

Abstract
Mild or low doses of oxidants are known to prime cells towards resistance against further damage. In cardiomyocytes, we found that pretreatment with 100 μM H2O2 prevents the cells from apoptosis induced by doxorubicin (Dox). Affymetrix microarray analyses of 28,000 genes reveal that H2O2 treated cells reduced expression of genes encoding cytochrome c, mitochondrial complex I, III, IV and V, and several contractile proteins. Elevated expression of antioxidant and detoxification genes appears as a dominant feature of the gene expression profile of H2O2 treated cells. Most of the genes in this category contain an Antioxidant Response Element (ARE) in their promoters. Measurements of ARE promoter-reporter gene activity indicate a dose and time-dependent activation of the ARE by H2O2. Since the Nrf2 transcription factor regulates ARE-mediated gene expression, we overexpressed Nrf2 to test whether activation of Nrf2 is sufficient to induce cytoprotection. High levels of Nrf2 expression were achieved via adenovirus mediated gene delivery. Transduced Nrf2 was present in the nuclei and caused an increase in the expression of NAD(P)H:quinone oxidoreductase 1 (NQO1), a representative downstream target of Nrf2. Unlike H2O2 pretreated cells, the cells expressing high levels of Nrf2 were not resistant to Dox-induced apoptosis. Therefore the cytoprotective effect of H2O2 pretreatment is not reliant upon Nrf2 activation alone as measured by resistance against Dox induced apoptosis.
Introduction
Substantial evidence supports the theory that oxidative stress plays an important role in heart failure. Oxidative metabolites can be detected in cardiac patients during angina and after emergency reperfusion procedures (1, 2). An increase in lipid peroxidation products has been found in heart failure patients and the level of this increase correlates with the severity of heart failure (3). Oxidative biomarkers have been detected in various experimental models of heart failure (4, 5). Paradoxically, elevated expression of antioxidant enzymes has been found in early stage heart failure (6-8). Recent studies have challenged the dogma that oxidants are detrimental and antioxidants can prevent or delay heart failure. While epidemiology studies found that dietary intakes rich in antioxidant vitamins are protective against cardiovascular disease (2, 9, 10), clinical trials of antioxidant vitamins have not yielded clear positive findings (11-13). Miller, et al. (14) showed recently that high doses of vitamin E supplement caused an increase in mortality, suggesting that antioxidant vitamins at high doses may even be harmful.
Adding to the complexity of oxidant paradox is the well-known phenomenon of preconditioning. In experimental animals, a brief period of ischemia usually produces two “windows” of protection: one at 2-3 hrs and one at 24-96 hrs after the initial stress (15-17). This preconditioning phenomenon has been linked to upregulation of cytoprotective enzymes such as superoxide dismutase (SOD) (18, 19). There is evidence that oxidants derived from the initial mild stress are responsible for this adaptation (20-23). A number of antioxidant and detoxification genes, including SOD, have been shown to be under the control of Nuclear Factor Erythroid-2 Related Factor 2 (Nrf2) through its interaction with the Antioxidant Response Element (ARE) in the promoter of these genes (24, 25). Nrf2, a bZIP transcription factor, is activated by various chemical or electrophilic stressors in many cell types. Activated Nrf2 forms a heterodimer with a partner for binding to the ARE (26-30). In cardiomyocytes, whether oxidants activate Nrf2 and the role of Nrf2 in stress response have not been addressed.
Recent development in microarray technology allows us to systematically evaluate the biological consequence of oxidative stress on the scale of the whole genome. The human genome project predicts that about 30,000 genes are expressed in a given cell type (31, 32). The Affymetrix microarray technique allows us to simultaneously measure levels of 28,000 transcripts. With microarray technology, one can address the questions of how many genes and what genes alter their expression pattern when cells encounter oxidative stress without the bias of prior knowledge. Functional genomics creates the opportunity to identify the network of genes changed by oxidants and to predict a centralized controller, such as a transcription factor, driving the expression of a cluster of genes within the network. This transcription factor serves as a target for testing whether activation of the cluster of genes is sufficient for the observed biological event of oxidative stress.
Apoptosis plays an important role in various forms of cardiac diseases, including heart failure. Doxorubicin (Dox), an anthracycline quinone commonly used as a cancer chemotherapeutic agent, is known to induce cardiomyopathy in subjected human populations and in experimental animals (33). Dox can produce oxidants by undergoing redox cycling and by reacting with enzymes of mitochondrial respiration (34, 35). In addition to producing reactive oxygen species, Dox is a DNA topoisomerase II inhibitor and a DNA interchelator (33, 36). With cardiomyocytes or other types of cells in culture, Dox serves as a reliable inducer of apoptosis. This system allows us to test whether oxidants or transcription factors activated by oxidants can protect cells from apoptosis.
Materials and Methods
Tissue Culture and H2O2 treatment
Ventricular cardiomyocytes (CMCs) and heart fibroblasts (HFs) were derived from the hearts of 1-2 day old Sprague-Dawley rats. CMCs were seeded in low glucose Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 100 units/mL penicillin G and 100 units/mL streptomycin. At the time of H2O2 treatment, over 90% of CMCs express myosin heavy chain (37, 38). HFs were retained by differential plating during the preparation of CMCs as described (39). HFs were cultured in high glucose DMEM containing 10% FBS in 100 mm dishes. Since HFs grow voluntarily, unlike CMCs or endothelial cells, the cells were subcultured once to reduce the possible contamination of CMCs and endothelial cells. The second passage of HFs was used for each experiment. Nearly all cells in the HF preparation express vimentin (a marker of fibroblasts), but not desmin (a marker for CMCs) or alpha-von Willebrand factor (a marker for endothelial cells) (39).
CMCs or HFs were placed in DMEM containing 0.5% FBS for 24 hrs from the 4th day of plating. Serum-starved cells were treated with 100 μM H2O2 for 1 hr or as indicated, followed by medium change to fresh 0.5% FBS DMEM to avoid nutrient deprivation due to oxidation of the medium by H2O2. The cells were recovered 24 hrs before Dox treatment or various measurements.
Caspase Assay
Detached cells were collected and combined with adherent cells for lysis in 200 μL buffer (0.5% Nonidet P-40, 0.5 mM EDTA, 150 mM NaCl, and 50 mM Tris, pH 7.5). Protein concentrations were determined using the Bradford assay (BioRad, Hercules, CA) for correcting caspase-3 activity per protein content. Caspase-3 activity was measured using the substrate of 40 μM N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Nacetyl-DEVD-AMC; Alexis Biochemicals, San Diego, CA). The resulting product AMC was measured using a 96-well fluorescence plate reader (Spectra Max Gemini XS, Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 365 nm and an emission wavelength of 450 nm.
Annexin V Staining
Cells were seeded onto coverglasses in 6-well plates. Detached cells in the supernatant were collected and combined with their corresponding group of cells remaining adherent to the coverglass. A drop (6 μl) of Annexin V-FLUOS (Roche Applied Science, Indianapolis, IN) in the labeling solution (10 mM HEPES/NaOH, Ph.7.4, 140 mm NaCl, 5 mM CaCl2) was added to the coverglasses. The images of Annexin V labeled cells were acquired under a Nikon E800m fluorescent microscope using a Hamamatsu C5180 digital camera.
RNA Isolation and Microarray Analysis
Cells were rinsed twice with PBS and harvested in TRIzol Reagent (Invitrogen) for total RNA extraction. The RNA was purified using an RNeasy kit from Qiagen before being processed and hybridized to Affymetrix RAE230A gene chips, one chip per sample as described previously (40). Raw data was analyzed using Microarray Suite 5.0.1. A cross-comparison of the control versus treated data from each experiment in silico lead to a total of four data sets for calculation of averages and standard deviations (40). Only genes that passed the criteria of being up or down-regulated by 1.2 fold or greater in three or four of the data sets, including two of which were the authentic experimental data sets, were judged as valid data and shown in the Results.
Gene Expression Network and Visual Display of Clustered Gene Expression Profile
Cytoscape 2.1 (www.cytoscape.org) was used in conjunction with Gene Ontology (GO) Molecular Function Categories of Affymetrix to visualize Microarray gene expression raw data. Each gene displaying a significant increase or decrease as determined by one representative Microarray analysis was categorized by its most specific Molecular Function in the GO tree. The GO categories were obtained from a batch query expression search of the Affymetrix database (www.affymetrix.com/analysis). A Java script removed each category containing fewer than 3 genes from the list if the genes in that category did not appear in any other GO Molecular Function categories. A Cytoscape network was then constructed from the remaining categories of genes. Each gene appears as a circular node on the network and is attached by a line (edge) to each GO Molecular Function category, labeled as a square or a diamond node. Categories and genes appearing in trials of both CMCs and HFs are shown with diamond nodes, bold labels and thick outlines to aid in visual comparison. Four major GO Molecular Function categories are displayed with colored lines to obviate the associated nodes. Each circular node is colored on a gradient representing the magnitude of Microarray measured fold change. If the fold change of the gene indicates down regulation, the node is colored in a blue gradient with white representing zero and saturated blue equivalent to 3 fold or more. If the gene is up regulated, it is colored on a red gradient with saturated red representing 3 fold or more.
RT-PCR
RNA was harvested as described above for reverse transcription using hexanucleotide random primers. PCR primers were designed using Primer 3 Input software according to the coding sequence of the genes and are presented in Table 1. PCR was carried out according to the temperatures and conditions recommended by the Oligonucleotide synthesis manufacturer (Integrated DNA technologies, Coralville, IA) with modification for optimal reaction.
Table 1.
Primers used for RT-PCR.
