

Abstract
The role of caspase-independent apoptotic events in heart failure is largely unknown. The present study examined the response of apoptotic factors, which can function independently of caspase machinery including AIF, EndoG, and HtrA2/Omi to high salt diet-induced pathologic heart failure and exercise-induced physiologic cardiac hypertrophy. Following ~4 months of a daily diet containing 6% salt, animals developed clinical evidence of heart failure accompanied by changes in AIF, EndoG, and HtrA2/Omi. Assessment of the mitochondria-free cytosolic fraction revealed cytosolic accumulations of AIF and processed HtrA2/Omi in the failed ventricle muscles. The subcellular translocation of AIF from mitochondria to cytosol and nuclei was supported by immunofluorescent analysis using confocal microscopy. However, according to our RT-PCR analyses, AIF and EndoG mRNA were decreased, rather than elevated, in the failed heart relative to control heart. No difference in any of the measured parameters of AIF, EndoG, and HtrA2/Omi was found in the ventricle muscle of either exercise-trained or 6 weeks high salt diet fed animals compared to controls. These findings are consistent with the hypothesis that caspase-independent events are involved in cardiac apoptosis during the late remodeling stage of pathologic heart failure.
Keywords: apoptosis, caspases, cardiac myocyte, heart failure, cardiac hypertrophy
INTRODUCTION
A growing body of evidence suggests that cardiac apoptosis plays an important role in the development of heart failure [1,2]. Specifically, it has been proposed that the activation of the apoptotic program is a transitional process during which the heart is remodeled from early cardiac hypertrophy to the decline of contractile performance [3]. It is important that accelerated cardiac apoptosis not only detrimentally influences the physiologic function of the heart by causing non-replaceable lose of vital cardiomyocytes, but that continuing cardiac cell death also exacerbates the situation by introducing further compensatory overload and hypertrophy in the existing cardiomyocytes, thus hastening the pathologic transition of the remaining cardiac tissue. Based on the tightly regulated biochemical nature of apoptosis, it is to be hoped that regimens effectively remedying fatal heart failure would emerge on the discovery of methods to rectify aberrant cardiac apoptosis. Consequently, it is crucial to fully understand the regulatory mechanisms that contribute to the promotion of the apoptotic signaling during heart failure.
In addition to the intrinsic mitochondrial and extrinsic death receptor apoptotic pathways, the signaling events contributing to apoptosis can be further sorted into caspase-dependent and caspase-independent pathways. With respect to the caspase-independent pathway, a cluster of cellular factors including apoptosis-inducing factor (AIF), endonuclease G (EndoG) and high temperature requirement protein A2 (HtrA2/Omi) have been shown to be able to induce apoptosis without the mediation of the caspases, the central apoptosis executing proteases [4–6]. Some suggest that AIF, a mitochondrial flavoprotein, has both oxidoreductase and apoptosis-inducing activities [7]. Although its precise physiologic functions are unclear, the apoptotic character of AIF has been well demonstrated and it is believed that its apoptotic trait might be attributable to a putative DNA binding site responsible for chromatin condensation and DNA fragmentation [8]. EndoG, an evolutionarily conserved, nuclear-encoded endonuclease, has also been shown to have chromosomal DNA cleavage ability in a caspase-independent manner [6]. In contrast, the mechanistic properties of serine protease HtrA2/Omi in causing apoptosis are relatively ambiguous. It has been thought that, similar to Smac/DIABLO, HtrA2/Omi promotes apoptosis by inhibiting the apoptosis-suppressing activities of inhibitor of apoptosis proteins (IAPs) through a caspase-associated process [9]. However, there are also data suggesting that HtrA2/Omi might be able to induce apoptosis by means of its proteolytic activity without caspase activation [10,4]. Due to the scarcity of the existing data on the apoptotic function of AIF, EndoG and HtrA2/Omi in pathologic cardiac conditions, the precise regulatory mechanisms and the interdependence of these cellular factors in caspase-independent pro-apoptotic signaling are largely unclear. Nevertheless, it is known that these caspase-independent factors are normally resident in mitochondrial intermembrane space. In response to apoptotic stimuli, their apoptotic functions are activated following their release to the cytosol by a less defined mechanism [4–6].
Although the activation of apoptosis and caspase-related apoptotic factors have been demonstrated in high salt (HS) diet-induced heart failure [11], it is uncertain whether the apoptotic factors that can function independently of caspase dependent mechanisms may also be involved. Therefore, this study investigated the responses of AIF, EndoG, and HtrA2/Omi to pathologic heart failure induced by HS diet and physiologic cardiac hypertrophy as induced by exercise training. The rationale of examining AIF, EndoG and HtrA2/Omi is based on their evidently shown capabilities in mediating the pro-apoptotic pathway and the activation of apoptosis without the involvement of caspases. We tested the hypothesis that caspase-independent apoptotic factors are responsive to heart failure induced by a pathologic stimulus, but not during physiologic cardiac hypertrophy.
