

Summary
Cancer stem cells are rare tumor cells characterized by their ability to self-renew and to induce tumorigenesis. They are present in gliomas and may be responsible for the lethality of these incurable brain tumors. In the most aggressive and invasive type, glioblastoma multiforme (GBM), an average of ±1 year spans the period between detection and death (1). The resistence of gliomas to current therapies may be related to the existence of cancer stem cells (2–6). We find that human gliomas display a stemness signature and demonstrate that HEDGEHOG (HH)-GLI signaling regulates the expression of stemness genes in and the self-renewal of CD133+ glioma cancer stem cells. HH-GLI signaling is also required for sustained glioma growth and survival, displaying additive and synergistic effects with temozolomide, the current chemotherapeutic agent of choice, which does not affect glioma stem cell self-renewal. Finally, interference of HH-GLI signaling with cyclopamine or through lentiviral-mediated silencing demonstrates that the tumorigenicity of human gliomas in nude mice requires an active pathway. Our results reveal the essential role of HH-GLI signaling in controlling the behavior of human glioma cancer stem cells and offer new therapeutic possibilities.
Results and discussion
Previous work provided evidence for a role of HH-GLI signaling in gliomas (7). SONIC HH (SHH) activates a signal transduction cascade that includes the action of the membrane proteins PATCHED1 (PTCH1) and SMOOTHENED (SMOH), leading to the action of GLI transcription factors (8). Analyses of a panel of fresh brain tumors (Fig. 1A, Fig. S1A) demonstrated the ubiquitous and varied expression of HH-GLI pathway components including GLI1, SHH and PTCH1, confirming the general presence of an active pathway (Fig. 1A, Figs. S1B, C, S2), as GLI1 transcription is the only reliable marker of pathway activity. GLI1 was the only gene overexpressed on average in all tumor types as compared with the average expression of three separate cortical regions of normal brain tissue, PTCH1 levels were lower in GBMs (9), and SHH was high only in a subset of low grade gliomas. Although some gliomas express SHH in tumor cells (7), a subset with low expression localized it in new vessels, whereas PTCH1 was indeed expressed in tumor cells (7; Fig. 1B).
Figure 1. Gene expression in human gliomas and requirement of HH-GLI signaling.
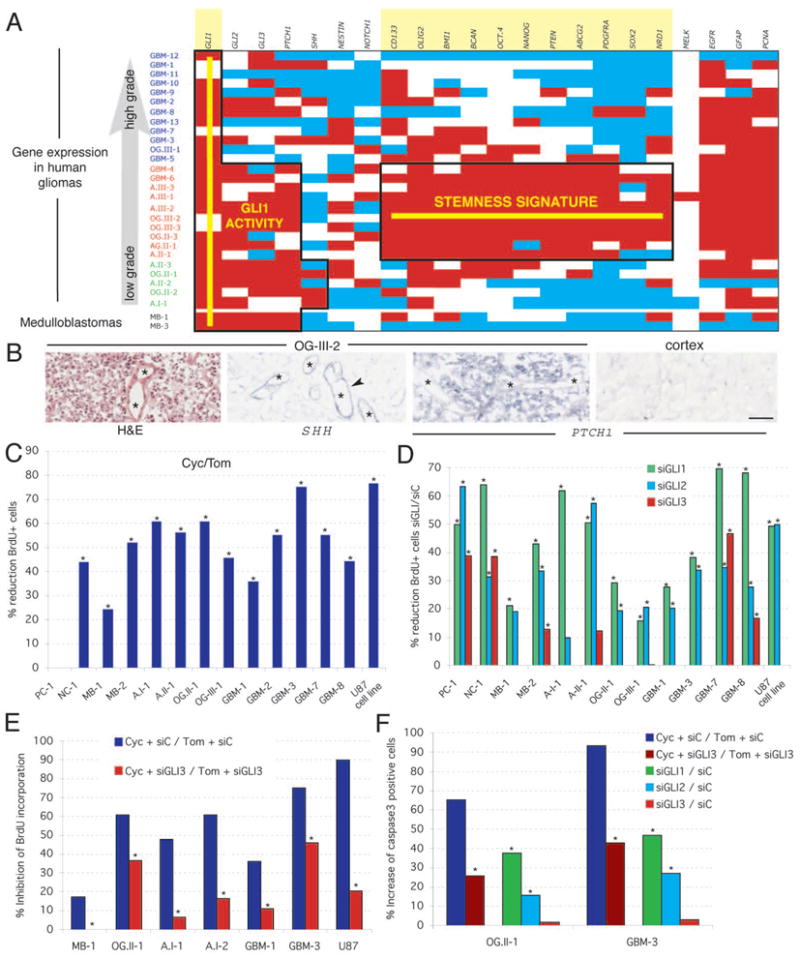