| Gene Name | Abbr | Primers |
|---|---|---|
| NAD(P)H dehydrogenase, quinone 1 | NQO1 | 5′-CATTCTGAAAGGCTGGTTTGA |
| 5′-CTAGCTTTGATCTGGTTGTCAG | ||
| Aldose reductase like protein | ARL | 5′-CGTCCTCCCCAGTAAAACAA |
| 5′-CTCTGGATGTGGAACCGAAT | ||
| Epoxide hydrolase 1 | Ephx1 | 5′-CGAGTTTGACTGGAGGAAGC |
| 5′-CTGGATGCCTCTGAGTAGCC | ||
| Microsomal glutatione S-transferase 2 | mGST2 | 5′-TGCAGTCTCCCTTCTGTGTG |
| 5′-CAGGAATCTGCTTGCTACCC | ||
| Microsomal glutatione S-transferase 1 | mGst1 | 5′-TCAAGCAGCTCATGGACAAC |
| 5′-GCAATGGTGTGGTAGATCCG | ||
| Glutathione S-transferase, pi 2 | GSTP2 | 5′-GGGCATCTGAAACCTTTTGA |
| 5′-AGGAGTTCCTGTCCCTTCGT | ||
| Transaldolase 1 | Taldo 1 | 5′-GTAAAACGCCAGAGGATGGA |
| 5′-CCCCAAACAGCACAAAAAGT | ||
| Monoamine oxidase A | MAOA | 5′-GCCAGGAACGGAAATTTGTA |
| 5′-TCTCAGGTGGAAGCTCTGGT | ||
| Cytochrome p450 1B1 | CYP1B1 | 5′-GAGCTCGCTGTCTACCCAAC |
| 5′-GATCTGAAAAACGTCGCCAT | ||
| Heme oxygenase-1 | HO1 | 5′-CACGCATATACCCGCTACCT |
| 5′-AAGGCGGTCTTAGCCTCTTC | ||
| UDP glycosyltransferase 1, A6 | UGT1A6 | 5′-TCTTCATTGGAGGGACCAAC |
| 5′-TTGGAACCCCATTGCATATT | ||
| Catalase | Cat | 5′-ACATGGTCTGGGACTTCTGG |
| 5′-CAAGTTTTTGATGCCCTGGT | ||
| Hydroxysteroid 11-beta dehydrogenase 1 |
HSD1 | 5′-AAAGCTTGTCACTGGGGCCAGCAAA |
| 5′-AGGATCCAGAGCAAACTTGCTTGCA | ||
| Aryl hydrocarbon receptor | Ahr | 5′-TGCGGGGCTCGAAAGAAGACAGAG |
| 5′-GGAGGTGGGTCCAGTCCAATGCAC | ||
| P-glycoprotein, multidrug resistance 1 | Pgy1 | 5′-AAAGCTGTCAAGGAAGCCAA |
| 5′-CAAGCGGTGAGCTATCACAA | ||
| Myosin heavy chain, polypeptide 4 | Myh4 | 5′-AGTGAGCAGAAGCGGAATGT |
| 5′-TGCCTCTCTTCGGTCATTCT | ||
| Myosin heavy chain, polypeptide 7 | Myh7 | 5′-CGCAACAGAGAACAAGGTGA |
| 5′-TCATCCAACTGCTGCTTGTC | ||
| Myosin, light polypeptide 3 | Myl3 | 5′-AATCCTACCCAGGCAGAGGT |
| 5′-GCATTATGGTTGGGAGATGG | ||
| Troponin I, type 1 | Tnni1 | 5′-TCATGCTGAAGAGCCTGATG |
| 5′-TGGACACCTTGTGTTTGGAA | ||
| Troponin I, type 3 | Tnni3 | 5′-TAAGATCTCCGCCTCCAGAA |
| 5′-AGAGTGGGCCGCTTAAACTT | ||
| Cytochrome c | CytC | 5′-AGACTCACCCGTGCTTCAGT |
| 5′-ACTCCCAATCAGGCATGAAC | ||
| cytochrome c oxidase subunit VIa polypeptide 2 |
Cox6a2 | 5′-CTGACCTTTGTGCTGGCTCT |
| 5′-TCACACCTTTATTGCGCTTG | ||
| cytochrome c oxidase subunit VIII-H |
Cox8h | 5′-ACATTCAGGGTGCCTCTTTG |
| 5′-CATGAAGCCAGCGACTATGA | ||
| mitochondrial NADH dehydrogenase 24kDa subunit |
Ndhase2 | 5′-CGTTCCCTGTCAGCCTAGAG |
| 5′-CACCTTGTTCATGGCAGAGA | ||
| Nf-E2 related factor-2 | Nrf2 | 5′-GCCAGCTGAACTCCTTAGAC |
| 5′-GATTCGTGCACAGCAGCA | ||
| glyceraldehyde-3-phosphate dehydrogenase |
GAPDH | 5′-AGACAGCCGCATCTTCTTGT |
| 5′-CCACAGTCTTCTGAGTGGCA |
Transfection and Luciferase Assay
ARE-luciferase reporter plasmid (0.2 μg) was cotransfected using Fugene 6 reagent (Roche) with a thymidine kinase promoter driven Renilla-luciferase plasmid (0.04 μg), which is used to correct for transfection efficiency. After 30 mins incubation at room temperature, Fugene/DNA mixtures were added to cells in 0% FBS DMEM for a 5-hrs incubation at 37°C. Cells were then placed in 10% FBS DMEM for an overnight recovery. At the end of treatments, cells were harvested in 1x Passive Lysis Buffer (Promega) For measurement of luciferase activity using a Dual Luciferase Assay System (Promega) and a Turner Designs Luminometer.
Western Blot Protocol
Laemmli lysis buffer [125 mM Tris, pH 6.8, 50% (v/v) glycerol, 2.4% (w/v) SDS, and freshly added 100 μM phenylmethylsulfonyl fluoride (PMSF) and 10 μg/mL aprotinin] was used for total protein extractions. Protein concentrations were determined using the Warburg–Christian method at an absorbance of 280 nm after adding 5% β-mercaptoethanol and boiling the samples (41). For cytosolic versus nuclear-enriched fractionation, cells were initially lysed with EB buffer (1% Triton X-100, 10 mM Tris, pH 7.4, 5 mM EDTA, 50 mM NaCl, 50 mM NaF, and freshly added 2 mM DTT, 1 mM Na2VO3, 1 mM PMSF, 100 ug/mL leupeptin and 10 μg/mL aprotinin). Samples were then centrifuged at 13,000 g at 4°C for 10 min, the supernatant was saved (cytosolic fraction) while the pellet was lysed in Laemmli lysis buffer. The lysate was then subjected to three rapid freeze-thaw cycles to obtain the nuclear-enriched fraction. Cell lysates containing 20 to 30 μg of protein were loaded in each well of a 10% SDS-polyacrylamide gel for electrophoresis and Western blot as described (40). Primary antibodies against Nrf2 (sc 722) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The NQO1 antibody was a kind gift of Dr. David Ross at the University of Colorado.
Adenoviral Infection
Nrf2 cDNA was inserted into GFP-expressing adenoviral constructs using the Cre-lox system. Therefore all constructs coexpress GFP along with the Nrf2 transgene, although they are not fused. The expression of these two genes is regulated by a CMV promoter. For infection, adenovirus at a Multiplicity of Infection (MOI) of 50 was added to CMCs in 6-well plates 2 days after seeding for a 5-hrs incubation. The media was then replaced with fresh 10% FBS DMEM for a 24 hr recovery before serum starvation (0.5% FBS DMEM) and H2O2 treatment.
DNA Degradation Measurement
Cardiomyocytes plated in 100 mm dishes (2×106 cells/dish) were scrapped off the dishes in 1 ml PBS for centrifugation to collect cell pellets. Genomic DNA was isolated using a DNA laddering kit (Cayman Chemical, Catalog. #660990). The isolated DNA was precipitated with 80% ice-cold ethanol, dried and resuspended in 10 μl ddH2O. DNA in solution was mixed with 10 μg ethidium bromide for separation on a 1.8% agarose gel by electrophoresis at 100V for 6 hrs. DNA smear was recorded using alphaDigiDoc system (alpha-Innotech) under an ultraviolet transilluminator.
Statistics
Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Bonferroni analysis for cross comparisons with Stata 8.2 software. Each group of means that is not significantly different from each other is indicated by a common letter symbol. Therefore, means in the “a” group are significantly different than means in the “b” group, and so on.
Results
In this study, we tested whether H2O2 pretreatment renders cardiomyocytes resistant to further damage by doxorubicin (Dox). H2O2 at 100 μM has been shown to induce minimal cell death but mild hypertrophy at 3-4 days after treatment (42), allowing us to challenge cells without causing significant cell death. At 24 hrs after H2O2 treatment, cells have stabilized from the initial insult. With Dox at 0.4 to 1.2 μM, a dose dependent increase of caspase-3 activity is detectable within 16 hrs of Dox treatment (Fig 1A). In comparison, cells pretreated with H2O2 showed a significant retardation in Dox induced caspase-3 activity (Fig 1A). Apoptotic cardiomyocytes detach from the culture matrix (40). Counting the fraction of detached cells showed that H2O2 pretreatment inhibited Dox from inducing cell death (Fig 1B). Annexin V binding assay also confirmed the protective effect of H2O2 pretreatment (Fig 1C). Our data indicate that H2O2 pretreatment induces a resistance against Dox-induced apoptosis.
Figure 1.
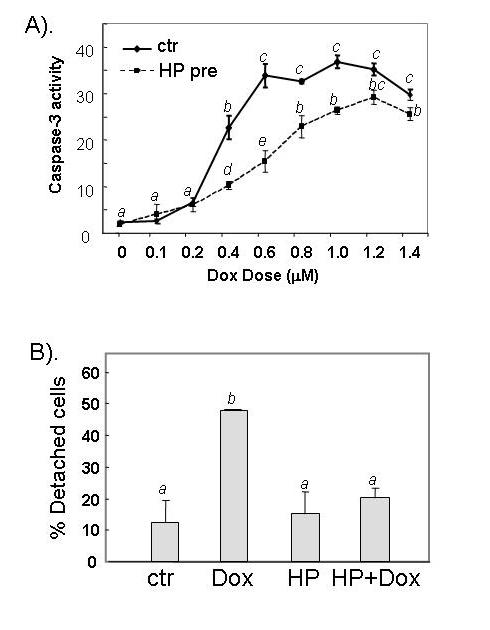
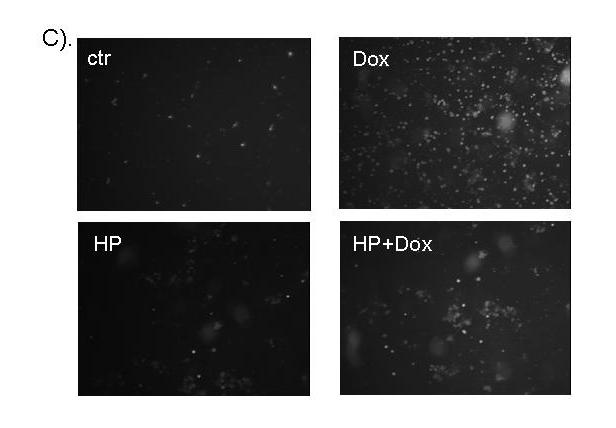
H2O2 pretreatment reduces Dox-induced apoptosis. Serum-starved CMCs were treated with 100 μM H2O2 for 1 hr. The cells recovered for 24 hr in fresh DMEM containing 0.5 % FBS before treatment with different doses of Dox (A) or 0.6 μM Dox (B & C). Caspase-3 activity (A), cell detachment (B) and Annexin V binding (C) were measured 16 hr after addition of Dox. The relative fluorescent unit (RFU) was corrected for protein content to indicate caspase-3 activity (A). At least 300 cells were scored from each view under a phase contrast microscope and three views were chosen randomly for scoring the proportion of detached versus attached cells (B). A letter indicates a significant difference (p<0.05) from the means labeled with a different letter as determined by ANOVA followed by Bonferroni analysis (A, B).
In an effort to understand the observed preconditioning effect, we compared gene expression profiles with or without oxidant treatment in CMCs. The results indicate that 2.2% or 342 of genes in CMCs had changed expression as a result of H2O2 treatment. Among those, 51 genes had increased expression and 291 genes had decreased expression (Table 2, note that EST or unknown genes were not listed). Among the upregulated genes, 19.6% (10 genes) can be classified after manual sorting as being related to antioxidant and detoxification responses (Table 2). Cytoscape-based gene network sorting of the Affymetrix data output also showed a common trend of upregulation of antioxidant and detoxification genes (Fig. 2A). Several outstanding groups of genes were downregulated, including cell cycle regulators, muscle or contractile-specific proteins, and metabolism enzymes (Table 2 and Fig. 2A). Four major components of mitochondrial respiration appear to be impaired by H2O2 treatment due to suppression of gene expression: Complex I (NADH dehydrogenases), Complex III (Ubiquinol-cytochrome C reductases), Complex IV (Cytochrome c oxidases) and Complex IV (ATP synthase). In addition, mitochondrial cytochrome c also appears to decrease expression with H2O2 treatment (Table 2).
Table 2.