MATERIALS AND METHODS
Animal models
Experiments were conducted using female Dahl salt-sensitive rats (Harlan, Indianapolis, IN) housed in pathogen-free conditions at ~20°C and a reverse daily 12:12 h light:dark cycle. The animal care standards were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all experimental procedures carried out with approval from the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Induction of physiologic cardiac hypertrophy and heart failure
The experimental procedures of the exercise-induced physiologic cardiac hypertrophy and the HS diet-induced pathologic heart failure were previously described [11,12]. In brief, at 6 weeks of age, rats in the exercise group exercised daily on a rodent treadmill (Columbus Instruments, Columbus, OH) following a modified training program which comprised a 3-wk progressive intensity-increasing period followed by a 3-wk maintenance period [13,14]. Animals exercised at the running speed of 20 m/min at a 5° incline for 90 min per session in the maintenance period. For HS diet and failure groups, animals were fed a 6% sodium chloride diet at 6 weeks of age to induce pressure-overload pathologic cardiac hypertrophy and subsequent heart failure. Animals developed pathologic cardiac hypertrophy by 12 weeks of age (6 weeks of HS diet) and clinical evidence of heart failure following 13–15 wk of HS diet [11,12]. All animals died after 15–17 weeks of HS diet. Age-matched sedentary animals fed normal rat chow served as control animals. Immediately after animals were sacrificed, left ventricle tissues were dissected and frozen in liquid nitrogen and stored at −80°C until used for analyses.
Echocardiography
Echocardiography was performed according to previously described protocol [11,12]. Rats were anesthetized with an intraperitoneal injection of ketamine HCl (50 mg/kg) and xylazine (10 mg/kg). Echocardiography was performed in prone decubitus position with a Hewlett-Packard Sonos 1500 sector scanner equipped with a 7.5 MHz phased-array transducer. Anterior and posterior wall thickness and LV internal dimensions were measured according to the leading-edge method of the American Society of Echocardiography.
Semi-quantitative RT-PCR analyses
Total RNA was extracted from the left ventricle myocardium with TRIzol Reagent (Invitrogen Life Technologies, Bethesda, MD). Frozen ventricle muscle was mechanically homogenized on ice in 1 ml of ice-cold TRIzol. Total RNA was solubilized in DEPC-treated RNase-free H2O and quantified in duplicate by measuring the optical density (OD) at 260 nm. Purity of RNA was assured by examining the OD260/OD280 ratio. Five micrograms of RNA was reverse transcribed by using Superscript First-strand Synthesis System (Invitrogen Life Technologies, Bethesda, MD) with oligo(dT) primers and Superscript II reverse transcriptase (RT) in a total volume of 20 μl according to standard methods. Control RT reaction was performed in which the RT enzyme was omitted and the control RT reaction was PCR amplified to ensure that DNA did not contaminate the RNA. One microliter of complementary DNA (cDNA) was then amplified by PCR using 2 pmol of forward and reverse primers of the gene of interest, 1 pmol of ribosomal 18S forward and reverse primers, 200 μM deoxyribonucleotide triphosphates (dNTPs), 1 x PCR buffer, and 1 unit of Taq DNA polymerase (Promega, Madison, WI) in a final volume of 20 μl. PCR was performed using a peltier thermocycler (PTC-200, Scientific Support Inc., Hayward, CA). PCR reactions were performed at an annealing temperature of 60°C for 30 cycles. Preliminary experiments were conducted with each gene to assure that the number of cycles represented a linear portion of the PCR optical density curve for the samples. The primer pairs were designed from sequences published in GenBank (Table 1) and PCR products were verified by restriction enzyme digestions. The cDNA from all samples were amplified simultaneously using aliquots from the same PCR mixture. After PCR amplification, reaction product was electrophoresed on 1.2% agarose gels and stained with ethidium bromide. Images were captured and the signals were quantified in arbitrary units as optical density (OD) x band area using ImageJ image analysis system. Ribosomal 18S primers acted as internal controls and all RT-PCR signals were normalized to the 18S signal of the corresponding RT product to eliminate the measurement error from uneven sample loading and provide a semi-quantitative measure of the relative changes in gene expression.
Table 1.
Primers used for PCR amplification of cDNA
| Product | GenBank accession # | Sequence | Product length, bp | Restriction enzyme | Restriction products, bp |
|---|---|---|---|---|---|
| AIF | AF375656 | Forward: 5′-GCCATTGCCTCTGCTGCAGAAG-3′
Reverse: 5′-GCCTCCCGTTGCAATCAAGCAC-3′ |
450 |
BamHI
BbsI |
321, 129
290, 160 |
| EndoG | AB221075 | Forward: 5′-AGTGCCAGTGTTGCCGGTTGTC-3′
Reverse: 5′-AGGCATGCTGGTTGAGGTGTGG-3′ |
410 |
NotI
PvuII |
319, 91
228, 182 |
| HtrA2/Omi | AA926045 | Forward: 5′-TGTGTTCTTCAGAGCCCAGGACTGC-3′
Reverse: 5′-CTACAGCTCCGAGAGCCAAGTTTCC-3′ |
402 |
BamHI
PvuII |
213, 189
226, 176 |
| 18S | M11188 | Forward: 5′-GTTATGGTTCCTTTGTCGCTCGCTC-3′
Reverse: 5′-TCGGCCCGAGGTTATCTAGAGTCAC-3′ |
209 | DpnI | 124, 85 |
bp= base pair.