A) Diagram of expression levels using white for expression within normal range, red for overexpression and blue for underexpression. Normal expression ranges and thresholds were established by quantifying the expression level of the 22 markers used in three samples of normal cortex (Fig.S2). Gliomas are ranked by grade and by gene expression pattern as GBM (glioblastoma mulitforme or grade IV astrocytomas; all primary GBMs), A: astrocytoma, OG: oligodendroglioma, AG: Anaplastic glioma. The roman numeral denotes WHO grade and the arabic numeral the tumor number within our series. Two medulloblastomas (MB) are used as controls for HH-GLI pathway activity (7). Domains of consistent (>50% of cases) GLI1 activity and of the stemness signature are boxed and the names of the genes are highlighted.
B) In situ hybridization of fresh-frozen OG-III-2 samples as indicated with an H&E stain (right) as control. Arrows point to expression in vessels (asterisks).
C,D) Inhibition of glioma cell proliferation in primary adherent cultures (passages 1–3) of fresh tumors. Percent reduction in BrdU incorporation as a measure of cell division is shown as the ratio of the result of treatment with cyclopamine over the result of treatment with tomatidine used as control (C), or as the ratio of treatment with each specified GLI siRNA over an unrelated siRNA control (siC; D). Error bars are not shown for histograms denoting ratios but significance is provided by asterisks (p<0.05). PC: pineocytoma; NC: neurocytoma.
E,F) Rescue of the effects of cyclopamine by siRNA GLI3. The incompleteness of the effect is likely due to partial siRNA lipofection compared with the ubiquitous effects of the drug. GLI1 or GLI2 siRNAs did not rescue the effects of cyclopamine (not shown). Asterisks denote significative (p<0.05) changes.
Scale bar = 200μm for (B).
Gliomas showed a stemness signature (Fig. 1A, Fig. S2) that included the expression of the stemness markers NANOG, OCT4, SOX and BMI1, centered over grade III gliomas but extending into neighboring grade IV (GBM) and grade II tumors (Fig. 1A). Tumors in transition may display a mixed character, such as a stemness signature and multiforme appearance in GBM-4 and GBM-6, and low grade pathology and a stemness signature in some grade II tumors (Fig. 1A). In contrast, there was only a poor correlation between grade and a set of previously identified glioma prognostic markers (Fig. S3). Two medulloblastomas used as control expressed GLI1 but did not overexpress the same stemness gene set (Fig. 1A). The stemness signature of grade III gliomas suggests the abundance of cancer stem cells in these tumors.
All but one adherent primary cultures of gliomas tested decreased cell proliferation as measured by BrdU incorporation after 48h treatment with cyclopamine, a specific SMOH inhibitor (10), as compared with tomatidine as control (Fig. 1C). Inhibition was dose-dependent and cumulative over time (Fig. S4A). The GBM cell line U87 behaved similarly (Fig. 1C), decreased GLI1 expression upon cyclopamine treatment (Fig. S4B), and showed complete cell death after 10 days of treatment (Fig. S4C). Drug removal after 5 days allowed culture recovery, indicating temporally distinct cytostatic and cytotoxic effects.