Microarray Detection of Genes Changing Expression with H2O2 Treatment CMCs or HFs were treated with 100 μM H2O2 for 1 hour and were placed in fresh culture medium for 24 hrs recovery before harvesting RNA for Affymetrix Gene Array Analyses (see Methods). The data indicates the averages and standard deviations of fold changes from 4 comparisons. The fold of increase or decrease (negative numbers in italic) for each comparison was determined by Microarray Suite 5.0.1 as statistically significant changes. The column in the table represents 1) Affymetrix sequence number, 2) UniGene number, 3) common name of the genes, 4) fold of changes in CMCs and 5) fold of changes in HFs.
| Antioxidant/detoxification enzymes | CMCs | HFs | ||
|---|---|---|---|---|
| 1387599a | Rn.11234 | NAD(P)H dehydrogenase, quinone 1 | 3.11±0.48 | 4.14±1.90 |
| 1370902 | Rn.23676 | aldose reductase-like protein | 3.06±0.52 | 4.02±1.26 |
| 1387669a | Rn.3603 | epoxide hydrolase 1 | 2.47±0.25 | 2.50±0.51 |
| 1372599 | Rn.7854 | microsomal glutathione S-transferase 2 | 2.31±0.26 | 2.26±0.68 |
| 1367612 | Rn.2580 | microsomal glutathione S-transferase 1 | 1.72±0.18 | 2.73±0.81 |
| 1388122 | Rn.44821 | glutathione S-transferase, pi 2 | 1.81±0.25 | 1.97±0.20 |
| 1369940 | Rn.3136 | transaldolase 1 | 1.99±0.36 | 2.31±0.26 |
| 1370678s | Rn.102240 | monoamine oxidase A gene | 1.92±0.35 | 1.60±0.05 |
| 1368990 | Rn.10125 | cytochrome P450 1B 1 (Cyp1b1) | 1.84±0.22 | 1.60±0.10 |
| 1370080 | Rn.3160 | Heme oxygenase | 2.22±0.49 | |
| 1387759s | Rn.26489 | UDP glycosyltransferase 1 family, A6 | 2.66±0.88 | |
| 1370613s | Rn.64664 | UDP glycosyltransferase 1 family, A7 | 2.21±0.40 | |
| 1369926 | Rn.1491 | glutathione peroxidase 3 | 1.87±0.13 | |
| 1367774 | Rn.10460 | glutathione S-transferase, α 1 | 1.81±0.16 | |
| 1372297 | Rn.15990 | glutathione S-transferase 8(GST class α) | 1.64±0.31 | |
| 1367995 | Rn.3001 | catalase | 1.58±0.19 | |
| 1370172 | Rn.10488 | superoxide dismutase 2 | 1.49±0.10 | |
| 1386958 | Rn.67581 | thioredoxin reductase 1 | 1.50±0.26 | |
| 1367870 | Rn.3578 | thioredoxin-like 2 | 1.38±0.05 | |
| 1367613 | Rn.2845 | peroxiredoxin 1 | 1.36±0.15 | |
| 1388300 | Rn.1916 | microsomal glutathione S-transferase 3 | −2.92±1.13 | |
| 1370708a | Rn.10021 | 3-α-hydroxysteroid dehydrogenase | −1.90±0.43 | |
| 1367979s | Rn.107152 | cytochrome P450, subfamily 51 | −1.47±0.13 | |
| 1387891 | Rn.17958 | peroxiredoxin 4 | −1.36±0.15 | |
| Metal binding proteins | ||||
| 1368420 | Rn.32777 | ceruloplasmin | 1.99±0.73 | |
| 1367565a | Rn.54447 | ferritin, heavy polypeptide 1 | 1.38±0.11 | |
| 1367559 | Rn.1905 | ferritin light chain 1 | 1.32±0.11 | |
| 1370282 | Rn.94754 | cysteine rich protein 2 | −1.76±0.33 | |
| Metabolic enzymes | ||||
| 1386953 | Rn.888 | hydroxysteroid 11-β dehydrogenase 1 | 2.38±0.78 | 3.37±1.58 |
| 1370154 | Rn.2283 | lysozyme | 1.73±0.36 | |
| 1367856 | Rn.11040 | glucose-6-phosphate dehydrogenase | 1.92±0.28 | |
| 1367982 | Rn.97126 | aminolevulinic acid synthase 1 | 1.80±0.34 | |
| 1387022 | Rn.6132 | aldehyde dehydrogenase family 1, A1 | 1.75±0.47 | |
| 1368976 | Rn.11414 | ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase, CD38 antigen |
1.72±0.29 | |
| 1386859 | Rn.5950 | transketolase | 1.55±0.22 | |
| 1372523 | Rn.8365 | glutamate-cysteine ligase catalytic subunit | 1.46±0.24 | |
| 1371521 | Rn.11126 | pyruvate dehydrogenase E1 α-like | 1.39±0.14 | |
| 1369931 | Rn.1556 | pyruvate kinase, muscle | 1.39±0.14 | |
| 1367734 | Rn.107801 | aldehyde reductase 1 (low Km aldose reductase) |
1.39±0.14 | |
| 1367739 | Rn.10325 |
Cytochrome c oxidase subunit VIII-H (heart/muscle) |
−4.71±3.19 | |
| 1367782 | Rn.5119 |
cytochrome c oxidase, subunit VIa, polypeptide 2 |
−2.97±1.37 | |
| 1367626 | Rn.10756 | creatine kinase, muscle | −2.44±0.96 | |
| 1369629 | Rn.3329 | adenosine kinase | −1.84±0.31 | |
| 1387773 | Rn.2202 | cytochrome c, somatic | −1.52±0.33 | |
| 1370276 | Rn.1817 |
ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit |
−1.53±0.21 | |
| 1370278 | Rn.3879 |
ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit |
−1.49±0.22 | |
| 1370284 | Rn.3454 |
ATP synthase, H+ transporting, mitochondrial F1 complex, epsilon subunit |
−1.45±0.21 | |
| 1370230 | Rn.5790 |
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 |
−1.42±0.13 | |
| 1371335 | Rn.107458 |
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit g |
−1.42±0.20 | |
| 1389964 | Rn.1318 |
NADH dehydrogenase (ubiquinone) (EC 1.6.5.3) acyl carrier chain, mitochondrial |
−1.53±0.21 | |
| 1371041 | Rn.11092 |
24-kDa subunit of mitochondrial NADH dehydrogenase |
−1.45±0.16 | |
| 1389012 | Rn.18013 |
NADH dehydrogenase (ubiquinone) 1 β subcomplex, 2 |
−1.46±0.24 | |
| 1389288 | Rn.14785 |
NADH dehydrogenase (ubiquinone) 1 α subcomplex, 2 (8kD, B8) |
−1.42±0.20 | |
| 1371381 | Rn.35208 |
Ubiquinol-cytochrome C reductase complex 7.2 kDa protein (HSPC119) |
−1.46±0.20 | |
| 1375197 | Rn.3254 |
Ubiquinol-cytochrome C reductase complex 6.4 kDa protein (Complex III subunit XI) |
−1.38±0.11 | |
| 1370218 | Rn.1785 | lactate dehydrogenase B | −1.83±0.54 | |
| 1370939 | Rn.6215 |
fatty acid Coenzyme A ligase, long chain 2 |
−1.80±0.54 | |
| 1367857 | Rn.28161 | fatty acid desaturase 1 | −1.69±0.23 | |
| 1386965 | Rn.3834 | Lipoprotein lipase | −1.58±0.11 | |
| 1367707 | Rn.9486 | fatty acid synthase | −1.45±0.17 | |
| 1388348 | Rn.4243 | fatty acid elongase 1 | −1.29 ± 0.05 | |
| 1370355 | Rn.1023 | stearoyl-Coenzyme A desaturase 1 | −1.78±0.25 | |
| 1367668a | Rn.83595 | stearoyl-Coenzyme A desaturase 2 | −1.76±0.25 | |
| 1370191 | Rn.6290 |
ornithine decarboxylase antizyme inhibitor |
−1.78±0.42 | |
| 1367667 | Rn.2622 | farensyl diphosphate synthase | −1.78±0.16 | |
| 1387271 | Rn.7279 | phytanoyl-CoA hydroxylase | −1.73±0.63 | |
| 1372462 | Rn.106162 | acetyl-CoA C-acetyltransferase, cytosolic | −1.71±0.37 | |
| 1370235 | Rn.3285 | diazepam binding inhibitor | −1.67±0.18 | |
| 1388403 | Rn.3490 |
Isocitrate dehydrogenase, cytoplasmic (Oxalosuccinate decarboxylase) |
−1.65±0.33 | |
| 1368878 | Rn.10780 | isopentenyl-diphosphate delta isomerase | −1.59±0.26 | |
| 1367932 | Rn.5106 |
3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 |
−1.42±0.14 | |
| 1389908 | Rn.73738 | β-galactosidase (Lactase) | −1.42±0.10 | |
| 1372665 | Rn.100813 | Phosphoserine aminotransferase 1 | −1.32±0.09 | |
| Endocrine factors, cytokines and binding factors | ||||
| 1367973 | Rn.4772 | small inducible cytokine A2 | 3.85±2.70 | 1.84±1.07 |
| 1370770s | Rn.44216 | Kit ligand, stem cell factor | 2.26±0.68 | 1.88±0.26 |
| 1368238 | Rn.9727 | pancreatitis-associated protein | 4.23±2.16 | |
| 1386879 | Rn.764 | lectin, galactose binding, soluble 3 | 3.22±1.24 | |
| 1371210s | Rn.39743 | MHC nonclassical class I antigen | 2.88±0.68 | |
| 1390507 | Rn.16103 | interferon stimulated gene (20kD) | 1.99±0.36 | |
| 1373882 | Rn.4256 | VEGF-D | 1.74±0.13 | |
| 1370082 | Rn.40136 | TGF β1 | 1.60±0.24 | |
| 1387450 | Rn.9952 | TGF α | 1.93±0.62 | |
| 1387316 | Rn.10907 | gro | 4.33±1.37 | |
| 1387029 | Rn.101777 | complement component factor H | 4.13±2.42 | |
| 1388255x | Rn.39743 | MHC nonclassical class I antigen, soluble precursor (RT1 class Ib gene) |
3.33±0.92 | |
| 1367850 | Rn.6050 | Fc receptor, IgG, low affinity III | 3.28±1.81 | |
| 1370154 | Rn.2283 | Lysozyme | 3.05±1.35 | |
| 1387687 | Rn.16316 | immunoglobulin superfamily, member 6 | 2.81±1.03 | |
| 1368490 | Rn.42942 | CD14 antigen | 1.56±0.22 | |
| 1368044 | Rn.45602 | secretogranin 2 | −3.87±2.38 | −2.73±1.15 |
| 1371500 | Rn.7961 | latent TGF β binding protein 1 | −1.83±0.15 | |
| 1387232 | Rn.10318 | bone morphogenetic protein 4 | −1.42±0.11 | |
| 1376425 | Rn.24539 | TGF β2 | −1.32±0.07 | |
| 1371500 | Rn.7961 |
latent TGF β binding protein 4 homologue (46%) |
−1.72±0.29 | |
| 1368448 | Rn.40921 | latent TGF β binding protein 2 | −1.46±0.20 | |
| 1367650 | Rn.1256 |
lipocalin 7 (glucocorticoid-inducible protein) |
−2.09±0.81 | |
| 1369519 | Rn.10918 | endothelin 1 | −1.56±0.50 | |
| 1368983 | Rn.10148 | heparin-binding EGF-like growth factor | −1.50±0.32 | |
| 1379375 | Rn.10999 | Platelet derived growth factor α | −1.39±0.14 | |
| Receptors | ||||
| 1369146a | Rn.91370 | Aryl hydrocarbon receptor | 1.96±0.20 | 1.79±0.19 |
| 1367940 | Rn.12959 | chemokine orphan receptor 1 | 2.33±0.71 | |
| 1368742 | Rn.10680 | complement component 5, receptor 1 | 2.03±0.89 | |
| 1387273 | Rn.10072 | interleukin 1 receptor-like 1 | 1.55±0.22 | |
| 1367619 | Rn.102356 | progesterone receptor membrane component 1 |
1.49±0.15 | |