Western immunoblot analyses
Protein abundances of AIF, EndoG, and HtrA2/Omi were examined in the mitochondrial and cytosolic S-100 fractions. Protein samples were boiled for 5 min at 95°C in loading buffer, loaded on each lane of a 12% polyacrylamide gel and separated by SDS-PAGE. The gels were blotted to methanol-treated PVDF membranes (Millipore, Bedford, MA) and stained with Ponceau S red (Sigma Chemical Co, St Louis, MO) to verify equal loading and transferring of proteins to the membrane in each lane. The membranes were blocked in 5% non-fat milk in Tris buffered saline with 0.05% Tween 20 (TBS-T) at room temperature for 1 h and then probed with an anti-AIF rabbit polyclonal antibody (BD Pharmingen, San Diego, CA), anti-EndoG rabbit polyclonal antibody (ProSci, Poway, CA), or anti-HtrA2/Omi rabbit polyclonal antibody (R&D, Minneapolis, MN) diluted in TBS-T with 2% milk at 4°C overnight. Secondary antibodies conjugated to horseradish peroxidase (Jackson Immuno Research, West Grove, PA) were used to detect the signals using Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Science, Boston, MA). The molecular sizes of the proteins were verified by using Precision Plus pre-stained standard (Bio-Rad, Hercules, CA). COXIV and GAPDH were probed in the blots as internal controls for loading. The signals were then visualized by exposing the membranes to X-ray films. Resulting bands were quantified as optical density (OD) × band area by ImageJ image analysis system and recorded in arbitrary units.
Subcellular protein fractionation
The fractionation method described by Clark et al. [15] was adopted with minor modifications to extract the mitochondrial and cytosolic S-100 fractions from frozen ventricular tissues. Briefly, ventricle muscle was homogenized on ice with 100 strokes of a glass homogenizer in ice-cold lysis buffer (10 mM NaCl, 1.5 mM MgCl2, 120 mM HEPES, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, and 250 mM sucrose) supplemented with a protease inhibitor cocktail. Following centrifuging at 1,050 g for 15 min at 4°C to pellet the nuclei and cell debris, the supernatants were collected and further centrifuged three times at 1,050 rpm for 15 min at 4°C to remove residual nuclei. The supernatants were then centrifuged at 15,000 g for 20 min at 4°C, and the resulting mitochondrial pellets were washed, resuspended, and stored as the mitochondrial protein fraction. The supernatants from the 15,000 g spin were then ultra-centrifuged at 100,000 g for 1 h at 4°C, and the final supernatants were stored as the pure S-100 cytosolic protein fraction. The purity of the extracted protein fractions was assessed by immunoblots with an anti-GAPDH (a cytosolic protein; RDI, Flanders, NJ) and anti-COXIV (a mitochondrial protein; Molecular Probes, Eugene, OR) antibody. The protein contents of the extracts were quantified in duplicate by Protein Assay (BioRad, Hercules, CA) based on the method of Bradford. Protein measurements were performed at 595 nm with a spectrophotometer.
Immunofluorescent and TUNEL staining
Frozen 5 μm-thick ventricle muscle cross sections from all groups were cut in a freezing cryostat at −20°C and placed on the same glass slide to control for processing differences (e.g., incubation time, temperature, etc.). The sections were air dried at room temperature, fixed in 4% paraformaldehyde in PBS, pH 7.4 at room temperature for 20 min, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate at 4°C for 3 min, and incubated with fluorescein-conjugated TUNEL reaction mixture (Roche Applied Science, Indianapolis, IN) in a humidified chamber at 37°C for 1 h in the dark. Positive control experiments were performed by treating the sections with DNaseI before the TUNEL reaction mixture incubation, whereas negative control experiments were done by omitting the TdT enzyme in the TUNEL reaction mixture on the tissue sections. The sections were then blocked in 10% donkey serum in PBS at room temperature for 30 min following permeabilization with 0.5% Triton in 0.02% saponin/PBS for 10 min. After washes in PBS, sections were incubated with an anti-AIF rabbit polyclonal antibody overnight at 4°C followed by an anti-rabbit IgG rhodamin conjugated secondary antibody incubation (Sigma, St Louis, MO) for 1 h at 4°C. Negative control experiments omitted the AIF antibody from the tissue sections. The sections were then incubated with To-Pro-3 staining solution (Molecular Probes, Eugene, OR) to visualize nuclei, and finally mounted with mounting medium (Vector Laboratories, Burlingame, CA). TUNEL, To-Pro-3 nuclear, and AIF staining were examined under a confocal fluorescence microscope (BioRad 1024 with Nikon E800). Images were obtained and stacked using a digital camera and software (Lasersharp 2000).
Statistical analyses
Statistical analyses were conducted using one-way ANOVA and Tukey’s tests for post hoc differences between group means. Statistical significance was accepted at P < 0.05. Data are given as means ± standard error of mean (SE).
RESULTS
Animal models of cardiac hypertrophy and heart failure
There was significant cardiac hypertrophy in both exercise (EX) and HS diet groups at 12 weeks of age (6 weeks after HS diet) compared to the age matched control. Echocardiographic analysis showed a significant increase in anterior wall thickness of the EX group and the HS group (Table 2). However, neither group had significant changes in fractional shortening consistent with cardiac hypertrophy with preserved cardiac function. Continuation of the HS diet resulted in a further increase in wall thickness and LV chamber diameter that progressed to a decrease in fractional shortening consistent with hypertensive dilated cardiomyopathy (Table 2). These rats also demonstrated clinical evidence of heart failure and ultimately death. Pathological examination of EX and HS rats at 12 weeks of age showed significant increases in heart weight without significant changes in other organ weights (Table 3). Again, with continued HS diet, there were significant decreases in body weight due to cardiac cachexia, a nearly two-fold increase in heart weight, and significant increases in the lung and liver weights consistent with left- and right-sided heart failure, respectively (Table 3).
Table 2.