GLI RNA interference (11) demonstrated the general requirement of GLI1 and GLI2 in brain tumor cell proliferation, with GLI1 knock-down having the strongest effects, as compared with an unrelated siRNA control (Fig. 1D). As for prostate tumors (11), most brain tumors tested had activation of the HH-GLI pathway at the level of SMOH in the membrane or above. However, since the one tumor that was insensitive to cyclopamine responded to GLI1 interference (Fig. 1D), activation here likely took place on or downstream of SMOH. The levels of inhibition reflected lipofection efficiencies and an independent set of siRNAs gave similar results (Fig. S5A, B). Interference with GLI1 and GLI2 also increased apoptosis (Fig. 1F). Long-term silencing in U87 cells increased in cell death after interference with GLI1 but not GLI2 (Fig. S5C), indicating their differential requirement for survival in these cells. GLI3, in contrast, had positive effects in only a minority of cases (Fig. 1D), consistent with its rare role as a minor activator and its more common role as a major repressor of GLI targets (8). GLI3 was found to mediate the effects of cyclopamine: cells transfected with GLI3 siRNA or a control siRNA and then treated with this drug showed that the anti-proliferative and pro-apoptotic effects of cyclopamine were reverted by inhibition of GLI3 (Fig. 1E, F).
To test for a possible role of HH-GLI signaling in cancer stem cells, we have used glioma stem cell cultures (gliomaspheres) (Fig. 2A, B), which self-renew and mimic the original tumor after transplantation into the brains of immunocompromised mice (5). Treatment with cyclopamine decreased their proliferation and increased apoptosis in a concentration-dependent manner (Fig. 2C, D). Inhibition of cell proliferation was recapitulated by transduction with a recombinant lentiviral vector expressing an shRNA specific for SMOH (LV-shSMOH) as compared with a control lentiviral vector (LV-control) (Fig. 2E). Inhibition of SMOH mRNA by the LV-shSMOH was at 67% in U87 cells and at 66% in GBM-8 72h post-transduction.
Figure 2. Requirement of HH-GLI function in glioma stem cell cultures and combined effects with temozolomide.
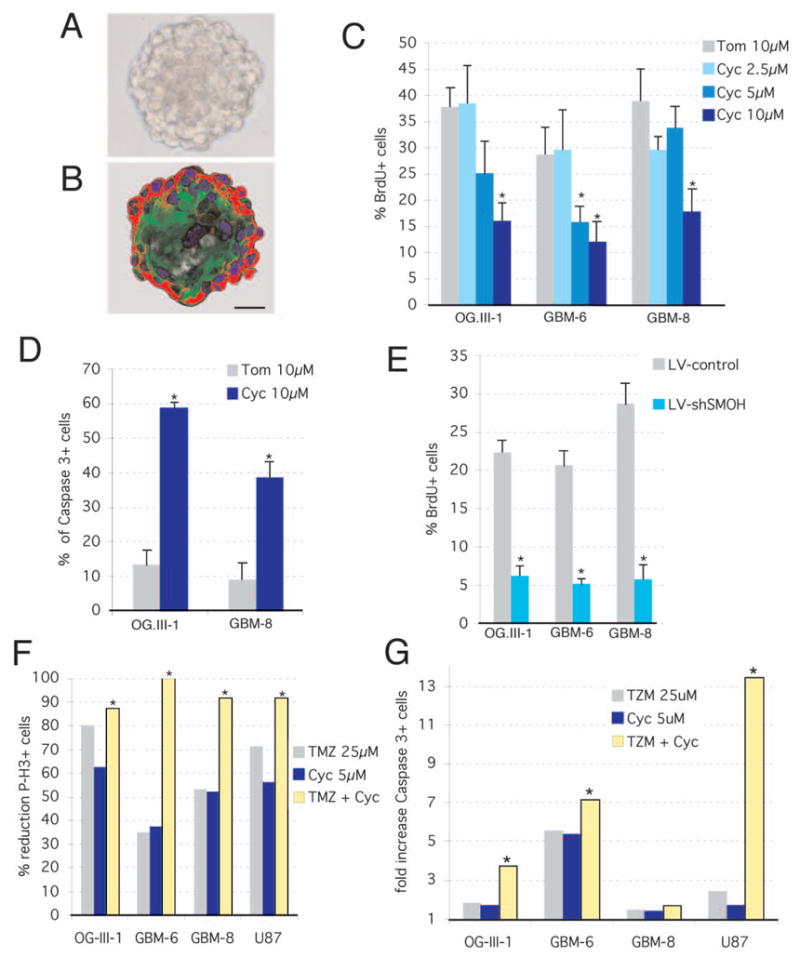
A,B) Phase-contrast and fluorescent images after immunolabeling with GFAP (green) and Nestin (red) antibodies, respectively, of GBM-8 gliomaspheres. C–E) Decrease in cell proliferation as measured by BrdU incorporation