| 1367636 | Rn.270 | insulin-like growth factor 2 receptor | 1.45±0.11 | |
| 1370249 | Rn.1820 | benzodiazepin receptor | 1.42±0.20 | |
| 1371840 | Rn.4102 |
endothelial differentiation sphingolipid G-protein-coupled receptor 1 |
−4.15±2.29 | |
| 1370462 | Rn.92304 |
Hyaluronan mediated motility receptor (RHAMM) |
−3.08±0.93 | |
| 1373661a | Rn.44431 | Chemokine receptor (LCR1) | −1.83±0.54 | |
| 1373803a | Rn.2178 |
growth hormone receptor (somatotropin receptor precursor) |
−1.71±0.18 | |
| 1371113a | Rn.98672 | transferrin receptor | −1.49±0.35 | |
| 1370541 | Rn.10055 |
nuclear receptor subfamily 1, group D, member 2 |
−1.45±0.06 | |
| 1368553 | Rn.10631 | activin A receptor type II-like 1 | −1.38±0.05 | |
| Signaling molecules | ||||
| 1370445 | Rn.10696 | phosphatidylserine-specific phospholipase A1 |
1.72±0.29 | 1.70±0.07 |
| 1387822 | Rn.21925 | G-protein α11 subunit | 1.52±0.11 | |
| 1368332 | Rn.25736 | guanylate binding protein 2, interferon- inducible |
2.25±0.23 | |
| 1368144 | Rn.1892 | regulator of G-protein signaling protein 2 | 2.20±0.61 | |
| 1372031 | Rn.14763 | Dab2: Disabled homolog 2, mitogen- responsive phosphoprotein |
1.75±0.47 | |
| 1370942 | Rn.23055 | RAS p21 protein activator 3 (49%) | 1.73±0.63 | |
| 1383180 | Rn.4016 | protein phosphatase 1, regulatory (inhibitor) subunit 2 |
1.53±0.21 | |
| 1369294 | Rn.10728 | bone marrow stromal cell antigen 1 (cyclic ADPribose metabolism) |
1.51±0.27 | |
| 1371353 | Rn.107103 | sequestosome 1 | 1.37±0.05 | |
| 1372017 | Rn.9090 | Direct IAP binding protein with low pI | 1.37±0.12 | |
| 1373658 | Rn.19950 | Rac GTPase-activating protein 1 | −5.44±2.20 | |
| 1375303 | Rn.17340 | enigma | −2.60±0.94 | |
| 1371984 | Rn.17340 | enigma homolog | −2.15±1.01 | |
| 1371873 | Rn.4268 | lecuine-rich acidic protein-like protein | −2.04±0.18 | |
| 1372084 | Rn.106043 |
tyrosine phosphatase type IVA, member 3 isoform 1 |
−1.63±0.37 | |
| 1368101 | Rn.2892 | calmodulin 3 | −1.52±0.00 | |
| 1370829 | Rn.8873 | farnesyltransferase β subunit | −1.70 ± 0.42 | |
| 1373082 | Rn.94808 | Pkia: Protein kinase inhibitor, α | −1.42±0.20 | |
| 1370012 | Rn.73051 | prostaglandin I2 synthase | −1.36±0.15 | |
| 1369097s | Rn.87228 | guanylate cyclase 1, soluble,β3 | −1.35±0.05 | |
| Cell cycle regulators | ||||
| 1387391 | Rn.10089 | cyclin-dependent kinase inhibitor 1A, p21 | 3.9±1.10 | 4.92±2.82 |
| 1383485 | Rn.91829 | Mdm2 | 1.99±0.36 | |
| 1367764 | Rn.5834 | cyclin G1 | 1.56±0.26 | |
| 1373823 | Rn.6116 | cell division control protein CKS2 | −4.08±1.24 | −1.83±0.19 |
| 1367671 | Rn.223 | Proliferating cell nuclear antigen | −2.01±0.25 | −1.52±0.15 |
| 1373557 | Rn.8341 | replication licensing factor MCM4 | −2.48±0.63 | −1.55±0.12 |
| 1375532 | Rn.3272 |
Inhibitor of DNA binding 2, dominant negative helix-loop-helix protein |
−1.83±0.55 | −1.47±0.13 |
| 1374775 | Rn.12774 | cell proliferation antigen Ki-67 | −20.5±15.0 | |
| 1367776 | Rn.6934 | cell division cycle 2 homolog A | −4.74±0.86 | |
| 1372685 | Rn.25026 | cyclin-dependent kinase inhibitor 3 | −4.71±1.18 | |
| 1370346 | Rn.9232 | Cyclin B1 | −3.79±1.35 | |
| 1389566 | Rn.23351 | strong similarity to cyclin B2 | −3.68±0.88 | |
| 1388395 | Rn.1040 |
Putative lymphocyte G0/G1 switch protein 2 |
−2.53±1.25 | |
| 1374449 | Rn.3246 |
cell division cycle associated 3; gene rich cluster, C8 gene; |
−2.31±0.29 | |
| 1371074a | Rn.33226 | mini chromosome maintenance deficient 6 | −2.25±0.64 | |
| 1367894 | Rn.772 | growth response protein (CL-6) | −1.91±0.20 | |
| 1370294a | Rn.9262 | cell cycle protein p55CDC | −1.81±0.37 | |
| 1372406 | Rn.12916 | replication licensing factor MCM3 | −1.63±0.17 | |
| 1367657 | Rn.1000 | B-cell translocation gene 1 | −1.29±0.11 | |
| 1369935 | Rn.3483 | cyclin D3 | −1.39±0.21 | |
| Cytoskeletal proteins and their regulators | ||||
| 1372750 | Rn.2743 | Follistatin | 1.91±0.20 | 2.30±0.87 |
| 1388460 | Rn.8945 | macrophage capping protein | 2.10±0.07 | |
| 1371618s | Rn.8216 | tubulin, β3 | 1.93±0.32 | |
| 1386857 | Rn.555 | stathmin 1 | −3.64±0.60 | −1.96±0.39 |
| 1367785 | Rn.31788 | Calponin 1 | −2.04±0.21 | −1.53±0.26 |
| 1371339 | Rn.11675 | cofilin 1 | −2.76±1.35 | |
| 1388718 | Rn.1646 | tropomodulin 1 | −2.39±0.76 | |
| 1387373 | Rn.48693 | myomegalin | −1.79±0.59 | |
| 1370875 | Rn.773 | villin 2 | −1.52±0.11 | |
| 1370949 | Rn.9560 |
myristoylated alanine rich protein kinase C substrate |
−1.39±0.21 | |
| 1368822 | Rn.95652 | follistatin-like | −1.33±0.16 | |
| Cell surface and extracellular matrix proteins | ||||
| 1367581a | Rn.8871 | secreted phosphoprotein 1 | 2.25±0.67 | 2.19±0.55 |
| 1374620 | Rn.91235 | Ceacam1 | 2.10±0.17 | |
| 1388985 | Rn.25004 | Collagen α1(V) homologue | 1.72±0.29 | |
| 1389966 | Rn.2157 | Collagen α3 (VI) homologue | 1.49±0.13 | |
| 1393891 | Rn.53843 | Collagen α1(VIII) homologue | 1.32±0.07 | |
| 1386879 | Rn.764 | lectin, galactose binding, soluble 3 | 2.06±0.86 | |
| 1368187 | Rn.13778 | glycoprotein (transmembrane) nmb | 2.05±0.76 | |
| 1367784a | Rn.1780 | clusterin | 1.91±0.20 | |
| 1368474 | Rn.11267 | Vascular cell adhesion molecule 1 | 1.80±0.25 | |
| 1370043 | Rn.5789 | activated leukocyte cell adhesion molecule |
1.37±0.16 | |
| 1392784 | Rn.52228 | growth arrest specific 6 | 1.35±0.11 | |
| 1369943 | Rn.10 | tissue-type transglutaminase | −2.04±0.48 | −1.81±0.25 |
| 1388111 | Rn.54384 | elastin | −1.80±0.39 | −3.23±2.46 |
| 1368171 | Rn.11372 | lysyl oxidase | −1.64±0.31 | −1.69±0.40 |
| 1387850 | Rn.44829 |
transmembrane protein with EGF-like and two follistatin-like domains 1 |
−3.21±1.23 | |
| 1388961 | Rn.39792 | integrin β1 binding protein (melusin) 2 | −3.00±1.84 | |
| 1370937a | Rn.54492 | integrin α7 | −2.00±0.71 | |
| 1373897 | Rn.11362 | lamin B1 | −2.99±0.31 | |
| 1373439 | Rn.6499 | lamin B receptor | −1.52±0.11 | |
| 1381504 | Rn.45067 |
Biglycan precursor (Bone/cartilage proteoglycan I) (PG-S1) |
−1.42±0.10 | |
| 1373401 | Rn.12723 | tenascin C | −2.49±1.37 | |
| 1369736 | Rn.19723 | Epithelial membrane protein 1 | −1.80±0.31 | |
| 1387280a | Rn.32261 | tumor-associated protein 1 | −1.57±0.32 | |
| 1367880 | Rn.774 | laminin, β2 | −1.39±0.21 | |
| 1369955 | Rn.117 | collagen, type V, α1 | −1.35±0.11 | |
| Muscle protein and contractile proteins | ||||
| 1368415 | Rn.9692 |
Myosin heavy chain, skeletal muscle, perinatal |
−14.38±4.05 | |
| 1367928 | Rn.48663 |
myosin heavy chain, polypeptide 7, cardiac muscle, β |
−3.87±2.42 | |
| 1371293 | Rn.23925 | Myosin light chain 1, atrial isoform | −4.83±1.98 | |
| 1367572 | Rn.1955 | myosin, light chain polypeptide 3 | −2.57±1.50 | |
| 1376789 | Rn.43838 | myosin-light-chain kinase | −2.12±0.82 | |
| 1388298 | Rn.6870 |
Myosin regulatory light chain 2, smooth muscle isoform |
−1.38±0.05 | |
| 1369928 | Rn.82732 | actin α 1 | −3.05±2.04 | |
| 1386931 | Rn.64141 | troponin 1, type 3 | −2.88±1.68 | |
| 1386873 | Rn.4035 | Troponin I, slow isoform | −2.55±0.94 | |
| 1388604 | Rn.38090 | Calsequestrin, cardiac muscle isoform | −3.82±1.91 | |
| 1371801 | Rn.12931 |
calcineurin-binding protein calsarcin-1; muscle-specific protein; FATZ related protein 2 |
−3.58±1.81 | |
| 1368988 | Rn.10111 | calsequestrin 2 | −2.36±1.03 | |
| 1398243 | Rn.11345 |
cysteine-rich protein 3; muscle LIM protein |
−3.17±1.69 | |
| 1370165 | Rn.4123 | small muscle protein, X-linked | −2.60±0.94 | |
| 1368252 | Rn.28875 | sarcomeric muscle protein | −2.30±0.82 | |
| 1372527 | Rn.3210 |
Rtn2: Reticulon 2 (Z-band associated protein) |
−2.27±0.76 | |
| 1390049 | Rn.34417 | four and a half LIM domains 1 | −2.12±0.93 | |
| 1371933 | Rn.48693 | myomegalin | −1.57±0.29 | |
| Channel proteins | ||||
| 1370583s | Rn.82691 | P-glycoprotein, multidrug resistance 1 | 5.02±1.52 | 4.72±1.64 |
| 1368207 | Rn.24997 | FXYD domain-containing ion transport regulator 5 |
1.73±0.36 | |
| 1367959a | Rn.4958 | sodium channel, voltage-gated, type I, β | 2.89±1.33 | |
| 1370516 | Rn.17317 | peptide/histidine transporter PHT2 | 2.14±0.79 | |
| 1373054 | Rn.97686 | Ctl1: Transporter-like protein | 1.43±0.23 | |
| 1367683 | Rn.2949 | karyopherin (importin) α2 | −1.62±0.0 | −1.64±0.31 |
| 1369960 | Rn.3828 |
FXYD domain-containing ion transport regulator 1 |
−2.60±1.27 | |
| 1368965 | Rn.10826 | monocarboxylate transporter | −2.36±0.65 | |
| 1386901 | Rn.3790 |
fatty acid binding/transport protein (cd36 antigen) |
−2.60±1.52 | |
| 1367660 | Rn.32566 | fatty acid binding protein 3 | −2.06±0.73 | |
| 1370281 | Rn.98269 | fatty acid binding protein 5, epidermal | −1.45±0.20 | |
| 1370850 | Rn.3402 | sodium channel β3 subunit | −2.63±1.51 | |
| 1369625 | Rn.1618 | aquaporin 1 | −2.38±1.27 | |