Echocardiographic findings
| C6 | C15 | E6 | H6 | H15 | |
|---|---|---|---|---|---|
| HR (beats/min) | 453 (+/− 27) | 483 (+/− 22) | 469 (+/− 35) | 459 (+/− 27) | 382 (+/− 9)* |
| AWd (mm) | 1.8 (+/− 0.1) | 1.7 (+/− 0.1) | 2.2 (+/− 0.1)* | 2.3 (+/− 0.1)* | 2.5 (+/− 0.1)* |
| PWd (mm) | 1.7 (+/− 0.1) | 1.6 (+/− 0.1) | 2.3 (+/− 0.1)* | 2.1 (+/− 0.1)* | 2.3 (+/− 0.1)* |
| LVDd (mm) | 4.9 (+/− 0.1) | 4.8 (+/− 0.2) | 4.8 (+/− 0.1) | 4.5 (+/− 0.1) | 5.6 (+/− 0.3)* |
| LVDs (mm) | 2.4 (+/− 0.3) | 2.4 (+/− 0.2) | 2.0 (+/− 0.2) | 2.0 (+/− 0.2) | 4.3 (+/− 0.1)* |
| FS (%) | 52 (+/− 5) | 49 (+/− 3) | 61 (+/− 3) | 58 (+/− 4) | 33 (+/− 2)* |
C6=control at 6 weeks, C12=control at 12 weeks, E6=exercise for 6 weeks, H6=HS diet for 6 weeks, H15=HS diet for 15 weeks. HR=heart rate, AWd=anterior wall thickness in diastole, PWd=posterior wall thickness in diastole, LVDd=left ventricular dimension in diastole, LVDs=left ventricular dimension in systole, FS=fractional shortening.
p<0.05; N=4.
Table 3.
Pathological findings
| C6 | C15 | E6 | H6 | H15 | |
|---|---|---|---|---|---|
| Body weight (g) | 211 (+/− 7) | 225 (+/− 2) | 224 (+/− 2) | 227 (+/− 1) | 198 (+/− 3)* |
| Heart weight (g) | 0.75 (+/− 0.01) | 0.77 (+/− 0.01) | 0.92 (+/− 0.01)* | 1.09 (+/− 0.03)* | 1.48 (+/− 0.12)# |
| Lung weight (g) | 1.5 (+/− 0.1) | 1.5 (+/− 0.1) | 1.5 (+/− 0.1) | 1.3 (+/− 0.1) | 2.4 (+/− 0.6)* |
| Liver weight (g) | 6.8 (+/− 0.1) | 7.0 (+/− 0.1) | 8.0 (+/− 0.5) | 10.2 (+/− 0.4)* | 10.6 (+/− 0.4)* |
| TL (mm) | 32 (+/− 0.7) | 35 (+/− 1.0) | 33 (+/− 1.0) | 36 (+/− 0.3)* | 37 (+/− 0.3)* |
| HW/BW (mg/g) | 3.5 (+/− 0.1) | 3.4 (+/− 0.1) | 4.1 (+/− 0.1)* | 4.8 (+/− 0.2)* | 7.4 (+/− 0.7)# |
| HW/TL (mg/mm) | 23.3 (+/− 0.5) | 21.9 (+/− 0.3) | 28.4 (+/− 1.0)* | 30.6 (+/− 0.9) * | 40.5 (+/− 3.0)# |
C6=control at 6 weeks, C12=control at 12 weeks, E6=exercise for 6 weeks, H6=HS diet for 6 weeks, H15=HS diet for 15 weeks. TL=tibial length, HW/BW=heart weight to body weight ratio, HW/TL=heart weight to tibial length ratio.
p<0.05,
p<0.01; N=4.
Total endogenous and subcellular protein levels
We found that there were high endogenous protein levels of AIF, EndoG, and HtrA/Omi in whole adult cardiac tissue (Figure 1). AIF was also expressed abundantly in the liver and to a lesser degree in skeletal muscle. EndoG was expressed at high levels in the heart and the liver. HtrA2/Omi was ubiquitously expressed in all tissues examined, but a high expression level of the processed form was observed specifically in heart (Figure 1).
Figure 1. Endogenous level of AIF, EndoG and HtrA2/Omi in different organs.

The representative Western blot of AIF, EndoG, and HtrA2/Omi from rats. Sm, skeletal muscle; Hr, heart; Br, brain; Lu, lung; Li, liver; Sp, spleen.
Assessment of the purity of the extracted protein fractions by anti-GAPDH (a cytosolic protein) and anti-COXIV (a mitochondrial protein) antibodies revealed good subcellular fractionation (Figure 2A). Western immunoblot analyses on the extracted fractions demonstrated that the protein abundances of AIF, EndoG and HtrA2/Omi in mitochondria, the main residing pool of these caspase-independent factors, were not different in the ventricular samples of the failed or exercise-trained animals when compared to the control animals (Figure 2B). However, the results from the S-100 cytosolic fraction showed that, in the failed ventricular sample, AIF protein content was markedly elevated in the cytosolic portion (Figure 2C and 2D). Since AIF is normally housed inside the mitochondria, this observation suggested that there was a release of AIF from mitochondria to cytosol in our HS diet-induced failed heart. There were no significant increases in cytosolic AIF in either physiologic or pathologic hypertrophied hearts. Furthermore, the cytosolic protein abundance of HtrA2/Omi was found to increase in only the failed heart, and not in either form of cardiac hypertrophy. The increase reached the level of statistical significance for the processed HtrA2/Omi but not precursor HtrA2/Omi (Figure 2C and 2D). EndoG was not significantly increased in any group compared to the control heart.