C,E) or increase in apoptosis measured by activated Caspase-3+ (D) in gliomaspheres treated with cyclopamine (Cyc) or tomatidine (Tom) (C,D) or transduced with LV-control or LV-shSMOH (E).
F,G) Synergistic or additive effects of intermediate doses of cyclopamine and TMZ on inhibition of cell proliferation and apoptosis as measured by phospho-Histone3+ (F) or Caspase3+ labeling (G), respectively. All combinations are significantly more effective (p<0.05; asterisks) as compared with cyclopamine alone and all but OG-III-1 as compared with TMZ alone (p<0.05). Scale bar = 140μm (A,B).
Long-term treatment and recovery assays showed that 20, but not 10, days of treatment with cyclopamine were sufficient to kill all gliomasphere cells as the culture did not recover after an additional 20 days without the drug (Fig. S6E). In contrast, the DNA-damaging chemotherapeutic agent temozolomide (TMZ; 1) also affected the proliferation and survival of glioma cells (Fig. S6A-D) but was unable to prevent recovery (Fig. S6F). Importantly, TMZ showed additive or synergistic effects in different glioma stem cell cultures with an intermediate dose (5μM) of cyclopamine on the inhibition of cell proliferation and the enhancement of apoptosis (Fig. 2F, G).
Intracraneal xenografts of untransduced, LacZ-lentiviral-transduced GBM gliomaspheres induced the development of massive infiltrative gliomas in nude mice after 2.5 months (Fig. 3A). These tumors expressed GLI1, SHH and PTCH1 (Fig. S7), consistent with the expression of these genes in gliomas by in situ (7). Using a protocol with 104 GFP-lentiviral-transduced gliomasphere cells injected into the striatum and left for 2 weeks prior to treatment for an additional 2 weeks we showed that cyclopamine had a beneficial effect. Systemic IP treatment with cyclopamine, but not with carrier alone, reduced glioma tumor volume (Fig. 3B) and cell proliferation (Fig. 3C). Similar effects were seen with subcutaneous xenografts of U87 glioma cells: Delivery of cyclopamine in and around the tumor inhibited all growth and the tumors disappeared (Fig. 3I, J, L), showing a size differential with controls as soon as the second day of treatment.
Figure 3. Inhibition of human glioma xenograft growth in vivo by interference with HH-GLI signaling.