| 1369065a | Rn.2305 |
ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 |
−1.87±0.70 | |
| 1371883 | Rn.34134 | solute carrier family 1, member 3 | −1.85±0.23 | |
| 1389967 | Rn.3366 |
ADP-ribosylation-like factor 6- interacting protein |
−1.50±0.15 | |
| 1398370 | Rn.45761 | rexo70 | −1.82±0.45 | |
| 1389986 | Rn.58137 | synaptic vesicle glycoprotein 2 b | −1.46±0.24 | |
| Protease or protease inhibitors | ||||
| 1368590 | Rn.52536 | matrix metalloproteinase 16 | 1.79±0.19 | |
| 1368530 | Rn.33193 | matrix metalloproteinase 12 | 2.15±0.82 | |
| 1368512a | Rn.53979 | aminopeptidase A | 2.24±0.92 | |
| 1387005 | Rn.11347 | cathepsin S | 2.24±0.92 | |
| 1368280 | Rn.11559 | cathepsin C | 2.01±0.54 | |
| 1368215 | Rn.43558 | ceroid-lipofuscinosis, neuronal 2 | 1.42±0.18 | |
| 1369977 | Rn.107213 | ubiquitin carboxy-terminal hydrolase L1 | 1.39±0.21 | |
| 1368544a | Rn.86956 | nucleolar protein 3 (apoptosis repressor with CARD domain) |
1.32±0.09 | |
| 1389408 | Rn.99540 |
neural precursor cell expressed, developmentally down-regulated gene 4A |
−5.78±0.52 | −3.55±1.36 |
| 1372440 | Rn.2271 |
serine (or cysteine) proteinase inhibitor, clade E, member 2 |
−1.60±0.48 | −1.80±0.25 |
| 1370064 | Rn.11045 | presenilin-2 | −1.42±0.20 | −1.38±0.05 |
| 1387804 | Rn.40636 |
muscle ring finger protein 1; ring finger protein 28 |
−1.97±0.56 | |
| 1368961 | Rn.22562 | Matrix metalloproteinase 23 | −1.28±0.05 | |
| 1368519 | Rn.29367 |
serine (or cysteine) proteinase inhibitor, member 1 |
−1.75±0.50 | |
| Nucleic acid metabolism | ||||
| 1368311 | Rn.9836 | O6-methylguanine-DNA methyltranferase | 1.97±0.17 | 1.36±0.15 |
| 1387659 | Rn.24783 | guanine deaminase | 1.91±0.27 | |
| 1371862 | Rn.22094 |
Ribonucleoside-diphosphate reductase M1 chain (Ribonucleotide reductase large chain). |
−2.64±0.15 | |
| 1387865 | Rn.6102 | Deoxyuridinetriphosphatase (dUTPase) | −1.69±0.15 | |
| 1373772 | Rn.6955 | DNA (cytosine-5)-methyltransferase 1 | −1.63±0.11 | |
| Chromosomal or DNA binding proteins | ||||
| 1388309 | Rn.83614 | non-histone chromosomal architectural protein HMGI-C |
1.75±0.22 | |
| 1374293 | Rn.40496 | Histone H2A.l | 1.49±0.22 | |
| 1367676 | Rn.2874 | high mobility group box 2 | −2.93±0.19 | −1.56±0.28 |
| 1368136 | Rn.3364 | lamina-associated protein 2 | −2.04±0.21 | −1.73±0.38 |
| 1386861 | Rn.3636 | H2A histone family, member Z | −1.62±0.09 | −1.30±0.04 |
| 1388827 | Rn.100938 |
H2A histone family, member V (predicted) |
−1.48±0.24 | |
| 1372516 | Rn.8601 | kinesin-like 4; Kid; | −2.89±0.34 | |
| 1388135 | Rn.40389 | p32-subunit of replication protein A | −2.00±0.11 | |
| 1371887 | Rn.13669 |
high-mobility group (nonhistone chromosomal) protein 4 |
−1.66±0.17 | |
| 1371352 | Rn.3517 | high mobility group protein 17 | −1.58±0.19 | |
| 1368042a | Rn.4121 | high mobility group box 1 | -1.39±0.14 | |
| 1388504 | Rn.3991 |
RAD21 homolog; protein involved in DNA double-strand break repair; nuclear matrix protein 1 |
−1.46±0.06 | |
| 1369996 | Rn.28212 | polymerase II | −1.37±0.05 | |
| 1367666 | Rn.23677 |
heterogeneous nuclear ribonucleoprotein H1 |
−1.36 ± 0.15 | |
| Transcription factors | ||||
| 1387087 | Rn.6479 | CCAAT/enhancer binding protein (C/EBP) β |
1.50±0.26 | |
| 1376569 | Rn.114645 | Kruppel-like factor 2 | −1.38±0.11 | −1.95±0.28 |
| 1389555 | Rn.14867 | transcription factor 19 (SC1) | −3.03±0.61 | |
| 1389528s | Rn.93714 |
v-jun sarcoma virus 17 oncogene homolog |
−1.72±0.26 | |
| 1388426 | Rn.95306 | sterol regulatory element binding factor 1 | −1.57±0.32 | |
| 1387769a | Rn.2760 |
Inhibitor of DNA binding 3, dominant negative helix-loop-helix protein |
−1.46±0.20 | |
| 1367664 | Rn.3789 | ankyrin-like repeat protein | −1.99±0.91 | |
| 1374335 | Rn.8701 | GATA binding protein 6 | −1.44±0.09 | |
| Protein synthesis and conformation | ||||
| 1371378 | Rn.4113 | Protein translation factor SUI1 homolog | −1.26±0.05 | |
| 1371421 | Rn.98472 |
translation elongation factor eEF-1 α chain |
−1.81±0.56 | |
| 1372431 | Rn.105932 | mitochondrial ribosomal protein L12 | −1.37±0.12 | |
| 1370007 | Rn.39305 |
protein disulfide isomerase related protein (calcium-binding protein, intestinal-related) |
−1.38±0.05 | |
| Stress genes | ||||
| 1387047 | Rn.20155 | heat shock 27kD protein family, 3 | −3.79±2.62 | |
| Miscellaneous | ||||
| 1388674 | Rn.36610 | tumor protein, translationally-controlled 1 | 3.08±0.11 | 2.46±0.70 |
| 1370459 | Rn.38451 | A5D3 protein | 2.46±0.0 | 1.45±0.16 |
| 1369008a | Rn.11005 | olfactomedin related ER localized protein | 1.78±0.12 | 1.57±0.06 |
| 1373932 | Rn.98491 | Cybb: Endothelial type gp91-phox gene | 2.93±1.42 | |
| 1368006 | Rn.24799 | lysosomal-associated protein transmembrane 5 |
2.39±0.76 | |
| 1389210 | Rn.14256 | lymphocyte cytosolic protein 1 | 2.36±0.84 | |
| 1390383 | Rn.101967 | Adipose differentiation-related protein | 1.73±0.41 | |
| 1371131a | Rn.2758 | upregulated by 1,25-dihydroxyvitamin D- 3 |
1.69±0.15 | |
| 1368013 | Rn.19672 | Smhs1 protein | 1.66±0.42 | |
| 1387946 | Rn.3251 | peptidylprolyl isomerase C-associated protein |
1.59±0.32 | |
| 1367881 | Rn.53971 | protein tyrosine phosphatase, non- receptor type substrate homolog |
1.51±0.27 | |
| 1387081 | Rn.6133 | reticulocalbin 2 | 1.42±0.18 | |
| 1390325 | Rn.11414 | ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase (CD38) |
1.38±0.05 | |
| 1387770 | Rn.3867 | Interferon α-inducible protein 27-like | 1.23±0.0 | |
| 1370697a | Rn.107975 | nexilin | −1.63±0.18 | −1.64±0.31 |
| 1374672 | Rn.3434 | cardiac ankyrin repeat kinase | −7.97 ± 7.22 | |
| 1370805 | Rn.8163 | melanocyte-specific gene 1 protein | −5.97±2.34 | |
| 1390137 | Rn.20235 | TRAF4 associated factor 1 homolog | −5.42±3.32 | |
| 1372886 | Rn.39396 |
Transforming acidic coiled-coil- containing protein 3 |
−3.71±1.36 | |
| 1377190 | Rn.12822 |
transcription activator of D-serine dehydratase |
−3.66±1.29 | |
| 1387957a | Rn.24200 | SH3-domain kinase binding protein 1 | −3.23 ± 1.50 | |
| 1372065 | Rn.4077 | ADP-ribosyltransferase 3 | −2.60±0.94 | |
| 1376084a | Rn.24582 | extra spindle poles like 1 | −2.47±0.54 | |
| 1372296 | Rn.4128 |
SH3 domain-binding glutamic acid-rich protein (21-glutamic acid-rich protein) |
−2.44±1.07 | |
| 1372646 | Rn.16593 | esophageal cancer related gene 4 protein | −1.99±0.34 | |
| 1368174 | Rn.10994 | EGL nine homolog 3 (C. elegans) | −1.85±0.39 | |
| 1368566a | Rn.6452 |
MIPP65 protein (mitochondrial phosphoprotein) |
−1.85±0.40 | |
| 1370828 | Rn.17310 | zinc finger, DHHC domain containing 2 | −1.85±0.28 | |
| 1372106 | Rn.7379 | pincher | −1.61±0.34 | |
| 1370176 | Rn.26957 |
amyotrophic lateral sclerosis 2 (juvenile) chromosome region, candidate 3 |
−1.57±0.29 | |
| 1374951 | Rn.45300 |
obscurin, cytoskeletal calmodulin and titin-interacting RhoGEF |
−1.50±0.26 | |
| 1371365 | Rn.3460 | ubiquitin carrier protein E2 | −1.48±0.24 | |
| 1370189 | Rn.8538 |
splicing factor, arginine/serine-rich (transformer 2 Drosophila homolog) 10 |
−1.42±0.18 | |
| 1371358 | Rn.3380 | synaptic glycoprotein SC2 (32%) | −1.42±0.20 | |
| 1370252 | Rn.13633 | Esau | −1.39±0.14 | |
| 1370062 | Rn.2084 | hypoxia induced gene 1 | −1.39±0.14 | |
| 1376749 | Rn.12119 | osteoinductive factor | −1.32±0.09 | |
| 1371298 | Rn.6171 | H19 fetal liver mRNA | −1.26±0.05 | |
| 1376198 | Rn.40141 |
visceral adipose tissue-specific transmembrane protein OL-16 |
−1.79 ± 0.74 | |
| 1370248 | Rn.839 |
FXYD domain-containing ion transport regulator 6 |
−1.52 ± 0.42 | |
| 1367604 | Rn.4267 | cysteine-rich protein 2 | −1.50 ± 0.23 | |
| 1373195 | Rn.9404 | orphan seven transmembrane receptor | −1.42 ± 0.10 | |
| 1370000 | Rn.41602 | NEFA precursor | −1.39 ± 0.21 | |
| 1367744 | Rn.11984 | melanoma antigen, family D, 2 | −1.33 ± 0.16 | |
Figure 2.
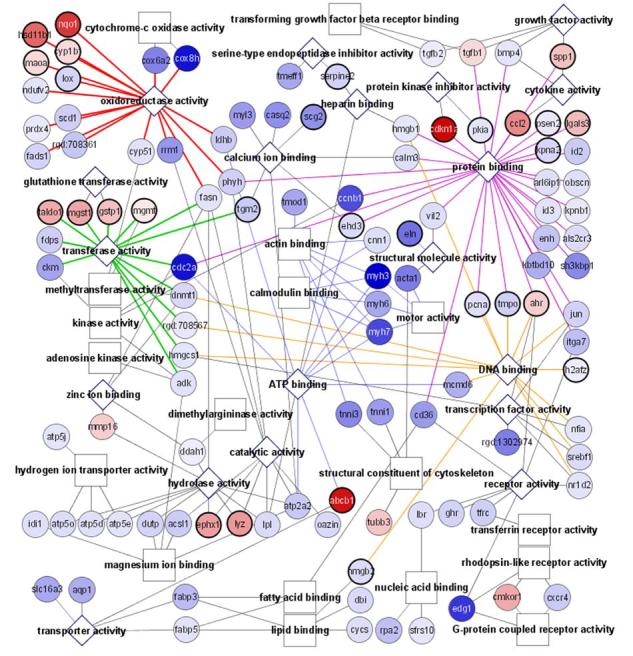
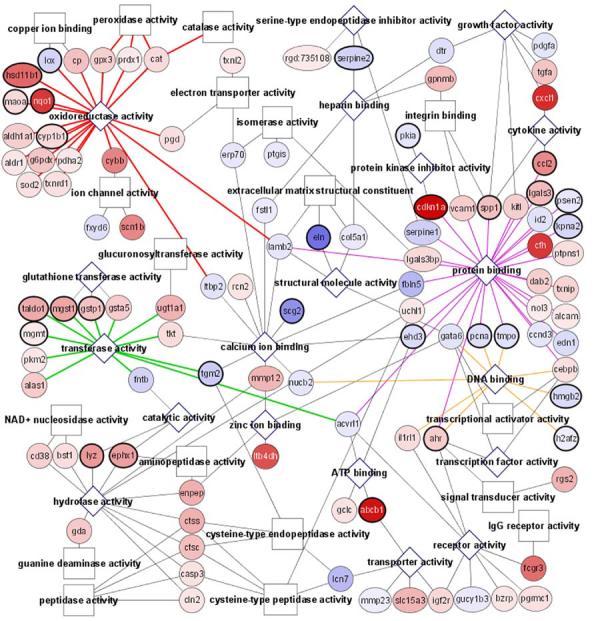
Functional display of the microarray data by the Cytoscape software analysis. Cardiomyocytes (CMCs) or heart fibroblasts (HFs) were treated with 100 μM H2O2 for 1 hr. Microarray analyses were performed using RNA collected 24 hrs after H2O2 treatment. The outcome of Cytoscape analyses of genes that changed with H2O2 treatment from CMCs (A) or HFs (B) is shown. Circles indicate individual genes, while boxes or diamonds indicate functional groups. Diamonds are shown when the functional group is found in both CMCs and HFs. Increasing shades of red indicate an increasing fold of upregulation, while increasing shades of blue show greater downregulation. A bold circle indicates a gene that is changed in both the CMCs and HFs with H2O2 treatment.