Figure 2. Subcellular localization of AIF, EndoG, and HtrA2/Omi.
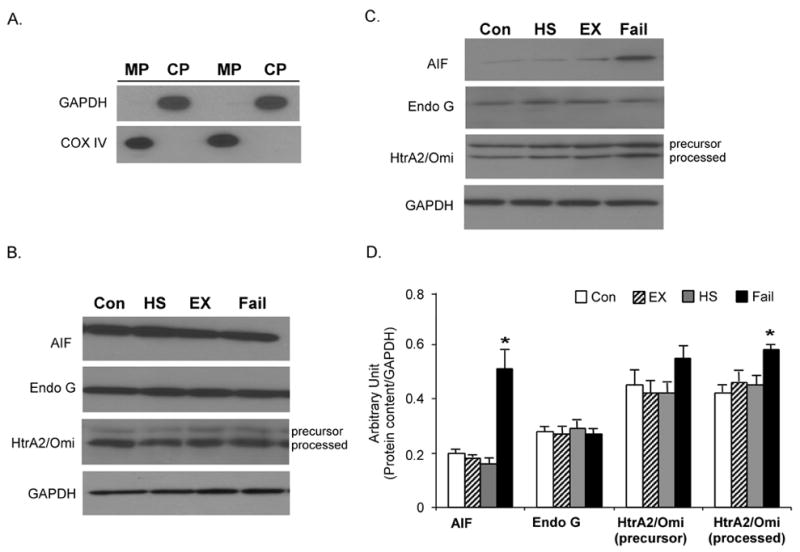
A. Verification of the protein fraction purity. The purity of the extracted protein fractions was examined by immunoblot of mitochondrial protein (MP) and cytosolic protein fractions (CP) probed with anti-COXIV and anti-GAPDH antibodies. The mitochondrial and cytosolic protein fractions were extracted from a same ventricle muscle and equal amount of protein was employed in the immunoblot. B. Mitochondrial protein content of AIF, EndoG, and HtrA2/Omi. The protein content of AIF, EndoG, and HtrA2/Omi was measured by Western immunoblot in the mitochondria fraction. The insets show representative blots. Con, control; Ex, exercise; HS, high salt; Fail, failed heart. C. Representative Western immunoblot of AIF, EndoG, and HtrA2/Omi in the mitochondria-free cytosolic fraction. GAPDH was used as an internal control. D. Quantification of cytosolic protein content of AIF, EndoG, and HtrA2/Omi. The data are presented as means ± SE. *P < 0.05, data are significantly different from control animals.
Relationship between AIF subcellular localization and apoptosis
In order to examine the relationship between AIF subcellular localization and apoptosis, we performed co-immunofluorescent staining for AIF and TUNEL. The immunostaining pattern of AIF in control heart tissue showed punctuated AIF staining suggesting predominately mitochondrial localization at baseline (Figure 3A). In contrast, failed heart demonstrated a more diffused immunostaining pattern for AIF (Figure 3B). There was also rare, but apparent, co-localization of the AIF staining and the TUNEL-positive nuclei suggesting the incidence of nuclear translocation of AIF in the failed heart sample (Figure 3B, 3D, 3F, 3H).
Figure 3. AIF immunofluorescent staining.
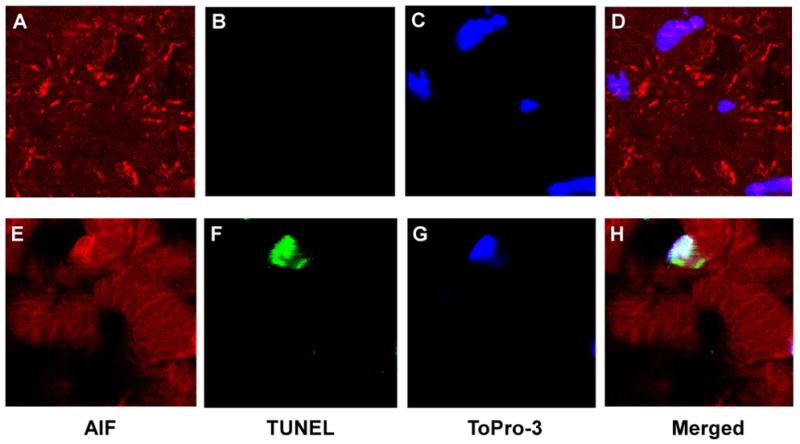
Immunofluorescent labeling of AIF was performed simultaneously with TUNEL staining. AIF (red) stained with secondary rhodamin, TUNEL (green) stained with fluorescein, and nuclei (blue) labeled using To-Pro-3. Image was obtained using ×2000 magnification.
Expression of mRNA contents
Our semi-quantitative RT-PCR analyses indicated that the amounts of mRNA transcript for AIF and EndoG in the failed heart were reduced relative to the control heart (Figure 4). Again, neither type of hypertrophied heart showed any significant change compared to the control . For HtrA2/Omi mRNA content, there was no significant change in all four groups examined.
Figure 4. mRNA content of AIF, EndoG, and HtrA2/Omi.