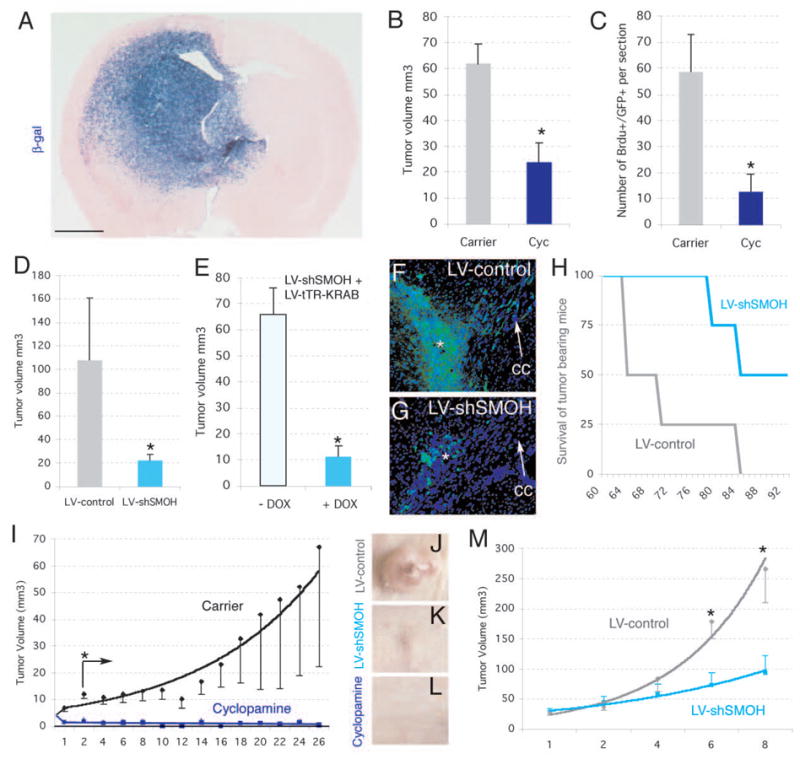
A) Development of invasive gliomas after intracerebral implantation of gliomaspheres transduced with a lentiviral vector expressing LacZ ±1 month after implantation of 105 cells (n=7 mice for GBM-8; n=7 mice for GBM-7 and n=7 mice for OG-III-1, not shown). Glioma stem cell-derived tumor cells infiltrate into the parenchyma (cells are blue after X-Gal cytochemistry).
B,C) Inhibition of intracraneal tumor growth after treatment with cyclopamine (n=3 mice for OG-III-1, n=3 mice for GBM-8, not shown) but not carrier alone (n=3 mice for OG-III-1, n=3 mice for GBM-8, not shown). Tumor volume (B) or BrdU+ cell number (C) were monitored in section reconstructions.
D) Tumor volume was also decreased after LV-shSMOH transduction (n=3 mice) but not after LV-control transduction (n=3 mice).
E) Infection with GBM-8 gliomasphere cells with LV-shSMOH and LV-tTR-KRAB (see supplemental information) reduced tumor volume only after induction by DOX (n=3 mice with DOX treatment and 3 mice without).
F,G) Representative examples of intracraneal gliomas ±1 month after implantation of 104 GFP+ gliomapsheres treated as described. The asterisk denotes the injection site. Note the strong migration of tumor cells contralaterally along the corpus callosum (cc, arrow) in control (G) but not LV-shSMOH-expressing cases (G).
H) Survival of mice harboring LV-control (n=4) or LV-shSMOH-transduced (n=4) cells. Mice were sacrificed at the first signs of terminal disease before extreme pain was apparent.
I-M). Inhibition of subcutaneous U87 gliomas by injection of cyclopamine (I, L; n=8 tumors) or interference with SMOH (LV-shSMOH; J,K,M; n=6 tumors), but not in U87 tumors treated with carrier alone (I and not shown; n=8 tumors) or infected with LV-control (J,M; n=6 tumors). Asterisks denote significative changes (p<0.05).
Scale bar= 0.25cm (A), 0.8mm (F,G) and 8mm for (J–L).