To address whether the response of inducing antioxidant/detoxification genes is unique to CMCs, we performed microarray analyses of HFs following H2O2 treatment. Since Affymetrix gene array techniques produce gene expression profiles for control and experimental groups, we can compare the difference of gene expression at the basal level between cell types. Comparison between untreated CMCs and HFs indicate a difference in about 10% or 2,466 genes. Many of these differences are predicted. For example CMCs express genes responsible for muscle cell differentiation and muscle contraction, such as myosins, troponins, α-actins, tropomyosins, desmin, phospholamban, calsequestrin, voltage-gated potassium channels and voltage-gated calcium channels. High levels of genes encoding mitochondrial enzymes observed in CMCs are consistent with the fact that CMCs contain more mitochondria per cell compared to HFs. Novel knowledge gained from the comparison includes the finding that CMCs express much higher levels of hsp27 (100 ± 6.3 fold), aquaporin 1 (11.7 ± 1.7), fibroblast growth factor 13 (8.0 ± 0.8), integrin alpha 7 (6.3 ± 0.6), DNA topoisomerase 1a (5.0 ± 1.2), and protein phosphase 2A (4.1 ± 0.7). Several genes identified depict the development and differentiation of CMCs, including an NK2 related transcription factor (7.8 ± 0.9), pituitary tumor-transforming 1 (6.6 ± 2.5), cyclin-dependent kinase inhibitor p57 (5.4 ± 0.6), and cyclin B1 (4.9 ± 0.4).
In HFs, H2O2 treatment caused 1.56% or 247 genes to change expression, among which 138 genes had increased and 109 genes had decreased expression (Table 2). About 14% (19 genes) of upregulated genes were in the antioxidant and detoxification response category, while metabolic enzymes, signaling molecules and cell surface/extracellular matrix proteins each made similar contributions (8%, 8% and 7.3% respectively, Table 2). Among the down regulated genes, cell surface and extracellular matrix proteins were prominent (Table 2).
Visual comparison between the gene expression profiles shows that more genes are upregulated by H2O2 treatment in HFs than in CMCs (Fig. 2A&B). These upregulated genes include those belonging to categories such as oxidoreductases, transferases, hydrolases, protein binding, and receptor proteins in both cell types (Fig. 2A&B). Most genes upregulated in CMCs in the category of antioxidant/detoxification enzymes were also upregulated in HFs (Table 2). HFs had an additional 10 genes upregulated in this category (Table 2).
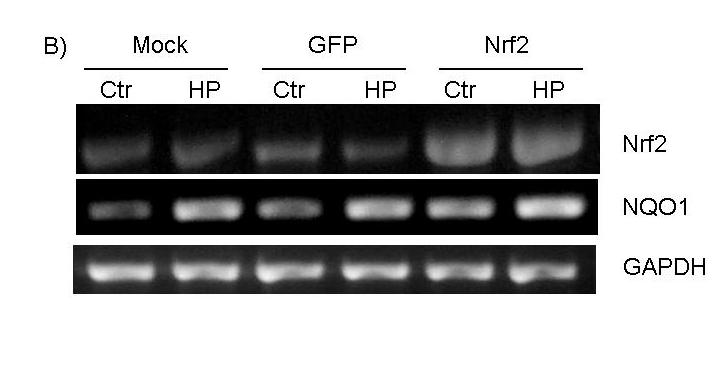
Microarray data were verified using RT-PCR analyses on selected genes. Emphasis was placed on the antioxidant/detoxification genes found to be upregulated in CMCs and HFs. These genes include NADP(H) quinone dehydrogenase 1 [NAD(P)H: quinone oxidoreductase 1, or NQO1], glutathione S-transferase pi 2 (GSTp2), microsomal glutathione S-transferase 1 (mGST1), microsomal glutathione S-transferase 2 (mGST2), UDP glycosyltransferase 1A6 (UGT1A6), heme oxygenase (HO1), catalase, epoxide hydrolase 1 (Ephx1), cytochrome p4501b1 (Cyp1b1), transaldolase (Taldo1), aldose reductase-like protein (Aldrlp), monoamine oxidase A (MaoA), hydroxysteroid 11-β dehydrogenase 1 (11-β-HSD1), aryl hydrocarbon receptor (AhR) and multidrug resistance P-glycoprotein 1 (Pgy1, Fig. 3A). We also verified several muscle genes and mitochondrial enzymes that showed decreased expression with H2O2 treatment in the microarray profile of CMCs (Fig. 3B). These include skeletal muscle perinatal myosin heavy chain (Myh4), myosin heavy chain 7 (Myh7), myosin light chain polypeptide 3 (Myl3), troponin I type 1 (Tnni1), troponin I type 3 (Tnni3), cytochrome c (CytC), cytochrome c oxidase subunit VIa polypeptide 2 (Cox6a2), cytochrome c oxidase subunit VIII-H (Cox8h), and 24kDa subunit of mitochondrial NADH dehydrogenase (Ndhase2, Fig. 3B). Overall, RT-PCR analyses were able to confirm the genes found by microarray analyses (Fig. 3A).
Figure 3.
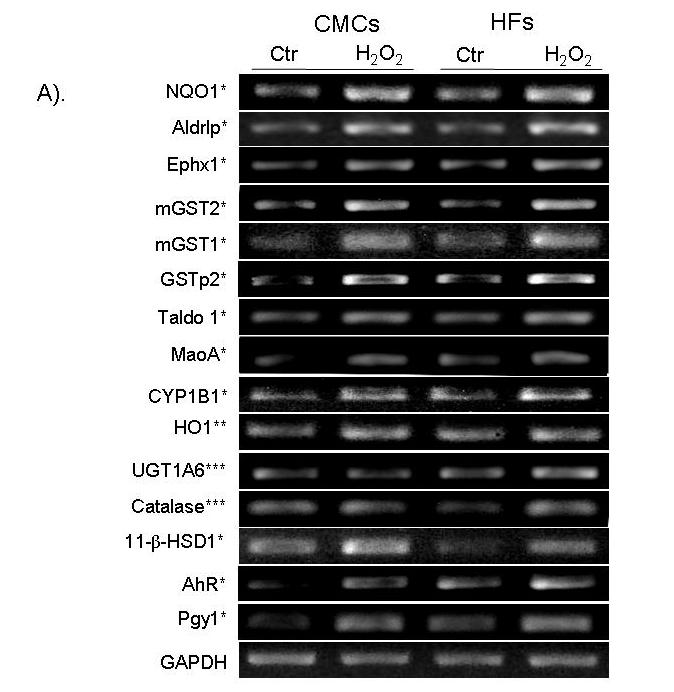

Confirmation of microarray results using semi-quantitative RT-PCR. RNAs were harvested for RT-PCR analyses as described in the Methods for upregulated genes in CMCs and HFs (A). An asterisk (*) indicates the genes found upregulated by H2O2 treatment in both CMCs and HFs by microarray analyses. Two (**) or three asterisks (***) indicate the upregulated genes were detected by microarray in only CMCs or HFs, respectively (A). The downregulated genes were measured in CMCs (B). GAPDH was used as a loading control (A, B).
Most of the genes in the category of antioxidant/detoxification enzymes that elevated expression after H2O2 treatment contain the cis-acting ARE in their promoters (24, 27-29). A luciferase reporter plasmid under the control of the ARE was used to investigate transcriptional activation of ARE-containing genes. This construct contains a core ARE sequence of TGACnnnGC in a 41 bp promoter sequence from the human NQO1 gene (43). Our results show that ARE-driven luciferase activity was significantly elevated 2 hr after 100 μM H2O2 treatment, peaking with a 7.5 fold induction at 4 hr post exposure (Fig. 4A). We have also performed a dose response study by harvesting cells at 4 hr after H2O2 treatment and found that 100 μM H2O2 was optimal for ARE activation (Fig. 4B). A mutant ARE (GC being replaced with AT in TGACnnnGC) was included as a negative control and H2O2 failed to activate the mutant ARE (Fig. 5A). Therefore the positive data from the promoter-reporter gene assay is reliant on a functional ARE for activation by H2O2.
Figure 4.

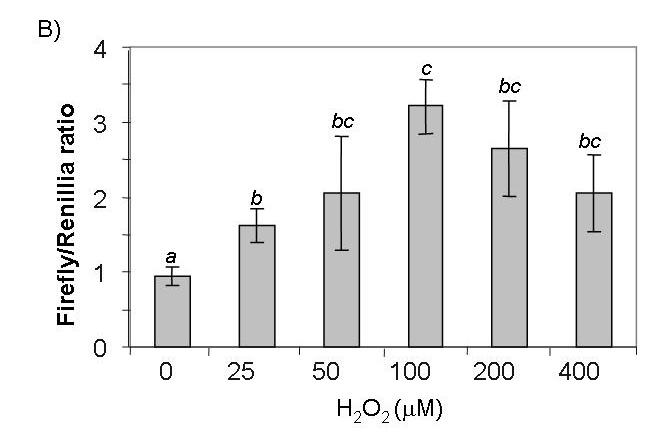
ARE-luciferase is induced by H2O2 in a time and dose-dependant manner. CMCs were cotransfected with 0.2 μg of ARE-luciferase and 0.04 μg of TK-driven renilla luciferase reporters. Serum-starved cells were treated with 100 μM (A) or various doses (B) of H2O2 for 10 min. Cells were placed in fresh medium and were harvested for luciferase assay at the indicated time points (A) or at 4 hrs (B) after H2O2 treatment. A letter indicates a significant difference (p<0.05) from the means labeled with a different letter as described in Figure 1.
Figure 5.
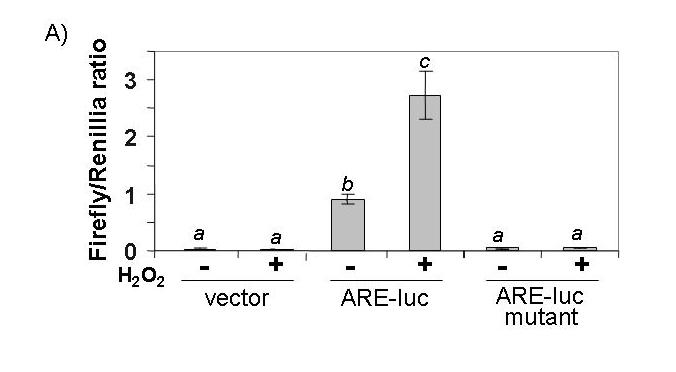
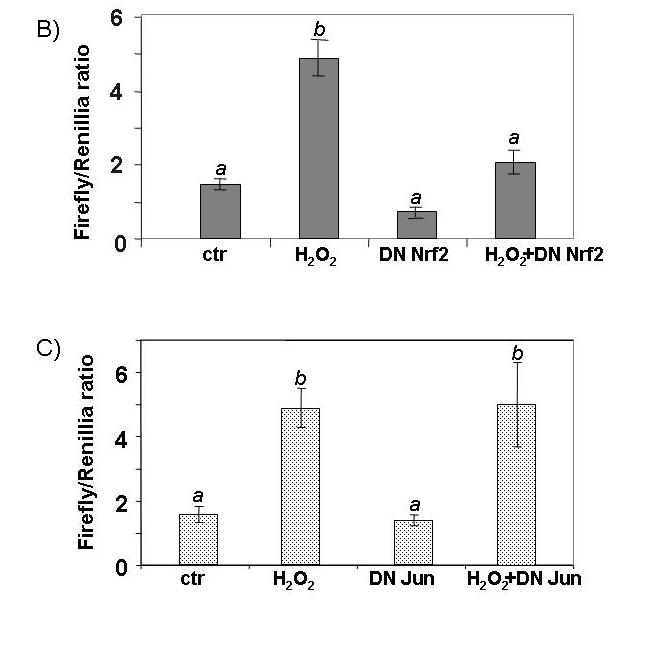
ARE-luciferase activity is dependant on a functional ARE and activity of Nrf2. CMCs were transfected with 0.2 μg of either ARE-luciferase, mutant ARE-luciferase or the empty vector (A). Equal amounts (0.05 μg) of empty vector (pEF2 for dominant-negative Nrf2 or pcDNA3.1HisC for dominant-negative c-Jun), dominant-negative Nrf2 or dominant-negative c-Jun (TAM67) were cotransfected with the ARE-luciferase (B, C). Transfected cells were treated with 100 μM H2O2 for 10 min and harvested 4 hrs later for measurements of luciferase activity. A letter indicates a significant difference (p<0.05) from the means labeled with a different letter as described in Figure 1.
