Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nature Genet. 1997;17:119–121. doi: 10.1038/ng0997-119. [DOI] [PubMed] [Google Scholar]
- 37.Liu LX, Spoerke JM, Mulligan EL, Chen J, Reardon B, Westlund B, Sun L, Abel K, Armstrong B, Hardiman G, King J, McCague L, Basson M, Clover R, Johnson CD. High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res. 1999;9:859–67. doi: 10.1101/gr.9.9.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahringer J, editor. Reverse genetics, (April 6, 2006) WormBook, ed. The C. elegans Research Community, WormBook. doi/10.1895/wormbook.1.47.1, http://www.wormbook.org.
- 39.Lubas WA, Frank DW, Krause M, Hanover JA. O-linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J BiolChem. 1997;272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 40.Hanover JA, Forsythe ME, Hennessey PT, Brodigan TM, Love DC, Ashwell G, Krause M. A Caenorhabditis elegans model of insulin resistance: altered macronutrient storage and dauer formation in an OGT-1 knockout. Proc Natl Acad Sci USA. 2005;102:11266–11271. doi: 10.1073/pnas.0408771102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen EM, Mango SE. The art and design of genetic screens: Caenorhabditis elegans. Nat Rev Genet. 2002;3:356–369. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- 42.Nonet ML. Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. Journal of Neuroscience Methods. 1999;89:33–40. doi: 10.1016/s0165-0270(99)00031-x. [DOI] [PubMed] [Google Scholar]
- 43.Lewis JA, Wu CH, Levine JH, Berg H. Levamisole-resistant mutants of the nematode Caenorhabditis elegans appear to lack pharmacological acetylcholine receptors. Neuroscience. 1980;5:967–989. doi: 10.1016/0306-4522(80)90180-3. [DOI] [PubMed] [Google Scholar]
- 44.Lewis JA, Fleming JT, McLafferty S, Murphy H, Wu C. The levamisole receptor, a cholinergic receptor of the nematode Caenorhabditis elegans. Mol Pharmacol. 1987;31:185–93. [PubMed] [Google Scholar]
- 45.Jones AK, Buckingham SD, Sattelle DB. Chemistry-to-gene screens in Caenorhabditis elegans. Nat Rev Drug Discov. 2005;4:321–30. doi: 10.1038/nrd1692. [DOI] [PubMed] [Google Scholar]
- 46.Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet. 2001;2001;28:160–164. doi: 10.1038/88878. [DOI] [PubMed] [Google Scholar]
- 47.Swan KA, Curtis DE, McKusick KB, Voinov AV, Mapa FA, Cancilla MR. High-throughput gene mapping in Caenorhabditis elegans. Genome Res. 2002;12:1100–5. doi: 10.1101/gr.208902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;12:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams BD. Genetic mapping with polymorphic sequence-tagged sites. In: Epstein HF, Shakes DC, editors. Caenorhabditis elegans: Modern Biological Analysis of an Organism. Academic Press; San Diego, CA: 1995. pp. 81–96. [Google Scholar]
- 50.Stinchcomb DT, Shaw JE, Carr SH, Hirsh D. Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol Cell Biol. 1985;5:3484–3496. doi: 10.1128/mcb.5.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilm T, Demel P, Koop HU, Schnabel H, Schnabel R. Ballistic transformation of Caenorhabditis elegans. Gene. 1999;229:31–35. doi: 10.1016/s0378-1119(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 53.Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–21. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 55.Withee J, Galligan B, Hawkins N, Garriga G. Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics. 2004;67:1165–76. doi: 10.1534/genetics.103.025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–98. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 57.Miller DM, III, Desai NS, Hardin DC, Piston DW, Patterson GH, Fleenor J, Xu S, Fire A. Two-color GFP expression system for C. elegans. Biotechniques. 1999;5:914–921. doi: 10.2144/99265rr01. [DOI] [PubMed] [Google Scholar]
- 58.Okkema PG, Krause M. Transcriptional regulation (December 23, 2005) WormBook, ed. The C. elegans Research Community, WormBook. doi: 10.1895/wormbook.1.45.1. doi/10.1895/wormbook.1.45.1, http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- 59.Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes Dev. 2004;18:132–7. doi: 10.1101/gad.1165404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 61.Hobert O. PCR fusion-based approach to create reporter constructs for expression analysis in transgenic C. elegans. BioTechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
- 62.Portman DS. Profiling C. elegans gene expression with DNA microarrays regulation. [January 20, 2006];WormBook, ed. The C. elegans Research Community, WormBook. doi: 10.1895/wormbook.1.104.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- 63.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–7. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 64.Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL. Genomic analysis of gene expression in C. elegans. Science. 2000;290:809–812. doi: 10.1126/science.290.5492.809. [DOI] [PubMed] [Google Scholar]