Similar results were obtained after the cell-autonomous knock-down of SMOH. LV-shRNA-SMOH reduced SMOH mRNA levels by 65–85% in U87 and GBM-8 cells, reduced GLI1 levels by ±45% in both, markedly reduced tumor volume, and reduced BrdU incorporation 2–3-fold (Fig. 3D, F, G, J, K, M and not shown). To further control any differences between the experimental and control injected cells, we rendered LV-shSMOH silent and conditionally inducible by treatment with doxycycline (DOX) through co-transduction with LV-tTR-KRAB (14). Two weeks after intracranial injection of 104 co-transduced GBM-8 cells, one half of the mice were treated with DOX for an additional 2 weeks while the other half were left untreated. DOX-treatment, inducing the silencing of SMOH, reduced tumor volume by ±80% as compared with mice that did not receive DOX (Fig. 3E). Long-term analysis of straight LV-shSMOH and LV-control transduced GBM-8 cells demonstrated the greater survival of LV-shSMOH harboring mice versus LV-control mice (Fig. 3H).
The stemness signature of grade III gliomas but not of most GBMs contrasts with the ability of both to form gliomaspheres. If GBM cancer stem cells are simply less abundant than those in grade III tumors, the selection of cancer stem cells in gliomasphere cultures could yield a similar enrichment. The expression patterns of 25 selected genes in 7 gliomasphere cultures (Figs. 1A, 4A, S8) identified a subgroup of genes displaying expression within a 2-fold variation from the mean, or over-expression, in all spheres. This included GLI1, PTCH1, NANOG, OCT4, SOX2, BMI1 and PCNA. The latter indicated similar proliferation of all sphere cultures. Different gliomaspheres therefore have active GLI1 function and might have similar numbers of cancer stem cells assuming similar expression per cell.
Figure 4. Effects of HH-GLI signaling on self-renewal and on CD133+ glioma cancer stem cells.
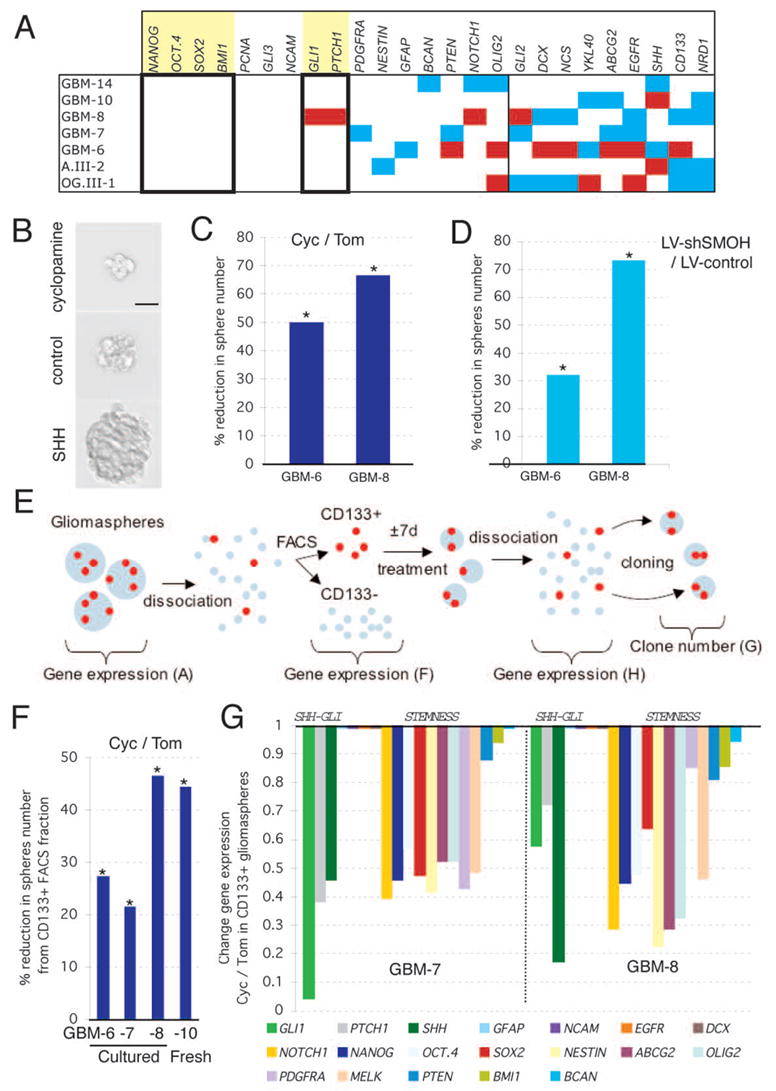
A) Profile of the expression of selected genes in 7 gliomasphere stem cell cultures. White denotes normal expression within 2-fold up and 50% down from the average of all values per gene without counting the outliers; red >2-fold overexpression and light blue <2-fold underexpression (Fig. S8). Boxed and highlighted are stemness genes that show normalcy in all samples, or enhanced expression like GLI1 and PTCH1.