To confirm a role of Nrf2 in H2O2 induced ARE activation, we used a dominant-negative Nrf2, which has the dimerization and DNA-binding domains but lacks the N-terminal half of the protein that contains a transactivation domain (44). Co-transfection of the dominant-negative Nrf2 with the ARE-luciferase construct reduced H2O2 induction of luciferase (Fig. 5B). Since the ARE consensus sequence resembles the AP-1 binding site, we excluded the possibility that the AP-1 transcription factor mediates ARE activation by co-transfection of a dominant negative c-Jun (Fig. 5B). This suggests that Nrf2 transcription factor serves as a key component in increasing the expression of the antioxidant/detoxification genes.
To test whether activation of the ARE pathway is sufficient to produce a preconditioning effect, we overexpressed Nrf2 in cardiomyocytes using a replication-deficient adenovirus. The adenovirus of Nrf2 coexpresses GFP (see Methods) and therefore the cells infected with GFP adenovirus serve as a negative control. A significant increase of Nrf2 protein was observed in total cell lysates of Nrf2 adenovirus infected cells (Fig. 6A). Nuclear localization of Nrf2 protein indicates that the transcription factor is active (Fig. 6A). An additional test to demonstrate that overexpressed Nrf2 is indeed functional is measurement of NQO1, a well-characterized target gene of the Nrf2 transcription factor. RT-PCR and Western blot analyses found that Nrf2 overexpression results in increased levels of NQO1 mRNA and protein (Fig. 6B&C).
Figure 6.
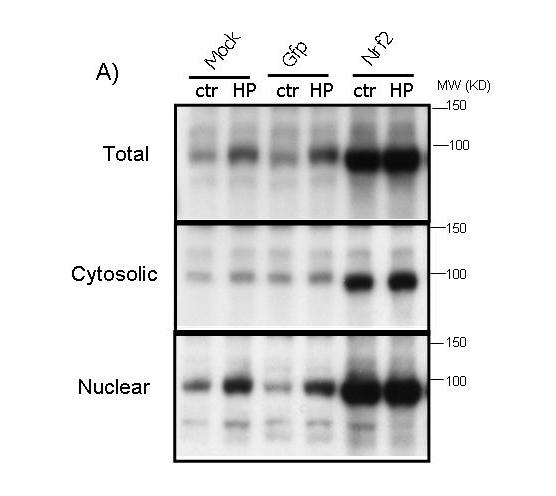
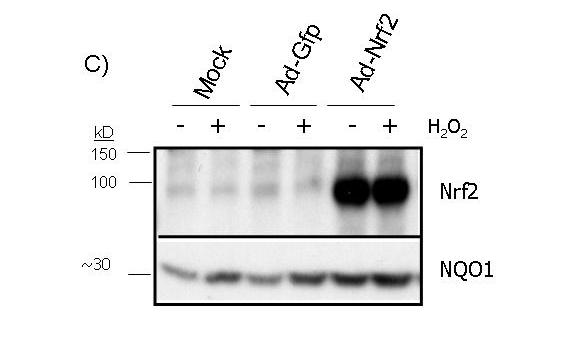
Overexpression of functional Nrf2 by recombinant adenoviral infection. CMCs were infected with replication-deficient adenovirus containing GFP alone or wild type Nrf2 that coexpresses GFP in a separate expression cassette. At 48 hrs after infection, cells were treated 10 mins with 100 μM H2O2 and were harvested 24 hrs later for Western blot (A, C) or RT-PCR (B). The cytosolic and nuclear fractions were harvested simultaneously for Western blot analyses as described in the Methods (A).
If the antioxidant/detoxification genes mediate the preconditioning effect of H2O2, overexpression of Nrf2 should produce resistance against Dox-induced apoptosis. Caspase 3 activity measurements from cells treated with various doses of Dox failed to reveal a resistance in Nrf2 overexpressing cells (Fig. 7A). In fact, a minor increase in caspase-3 activity was observed with Nrf2 overexpression (Fig. 7A). Counting the fraction of detached cells failed to show a protective effect of Nrf2 overexpression against Dox induced apoptosis (Fig.7B). Lack of protection with Nrf2 overexpression was also shown with Annexin V binding assay (Fig. 8). Nrf2 adenovirus infected cells did not show reduction in Annexin V staining compared to GFP adenovirus infected cells (Fig. 8 C & E). The overlap between green fluorescence (GFP) and red Annexin V staining in Nrf2 overexpressing cells argues against a protective effect of Nrf2 (Fig. 8E-H). Apoptosis is known to result in DNA cleavage. Agarose gel electrophoresis of DNA extracted from Dox treated cells show a smear due to DNA degradation (Fig 9). While pretreatment of H2O2 prevented DNA degradation, overexpresion of Nrf2 failed to do so (Fig 9).
Figure 7.
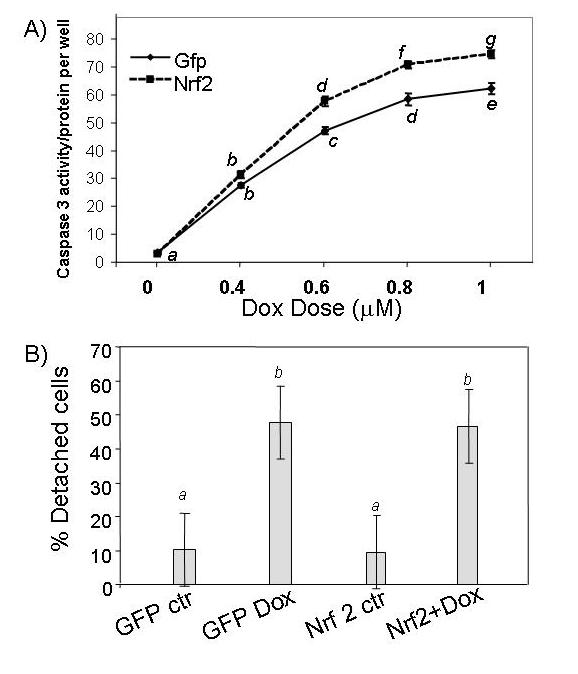
Ad-Nrf2 infection does not prevent Dox-induced Caspase-3 or cell detachment. Adenoviral-infected CMCs were subjected to 16 hr Dox treatment at the dose indicated for Caspase-3 activity assay (A). The portion of detached cells was counted under a microscope in the groups treated with 0.6 μM Dox for 16 hrs (B). A letter indicates a significant difference (p<0.05) from the means labeled with a different letter as described in Figure 1.
Figure 8.
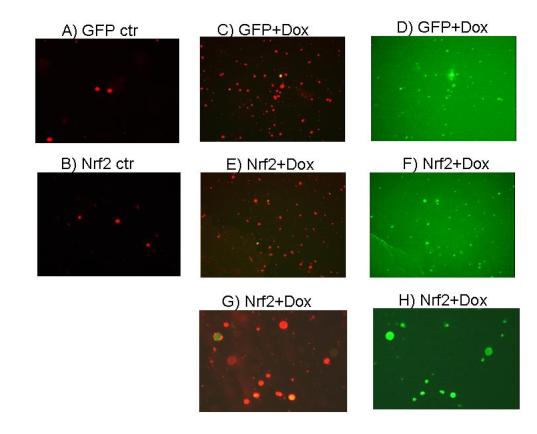
Annexin V binding in Ad-Nrf2 infected cells following Dox treatment. CMCs were infected with GFP or GFP-Nrf2 adenovirus. The cells were subjected to 16-hrs treatment of 0.6 μM Dox. Detached cells were collected for Annexin V staining. The images of Annexin V positive cells (red) or GFP positive (green) cells were recorded under a dual fluorescence channel (A, B, C, E and G) or a green fluorescence channel (D, F and H). A and B: GFP or GFP-Nrf2 adenovirus infected cells without Dox treatment. C and D: corresponding views of detached cells collected from Dox treated GFP adenovirus infected group. E-H: detached cells from Dox treated Nrf2 adenovirus infected group.
Figure 9.
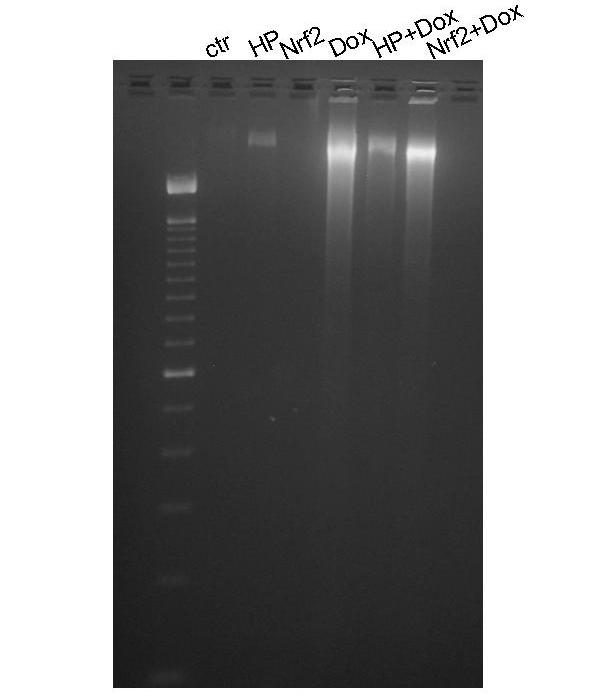
Nrf2 failed to prevent DNA degradation induced by Dox. CMCs with or without H2O2 pretreatment or Nrf2 adenovirus infection were treated with Dox for 16 hrs. DNA was extracted for ethidium bromide staining and separation by agarose gel electrophoresis.
Discussion
This study has evaluated the changes of gene expression as a result of oxidant exposure at the global level and investigated the role of antioxidant/detoxification genes in a preconditioning-like phenotype induced by H2O2. Microarray technology was used to determine the expression levels of 28,000 genes after H2O2 treatment. Manual category grouping or Cytoscape-based functional genomics of Affymetrix data output suggests that induction of antioxidant/detoxification genes predominates the response of CMCs to a mild dose of H2O2. Reduced expression of mitochondrial respiratory chain proteins and several muscle genes are also important features of CMCs surviving H2O2 treatment. Induction of antioxidant/detoxification response is not CMC cell type specific since HFs also show elevated expression of a cluster of antioxidant/detoxification genes.
Traditionally it is thought that induction of antioxidant and detoxification enzymes and increases in the reservoir of glutathione are important for the preconditioning effect. Promoter activity assays confirmed that the ARE is activated by H2O2 and that the Nrf2 transcription factor is responsible for ARE activation in CMCs. Nrf2 requires heterodimerization with a partner in order to bind to the ARE (27-30). This heterodimerization usually involves small Maf proteins, but regulation of ARE activity has also been attributed to the binding of Nrf2 by c-Jun, Bach-1 or Bach-2 proteins. Although we have eliminated the possibility of c-Jun by a dominant negative c-Jun cotransfection assay (Fig. 5C), the specific binding partner that contributes to H2O2-induced ARE activation remains to be elucidated. Regardless of the mechanism of ARE activation, we have determined the functional consequence of ARE activation by elevating the level of Nrf2 in CMCs. The transduced Nrf2 is fully functional as demonstrated by nuclear localization and increased expression of the NQO1 gene at mRNA and protein levels. However, unlike H2O2 pretreatment, overexpression of Nrf2 cannot produce resistance against Dox induced apoptosis. Our data suggest that activation of Nrf2-ARE pathway alone is not sufficient to induce the preconditioning effect.