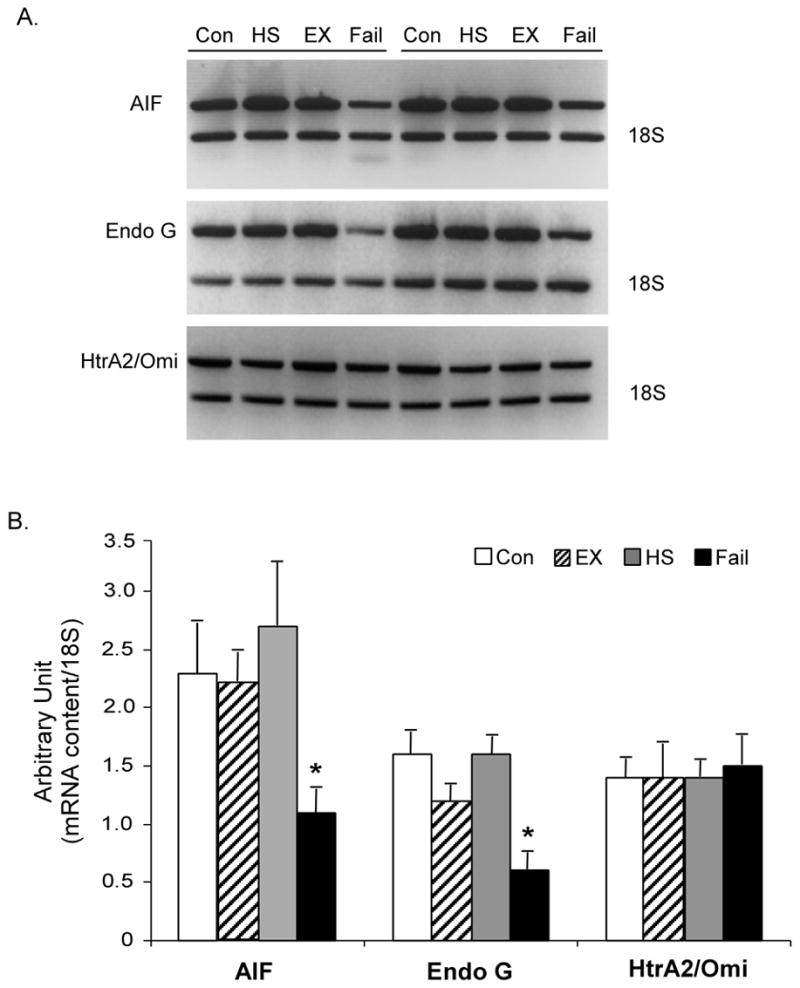
A. Representative semi-quantitative RT-PCR of AIF, EndoG, and HtrA2/Omi. 18S RNA is shown for internal control. B. Quantification of the mRNA expression of AIF, EndoG, and HtrA2/Omi. The data are presented as means ± SE. *P < 0.05, data are significantly different from control animals.
DISCUSSION
Although the presence of apoptosis during heart failure has been clearly demonstrated [1,16], the exact mechanisms responsible for the activation of the apoptotic signaling pathway leading to the death of cardiomyocytes remain to be fully identified. In this study, we have provided descriptive evidence suggesting that caspase-independent apoptotic events are also activated in the late stage of HS-induced heart failure and are involved in the regulation of cardiac apoptosis. Our previous study showed that the caspase-dependent mechanism may be activated in pathologic cardiac hypertrophy, but not in physiologic hypertrophy [11]. Since no significant increase in the caspase-independent cascade was observed in pathologic cardiac hypertrophy, our current findings suggest that activation of caspase-independent pathway likely occurs later in the development of heart failure. Furthermore, we demonstrated that apoptotic factors AIF and HtrA2/Omi were accumulated in the cytosol in heart failure. The incidence of mitochondrial release and nuclear translocation of AIF in the failed heart seemed to be supported by the result of TUNEL/immunofluorescent staining. These findings are consistent with the hypothesis that caspase-independent apoptotic factors contribute to the activation of cardiac apoptosis in heart failure. Another notable finding in this study is that the mRNA level of AIF and EndoG were markedly decreased in the failed heart. This finding supports our previous observation of secondary anti-apoptotic changes or activation of the survival gene program in the very end stage of heart failure [11].
In this study, we first revealed relatively high endogenous protein level of AIF, EndoG, and HtrA2/Omi in the heart compared to other organs. This leads to the speculation that these factors might have certain physiologic functions in mature cardiomyocytes. Caspase-independent apoptotic signaling is principally driven by these specific, mitochondrial apoptotic factors that are released to cytosol in response to apoptotic stimulation. Thus, the accumulation of AIF and HtrA2/Omi in the cytosolic fraction of failed heart lead to a suggestion that the caspase-independent machinery may have contributed to the execution of apoptosis during heart failure. Indeed, there have been a few studies that examined the role of caspase-independent factors in different cardiac pathological conditions. Liu and colleagues [17] demonstrated that ischemia-reperfusion results in translocation of HtrA2/Omi from mitochondria to cytosol in the mouse heart. By using a specific HtrA2/Omi inhibitor (ucf-101), the important role of HtrA2/Omi in cardiac ischemia-reperfusion injury was exhibited based on the improvements of TUNEL-mediated apoptosis staining, incidence of DNA ladder fragmentation, and infarct size following the inhibition of HtrA2/Omi [17]. In a recent study conducted by Bahi and coworkers [18], the release of EndoG together with cytochrome c and AIF from mitochondria to cytosol in rat postnatal differentiated cardiomyocytes was shown to be induced by cardiac ischemia. By knocking down the expression of EndoG using lentivirus-mediated shRNA, they demonstrated the essential role of EndoG in the ischemia-induced caspase-independent DNA degradation/processing in differentiated neonatal cardiomyocytes [18].