B) Representative image of gliomaspheres (GBM-8) treated with cyclopamine (top), tomatidine (middle) or SHH (bottom) for 7 days in a cloning assay.
C,D) Reduction of the number of secondary gliomaspheres, as a measure of self-renewal, after interference with SMOH function through treatment with cyclopamine (C) or RNA interference (D). Ratios are shown as percentage of reduction. Asterisks denote significant changes (p<0.05) over controls as indicated.
E) Schematic diagram of the dissociation, FACS analyses, treatment and cloning methods used to determine the self-renewal properties and expression profile of CD133+ cells (see F–H).
F) Inhibition of clonogenicity of CD133+ cells isolated from three glioma stem cell cultures and one fresh tumor. Ratios are cyc/tom and asterisks denote significant changes (p<0.05).
G) Gene expression changes in gliomaspheres derived from CD133+ cells treated for 7d with cyclopamine or tomatidine used in (F). Changes are shown as cyc/tom ratios and columns show the decrease in gene expression. E.g. in GMB-7 GLI1 is reduced to 5% of its normal expression. Note that the expression of GFAP, NCAM, EGFR, DOUBLECORIN (DCX) and BCAN is not altered.
Scale bar = 80μm for (B).
To test for a link between HH-GLI signaling and stemness we quantified the ability of dissociated, single gliomasphere cells to generate secondary spheres as a measure of self-renewal (Fig. 4B, E). Treatment of dissociated gliomasphere cells with cyclopamine for 7 days, prior to renewed dissociation and cloning without the drug, resulted in a ±23–65% reduction in clones as compared to tomatidine treatment (Fig. 4C). Because 7 days allows for ±5 cell divisions (calculated from BrdU incorporation data), giving rise to a 32-cell gliomasphere derived from a single cell (assuming constant division rates), and the cloning rate was at 6–7%, there are on average 2 stem cells per 32-cell sphere. Since cyclopamine treatment for 7d also decreased sphere volume by ±50% (Fig. 4B), as it also affected growth, we deduced that cyclopamine treatment reduced the number of cancer stem cells to 0.25 per sphere on average, thus rendering most of them non-viable, consistent with the lack of recovery after long-term cyclopamine treatment (Fig. S6E). Transient inhibition of SMOH (with DOX) for 7 days prior to cloning mimicked the results obtained with cyclopamine (Fig. 4D). Similar treatment with recombinant SHH, in contrast, increased the number of clones by ±145–240% (Fig. S9A). As SHH treatment increased sphere volume ±3–4-fold on average (Fig. 4B), it induced an increased in the number of glioma stem cells to ±12–15 per sphere. Similar experiments with TMZ showed that it does not significantly affect cancer stem cell self-renewal (p=0.3, 50μM TMZ versus DMSO-treatment).