H2O2 treatment induces many changes at gene expression levels not relevant to the Nrf2-ARE pathway. In the literature, oxidants have been shown to activate a number of transcription factors, including NF-kB, which serves as signaling integrator to regulate gene expression programs downstream of oxidative stress. Activation of NF-kB has been shown to regulate cell survival and cardiac hypertrophy (45, 46). Our microarray data indicate elevated expression of several cytokines and chemokines, suggesting a possible role of NF-kB in H2O2 induced cytoprotection. Whether NF-kB pathway collaborates with Nrf2-ARE pathway in regulating cell survival response in the paradigm of oxidative stress remains to be investigated. Regardless although the adaptive response induced by H2O2 may be important for cell survival, suppressed expression of mitochondrial and contractile genes may explain reduced contractility and pumping function in heart failure.
In apoptosis induced by chemicals and cytokines or death receptor ligands, the mitochondria play a key role in caspase activation. Mitochondrial fission, opening of mitochondrial membrane permeability transition pores and opening of channels formed by bax family members, cause the release of cytochrome c from the mitochondrial intramembrane space (47, 48). Cytochrome c in the cytosol triggers formation of the apoptosome and therefore activation of caspase-9, an initiator caspase (49, 50). Our microarray analyses reveal that a mild dose of H2O2 somewhat selectively depresses the expression of cytochrome c and several components of the mitochondrial respiratory chain. There is a possibility that lack of functional mitochondria or a reduced level of cytochrome c protein is responsible in part for the gain of resistance against Dox induced apoptosis in CMCs pretreated with H2O2.
Recent views on Nrf2 and ARE-induced gene activity have implicated this pathway as a universal organ protection mechanism (51). Nrf2 activity has been linked to stress resistance in diverse organs such as the brain, kidney, lungs and liver. Several chemopreventative compounds have been shown to induce antioxidant and detoxification genes (reviewed in (52)). Although Nrf2 has been studied in many tissues, the effect of Nrf2 in cardiac injury has not been considered prior to this study. The concentration of oxidants produced under various pathophysiological conditions is usually low and not sufficient to kill the majority of the cells. Using a dose of H2O2 that creates a scenario where the majority of the cells survive the treatment, we found that H2O2 activates the Nrf2 transcription factor, induces the expression of antioxidant/detoxification genes and elicits a resistance against Dox-induced apoptosis. The fact that Nrf2 overexpression failed to reproduce the resistance against Dox-induced apoptosis challenges the belief that induction of antioxidant/detoxification genes is sufficient for cytoprotection.
Acknowledgement
Works from our laboratory have been supported by the Burroughs Wellcome Foundation, American Heart Association, American Federation for Aging Research, Arizona Disease Control Research Commission, NIH R01 ES010826, and NIH RO1 HL076530-01 (QMC). We thank the Genomics Core facility of Arizona Cancer Center and Southwest Environmental Health Sciences Center (ES06694) for Microarray Analyses. We thank Dr. David Ross for NQO1 antibody. Sally Purdom was supported by NIH T32 ES007091.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cipollone F, Ciabattoni G, Patrignani P, Pasquale M, Di Gregorio D, Bucciarelli T, et al. Oxidant stress and aspirin-insensitive thromboxane biosynthesis in severe unstable angina. Circulation. 2000;102(9):1007–13. doi: 10.1161/01.cir.102.9.1007. [DOI] [PubMed] [Google Scholar]
- 2.Riley SJ, Stouffer GA. Cardiology Grand Rounds from the University of North Carolina at Chapel Hill. The antioxidant vitamins and coronary heart disease: Part II. Randomized clinical trials. Am J Med Sci. 2003;325(1):15–9. doi: 10.1097/00000441-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, et al. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol. 1998;3(6):1352–6. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- 4.Singh N, Dhalla AK, Seneviratne C, Singal PK. Oxidative stress and heart failure. Mol Cell Biochem. 1995;147(12):77–81. doi: 10.1007/BF00944786. [DOI] [PubMed] [Google Scholar]
- 5.Sawyer DB, Colucci WS. Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ Res. 2000;86(2):119–20. doi: 10.1161/01.res.86.2.119. [DOI] [PubMed] [Google Scholar]
- 6.Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol. 1996;28(2):506–14. doi: 10.1016/0735-1097(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 7.Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol. 1996;148(1):291–300. [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta M, Singal PK. Higher antioxidative capacity during a chronic stable heart hypertrophy. Circ Res. 1989;64(2):398–406. doi: 10.1161/01.res.64.2.398. [DOI] [PubMed] [Google Scholar]
- 9.Todd S, Woodward M, Bolton-Smith C, Tunstall-Pedoe H. An investigation of the relationship between antioxidant vitamin intake and coronary heart disease in men and women using discriminant analysis. J Clin Epidemiol. 1995;48(2):297–305. doi: 10.1016/0895-4356(94)00177-r. [DOI] [PubMed] [Google Scholar]
- 10.Kritharides L, Stocker R. The use of antioxidant supplements in coronary heart disease. Atherosclerosis. 2002;164(2):211–9. doi: 10.1016/s0021-9150(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 11.Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS) Lancet. 1996;347(9004):781–6. doi: 10.1016/s0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 12.Azen SP, Qian D, Mack WJ, Sevanian A, Selzer RH, Liu CR, et al. Effect of supplementary antioxidant vitamin intake on carotid arterial wall intima-media thickness in a controlled clinical trial of cholesterol lowering. Circulation. 1996;94(10):2369–72. doi: 10.1161/01.cir.94.10.2369. [DOI] [PubMed] [Google Scholar]
- 13.Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. New Engl J Med. 1993;328(20):1450–6. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 14.Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar G. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 15.Eisen A, Fisman EZ, Rubenfire M, Freimark D, McKechnie R, Tenenbaum A, et al. Ischemic preconditioning: nearly two decades of research. A comprehensive review. Atherosclerosis. 2004;172(2):201–10. doi: 10.1016/S0021-9150(03)00238-7. [DOI] [PubMed] [Google Scholar]
- 16.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001;104(25):3158–67. doi: 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- 17.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001;104(24):2981–9. doi: 10.1161/hc4801.100038. [DOI] [PubMed] [Google Scholar]
- 18.Zhou X, Zhai X, Ashraf M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation. 1996;93(6):1177–84. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]
- 19.Jin ZQ, Zhou HZ, Cecchini G, Gray MO, Karliner JS. MnSOD in mouse heart: acute responses to ischemic preconditioning and ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2005;288(6):H2986–94. doi: 10.1152/ajpheart.01144.2004. [DOI] [PubMed] [Google Scholar]
- 20.Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxidants Redox Signal. 2004;6(2):449–69. doi: 10.1089/152308604322899521. [DOI] [PubMed] [Google Scholar]
- 21.Tang XL, Takano H, Rizvi A, Turrens JF, Qiu Y, Wu WJ, et al. Oxidant species trigger late preconditioning against myocardial stunning in conscious rabbits. Am J Physiol Heart Circ Physiol. 2002;282(1):H281–91. doi: 10.1152/ajpheart.2002.282.1.H281. [DOI] [PubMed] [Google Scholar]
- 22.Toufektsian MC, Morel S, Tanguy S, Jeunet A, de Leiris J, Boucher F. Involvement of reactive oxygen species in cardiac preconditioning in rats. Antioxidants Redox Signal. 2003;5(1):115–22. doi: 10.1089/152308603321223603. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita N, Hoshida S, Taniguchi N, Kuzuya T, Hori M. A “second window of protection” occurs 24 h after ischemic preconditioning in the rat heart. J Mol Cell Cardiol. 1998;30(6):1181–9. doi: 10.1006/jmcc.1998.0682. [DOI] [PubMed] [Google Scholar]
- 24.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278(14):12029–38. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 2005;579(14):3029–36. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 26.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37(2):139–43. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 27.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10(11):549–57. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radical Biol Med. 2004;37(4):433–41. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 29.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radical Biol Med. 2004;36(10):1199–207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Ann Rev Pharm Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 31.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 32.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2004;291(5507):1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 33.Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207(12):77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- 34.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261(7):3068–74. [PubMed] [Google Scholar]
- 35.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261(7):3060–7. [PubMed] [Google Scholar]
- 36.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharm. 1999;57(7):727–41. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 37.Tu V, Bahl J, Chen Q. Signals of Oxidant-Induced Hypertrophy of Cardiac Myocytes: Key Activation of phosphatidylinositol 3-kinase and p70S6 kinase. J Pharm Exp Therap. 2002;300(3):1101–1110. doi: 10.1124/jpet.300.3.1101. [DOI] [PubMed] [Google Scholar]
- 38.Coronella-Wood J, Terrand J, Sun H, Chen QM. c-Fos phosphorylation induced by H2O2 prevents proteasomal degradation of c-Fos in cardiomyocytes. J Biol Chem. 2004;279(32):33567–74. doi: 10.1074/jbc.M404013200. [DOI] [PubMed] [Google Scholar]
- 39.Purdom S, Chen QM. Epidermal growth factor receptor-dependent and -independent pathways in hydrogen peroxide-induced mitogen-activated protein kinase activation in cardiomyocytes and heart fibroblasts. J Pharm Exp Therap. 2005;312(3):1179–86. doi: 10.1124/jpet.104.077057. [DOI] [PubMed] [Google Scholar]
- 40.Chen Q, Alexander D, Sun H, Xie L, Lin Y, Terrand J, et al. Corticosteroids Inhibit Cell Death Induced by Doxorubicin in Cardiomyocytes: Induction of Anti-apoptosis, Antioxidant and Detoxification Genes. Mol Pharm. 2005;67(6):1861–73. doi: 10.1124/mol.104.003814. [DOI] [PubMed] [Google Scholar]
- 41.Chen Q, Liu J, Merrett J. Apoptosis or Senescence-Like Growth Arrest: Influence of Cell Cycle Position, p53, p21 and bax in H2O2 Response of Normal Human Fibroblasts. Biochem J. 2000;347:543–551. doi: 10.1042/0264-6021:3470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Q, Tu V, Wu Y, Bahl J. Hydrogen Peroxide Dose Dependent Induction of Cell Death or Hypertrophy in Cardiomyocytes. Arch Biochem Biophys. 2000;373(1):242–248. doi: 10.1006/abbi.1999.1558. [DOI] [PubMed] [Google Scholar]
- 43.Moehlenkamp JD, Johnson JA. Activation of antioxidant/electrophile-responsive elements in IMR-32 human neuroblastoma cells. Arch Biochem Biophys. 1999;363(1):98–106. doi: 10.1006/abbi.1998.1046. [DOI] [PubMed] [Google Scholar]
- 44.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274(37):26071–8. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 45.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-kappaB: a key to survival. J Cell Sci. 2004;117(Pt 22):5197–208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 46.Force T, Haq S, Kilter H, Michael A. Apoptosis signal-regulating kinase/nuclear factor-kappaB: a novel signaling pathway regulates cardiomyocyte hypertrophy. Circulation. 2002;105(4):402–4. [PubMed] [Google Scholar]
- 47.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 48.Shi Y. A structural view of mitochondria-mediated apoptosis. Nature Struct Biol. 2001;8(5):394–401. doi: 10.1038/87548. [DOI] [PubMed] [Google Scholar]
- 49.Czerski L, Nunez G. Apoptosome formation and caspase activation: is it different in the heart? J Mol Cell Cardiol. 2004;37(3):643–52. doi: 10.1016/j.yjmcc.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 50.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Ann Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 51.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, et al. Nrf2, a multi-organ protector? FASEB J. 2005;19(9):1061–6. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Gordon GB. A strategy for cancer prevention: stimulation of the Nrf2-ARE signaling pathway. Mol Cancer Therap. 2004;3(7):885–93. [PubMed] [Google Scholar]