- 65.Jiang M, Ryu J, Kiraly M, Duke K, Reinke V, Kim SK. Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2001;98:218–223. doi: 10.1073/pnas.011520898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–92. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- 67.Reinke V, Smith HE, Nance J, Wang J, van Doren C, Begley R, Jones SJM, Davis EB, Scherer S, Ward S, Kim SK. A global profile of germline gene expression in C. elegans. Mol Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- 68.Portman DS, Emmons SW. Identification of C. elegans sensory ray genes using whole-genome expression profiling. Dev Biol. 2004;270:499–512. doi: 10.1016/j.ydbio.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 69.Kwon JY, Hong M, Choi MS, Kang S, Duke K, Kim S, Lee S, Lee J. Ethanol-response genes and their regulation analyzed by a microarray and comparative genomic approach in the nematode Caenorhabditis elegans. Genomics. 2004;83:600–614. doi: 10.1016/j.ygeno.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 70.Romagnolo B, Jiang M, Kiraly M, Breton C, Begley R, Wang J, Lund J, Kim SK. Downstream targets of let-60 Ras in Caenorhabditis elegans. Dev Biol. 2002;247:127–136. doi: 10.1006/dbio.2002.0692. [DOI] [PubMed] [Google Scholar]
- 71.Wang P, Zhao J, Corsi AK. Identification of novel target genes of CeTwist and CeE/DA. Dev Biol. 2006;293:486–98. doi: 10.1016/j.ydbio.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 72.Christensen M, Estevez A, Yin X, Fox R, Morrison R, McDonnell M, Gleason C, Miller DM, III, Strange K. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron. 2002;33:503–514. doi: 10.1016/s0896-6273(02)00591-3. [DOI] [PubMed] [Google Scholar]
- 73.Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM., III A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics. 2005;6:42. doi: 10.1186/1471-2164-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 75.Reimers M, Heilig M, Sommer WH. Gene discovery in neuropharmacological and behavioral studies using Affymetrix microarray data. Methods. 2005;37:219–28. doi: 10.1016/j.ymeth.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 76.Liang Y, Kelemen A. Associating phenotypes with molecular events: recent statistical advances and challenges underpinning microarray experiments. Funct Integr Genomics. 2006;6:1–13. doi: 10.1007/s10142-005-0006-z. [DOI] [PubMed] [Google Scholar]
- 77.Harris HE, Epstein HF. Myosin and paramyosin of Caenorhabditis elegans: biochemical and structural properties of wild-type and mutant proteins. Cell. 1977;10:709–19. doi: 10.1016/0092-8674(77)90105-2. [DOI] [PubMed] [Google Scholar]
- 78.Ono S. Purification and biochemical characterization of actin from Caenorhabditis elegans: its difference from rabbit muscle actin in the interaction with nematode ADF/cofilin. Cell Motil Cytoskeleton. 1999;43:128–36. doi: 10.1002/(SICI)1097-0169(1999)43:2<128::AID-CM4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 79.Kramer JM. Extracellular matrix. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor Laboratory Press; 1997. pp. 471–500. [PubMed] [Google Scholar]
- 80.Johnstone IL. Cuticle Cuticle collagen genes. Expression in Caenorhabditis elegans. Trends Genet. 2000;16:21–7. doi: 10.1016/s0168-9525(99)01857-0. [DOI] [PubMed] [Google Scholar]
- 81.Cox GN, Kusch M, Edgar RS. Cuticle of Caenorhabditis elegans: its isolation and partial characterization. J Cell Biol. 1981;90:7–17. doi: 10.1083/jcb.90.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chitwood DJ. Biochemistry and function of nematode steroids. Crit Rev Biochem Mol Biol. 1999;34:273–284. doi: 10.1080/10409239991209309. [DOI] [PubMed] [Google Scholar]
- 83.Kurzchalia TV, Ward S. Why do worms need cholesterol? Nature Cell Biol. 2003;5:684–688. doi: 10.1038/ncb0803-684. [DOI] [PubMed] [Google Scholar]
- 84.Choi BK, Chitwood DJ, Paik YK. Proteomic changes during disturbance of cholesterol metabolism by azacoprostane treatment in Caenorhabditis elegans. Molec Cell Proteomics. 2003;2:1086–1095. doi: 10.1074/mcp.M300036-MCP200. [DOI] [PubMed] [Google Scholar]
- 85.Lee J, Nam S, Hwang SB, Hong M, Kwon JY, Joeng KS, Im SH, Shim J, Park MC. Functional genomic approaches using the nematode Caenorhabditis elegans as a model system. J Biochem Mol Biol. 2004;37:107–113. doi: 10.5483/bmbrep.2004.37.1.107. [DOI] [PubMed] [Google Scholar]
- 86.Duchaine TF, Wohlschlegel JA, Kennedy S, Bei Y, Conte D, Jr, Pang K, Brownell DR, Harding S, Mitani S, Ruvkun G, Yates JR, III, Mello CC. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 87.Harfe BD, Vaz Gomes A, Kenyon C, Liu J, Krause M, Fire A. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev. 1998;12:2623–35. doi: 10.1101/gad.12.16.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corsi AK, Brodigan TM, Jorgensen EM, Krause M. Characterization of a dominant negative C. elegans Twist mutant protein with implications for human Saethre-Chotzen syndrome. Development. 2002;129:2761–72. doi: 10.1242/dev.129.11.2761. [DOI] [PubMed] [Google Scholar]
- 89.Corsi AK, Kostas SA, Fire A, Krause M. Caenorhabditis elegans twist plays an essential role in non-striated muscle development. Development. 2000;127:2041–51. doi: 10.1242/dev.127.10.2041. [DOI] [PubMed] [Google Scholar]
- 90.Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6:1647–56. doi: 10.1093/hmg/6.10.1647. [DOI] [PubMed] [Google Scholar]