Compared to EndoG and HtrA2/Omi, AIF has been relatively well studied. However, the role of AIF in cardiac cell death is still unclear since it has been shown in the literature that AIF may function as both pro-apoptotic and pro-survival factor. AIF was first discovered as a mitochondrial effector of apoptotic cell death working independently of the caspase machinery [19]. With a focus on the pro-apoptotic role of AIF in cardiovascular diseases, the involvement of AIF have been consistently demonstrated in several different cardiac cell death settings, as, for example, in ischemia-reperfusion, oxidative stress (i.e., hydrogen peroxide) exposure in vitro, and heart failure induced by transverse aortic constriction/banding [20–22]. Kim and colleagues [21] demonstrated the accumulation of AIF in the cytosolic and nuclear fractions of heart following ischemia-reperfusion whereas AIF was found to be confined only in mitochondria in control heart. They also reported that the ischemia-reperfusion-induced release of AIF and the extent of DNA fragmentation were attenuated by ischemic precondition. Chen and coworkers [20] showed that, in murine cardiomyocytes, challenge with hydrogen peroxide resulted in the release of cytochrome c and mitochondrial-to-nuclear translocation of AIF. By comparing wild-type and PARP-1-deficient myocytes, they also provided evidence showing that PARP-1 might directly or indirectly be involved in the regulation of AIF translocation subjected to oxidative stress [20]. By using a mouse heart failure model induced by transverse aortic banding, the mitochondrial-to-nuclear translocation of AIF was also exhibited while the extent of translocation was attenuated by inhibition of the activation of PARP-1 either through the isoindolinone-based PARP inhibitor (INO-1001) or PARP-1 genetic deficient mice [22]. While these data have related AIF to the incidence of cardiac apoptosis, it is worthy noting that PARP-1 has been suggested to play an important mediating role in the regulation of the release and translocation of AIF and therefore subsequent activation of caspase-independent apoptosis in ischemia-reperfusion and heart failure [23,22]. Although the exact role of PARP-1/AIF in cardiac apoptosis is not fully understood, it has been suggested that energy deficit (i.e., depletion of NAD+and ATP) resulting from over-activation of PARP-1 due to oxidative stress-induced DNA damage might be one of the events which initiates the release and translocation of AIF during ischemia-reperfusion [23]. Nevertheless, the role of PARP in cardiac caspase-independent apoptosis was not investigated in the present high salt diet-induced heart failure model.
In regard to the pro-survival role of AIF, there has been some evidence indicating that AIF may have an important role in mitochondrial respiration or even act as a cardiac antioxidant. Deletion of AIF by homologous recombination or the suppression of AIF expression by siRNA resulted in high lactate production and increased dependency on the glycolytic energy generating process due to a remarkable reduction of respiratory chain complex I activity [7][24]. Joza et al. showed that animals that were cardiac and skeletal muscle-specifically deficient in AIF developed severe dilated cardiomyopathy, heart failure, and skeletal atrophy accompanied by impaired activity and protein expression of respiratory chain complex I, defective mitochondrial respiratory function, a metabolic switch toward glucose metabolism, and lactic acidosis. Van Empel et al. [25] demonstrated that cardiomyocytes isolated from Hq mice, an AIF-deficient mouse mutant, were more sensitive to hydrogen peroxide-induced cell death. The Hq hearts also showed more damage from ischemia-reperfusion and aortic banding-induced heart failure. They provided data showing that AIF deficiency reduced the ability of cardiac mitochondria to decompose hydrogen peroxide, suggesting that AIF may function as a cardiac antioxidant independent of mitochondrial complex activity [25]. A subsequent study [26] substantiated the suggestion that AIF functions as an important cardiac free radical scavenger by showing that EUK-8, a superoxide dismutase and catalase mimetic, protected against pressure overload-induced heart failure in AIF-deficient Hq mice. Nonetheless, further investigation is needed to fully understand the ambiguous role of AIF in the development of heart failure.
According to the data indicating that the caspase-independent machinery was activated in the end stage of heart failure, it was expected that the transcriptional profile of caspase-independent apoptotic factors would be elevated. However, surprisingly, our RT-PCR analyses indicated that the transcript abundance of AIF and EndoG in the failed heart was noticeably reduced relative to the control heart, while that of HtrA2/Omi was unchanged. Our fractionated protein data suggest that high salt diet-induced heart failure is associated with the subcellular redistribution of caspase-independent factors as shown by the accumulation of protein abundance of AIF and HtrA2/Omi in the mitochondria-free cytosolic fraction in the failed heart. However, it is noted that no difference was found in the protein content of AIF, EndoG and HtrA2/Omi in the mitochondrial fraction. Since mitochondrion is the major site for the existence of these proteins, it appears that these caspase-independent factors do not respond by changing the overall protein expression in the late developmental stage of heart failure, at least at the time point being examined in the present study. To explain the discrepancy of the mRNA and protein abundances, we speculate that there might be a time lag between the responses of transcript and protein expression and thus consistency was not picked up at our selected examination time point. We interpret this transcriptional response as a molecular adaptation of a secondary switch to a pro-survival milieu in the very end stage of heart failure. Indeed, the reduction of caspase-independent apoptotic factors mRNA level in the present study is in agreement with the previously reported anti-apoptotic changes including decreases in FAS, caspase-2, caspase-3 and Bax and increases in Bcl-2, Bcl-xL and XIAP during the late stage of HS-induced heart failure [11]. Nonetheless, the exact physiological function of these anti-apoptotic changes is still largely unknown.
In conclusion, we have provided descriptive evidence indicating that caspase-independent apoptotic factors are associated with activation of the apoptotic program in HS diet-induced heart failure, a reliable animal model in reproducing clinical evidence of heart failure. Our data shows the activation of caspase-independent events including the release of apoptotic factors AIF and HtrA2/Omi from mitochondria to cytosol and AIF nuclear translocation during the end stage of HS diet-induced heart failure. Nevertheless, additional research is needed to fully understand the precise mechanisms of caspase-independent signaling in the activation of cardiac apoptosis in heart failure. Based on the previously reported early activation of caspase-dependent events in pathologic cardiac hypertrophy and the present result that no significant elevation of the caspase-independent factors was observed in pathologic cardiac hypertrophy, it appears that activation of caspase-independent pathway likely occurs later in the development of heart failure when compared to caspase-dependent pathway. However, the exact time frame of the activation of caspase-dependent and -independent cascades in contributing to the development of pathologic heart failure is still largely unknown. Further investigation aimed at inhibiting both caspase-dependent and -independent pathways in the setting of heart failure and other cardiac apoptosis-associated pathological situations is needed.
Acknowledgments
This study was supported in part by the grants from National Heart, Lung, and Blood Institute HL65742 and HL67091 (PMK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kang PM, Izumo S. Apoptosis and heart failure: A critical review of the literature. Circ Res. 2000;86(11):1107–13. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 2.Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92(2):139–50. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- 3.van Empel VP, Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67(1):21–9. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Blink E, Maianski NA, Alnemri ES, Zervos AS, Roos D, Kuijpers TW. Intramitochondrial serine protease activity of Omi/HtrA2 is required for caspase-independent cell death of human neutrophils. Cell Death Differ. 2004;11(8):937–9. doi: 10.1038/sj.cdd.4401409. [DOI] [PubMed] [Google Scholar]
- 5.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410(6828):549–54. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 6.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412(6842):95–9. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 7.Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, et al. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25(23):10261–72. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111(2):147–50. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]
- 9.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277(1):432–8. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004;11(2):208–16. doi: 10.1038/sj.cdd.4401343. [DOI] [PubMed] [Google Scholar]
- 11.Kang PM, Yue P, Liu Z, Tarnavski O, Bodyak N, Izumo S. Alterations in apoptosis regulatory factors during hypertrophy and heart failure. Am J Physiol Heart Circ Physiol. 2004;287(1):H72–80. doi: 10.1152/ajpheart.00556.2003. [DOI] [PubMed] [Google Scholar]
- 12.Kong SW, Bodyak N, Yue P, Liu Z, Brown J, Izumo S, et al. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats. Physiol Genomics. 2005;21(1):34–42. doi: 10.1152/physiolgenomics.00226.2004. [DOI] [PubMed] [Google Scholar]
- 13.Jin H, Yang R, Li W, Lu H, Ryan AM, Ogasawara AK, et al. Effects of exercise training on cardiac function, gene expression, and apoptosis in rats. Am J Physiol Heart Circ Physiol. 2000;279(6):H2994–3002. doi: 10.1152/ajpheart.2000.279.6.H2994. [DOI] [PubMed] [Google Scholar]
- 14.Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, et al. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. 2001;50(3):495–508. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- 15.Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74(2):740–53. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- 16.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336(16):1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 17.Liu HR, Gao E, Hu A, Tao L, Qu Y, Most P, et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111(1):90–6. doi: 10.1161/01.CIR.0000151613.90994.17. [DOI] [PubMed] [Google Scholar]
- 18.Bahi N, Zhang J, Llovera M, Ballester M, Comella JX, Sanchis D. Switch from caspase-dependent to caspase-independent death during heart development: essential role of endonuclease G in ischemia-induced DNA processing of differentiated cardiomyocytes. J Biol Chem. 2006;281(32):22943–52. doi: 10.1074/jbc.M601025200. [DOI] [PubMed] [Google Scholar]
- 19.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, Zsengeller Z, Xiao CY, Szabo C. Mitochondrial-to-nuclear translocation of apoptosis-inducing factor in cardiac myocytes during oxidant stress: potential role of poly(ADP-ribose) polymerase-1. Cardiovasc Res. 2004;63(4):682–8. doi: 10.1016/j.cardiores.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Kim GT, Chun YS, Park JW, Kim MS. Role of apoptosis-inducing factor in myocardial cell death by ischemia-reperfusion. Biochem Biophys Res Commun. 2003;309(3):619–24. doi: 10.1016/j.bbrc.2003.08.045. [DOI] [PubMed] [Google Scholar]
- 22.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, et al. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J Pharmacol Exp Ther. 2005;312(3):891–8. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- 23.van Wijk SJ, Hageman GJ. Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic Biol Med. 2005;39(1):81–90. doi: 10.1016/j.freeradbiomed.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 24.Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, et al. AIF deficiency compromises oxidative phosphorylation. Embo J. 2004;23(23):4679–89. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Empel VP, Bertrand AT, van der Nagel R, Kostin S, Doevendans PA, Crijns HJ, et al. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96(12):e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- 26.van Empel VP, Bertrand AT, van Oort RJ, van der Nagel R, Engelen M, van Rijen HV, et al. EUK-8, a Superoxide Dismutase and Catalase Mimetic, Reduces Cardiac Oxidative Stress and Ameliorates Pressure Overload-Induced Heart Failure in the Harlequin Mouse Mutant. J Am Coll Cardiol. 2006;48(4):824–32. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]