We used CD133 expression to purify glioma-initiating cancer stem cells by FACS (5–6) (Fig. 4E). The percentage of secondary clones from CD133+ FACS-sorted GBM-8 cells was 40-fold higher as compared with CD133− cells. The percentage of secondary spheres from CD133+ FACS-sorted cell from 4 gliomasphere cultures was lower than the percentage of CD133+ cells (Fig. 4F), raising the possibility that not all CD133+ cells are cancer stem cells as determined by clonogenic behavior. Analysis of a subset of genes in CD133+ versus CD133− cells showed that SHH was enriched in all samples whereas NOTCH1 was enriched in all efficient cultures (Fig. 4F; Fig. S10), revealing a correlation between efficiency of sphere formation (quality) and stemness marker expression (Fig. S10).
Cyclopamine treatment of CD133+ cells for 7 days prior to cloning without the drug (see Fig. 4E) reduced the number of clones by ±20–45% (Fig 4F). This result was observed in sphere cultures after several passages as well as in CD133+ cells freshly purified from a patient’s GBM (Fig. 4F). Conversely, SHH treatment increased secondary sphere formation by 120–140% (Fig. S9B). The decrease in clonogenicity resulting from inhibition of endogenous HH-GLI signaling was correlated with a coordinated decrease in GLI1, PTCH1 and SHH expression at the time of cloning and with the specific and coordinated downregulation of stemness genes (Fig. 4G). Treatment of freshly FACS-sorted CD133+ cells with cyclopamine for only 4h resulted in a decrease in GLI1, PTCH1, NANOG, SOX2 and OCT4 expression by 50%, 40%, 10%, 57% and 20% for GBM-8 (and in GMB-6, not shown), respectively, as compared with cyclopamine-treated CD133- cells, strongly suggesting the direct inhibition of HH-GLI signaling and stemness in CD133+ cells.
Here we demonstrate that human gliomas and their cancer stem cells require HH-GLI pathway activity for proliferation, survival, self-renewal and tumorigenicity. The widespread expression of GLI1 in gliomas is consistent with its isolation from a glioma cell line (15) and its expression in brain tumors in situ (7). The discrepancy with previous work, suggesting that GLI1 was not overexpressed in gliomas (16), can be partly explained by the fact that the cerebellum was used as the baseline, which we now know expresses GLI1 at the highest levels (17). We have also uncovered a stemness signature that we interpret to denote the abundance of cancer stem cells in grade III gliomas. Cancer stem cells in GBMs may become diluted by the increased number of more differentiated tumor-derived cells, which gives them their multiforme character, and by the enhanced participation of non-tumor cells in GBMs, for instance through increased angiogenesis. Interestingly, angiogenesis in GBMs could not only provide required nutrients to the tumor but also enhance its growth and cancer stem cell self-renewal in cases when new vessels express SHH. Whereas glioma cancer stem cells express markers of endogenous stem cells (4,18,19), the origin of the former is unclear. They could arise through a dedifferentiation process or derive from precursors or endogenous brain stem cells (18,20–22). Paralleling the derivation of medulloblastomas from SHH-dependent cortical cerebellar cells (7,17,23), glioma stem cells could derive from endogenous GLI1+ cells, including those found in stem cell niches (7,17,24–27).
Our results argue for the development of antagonists of GLI1,2 and agonists of GLI3R as anti-cancer agents. Since cyclopamine induces both of these changes simultaneously and shows additive and synergistic effects with TMZ, a combinatorial approach with these agents may be advantageous to treat the tumor bulk and its cancer stem cells. The findings that HH-GLI signaling is involved in regulating the proliferation of both endogenous brain stem cells (24–27) and glioma cancer stem cells (this work), and due to the lack of obvious secondary effects in cyclopamine-treated adult mice (25,28, this work), we suggest that such treatments may spare normal quiescent stem cells in their niches, likely allowing regeneration of any damage to normal adult tissues after cessation of treatment.
Supplementary Material
Acknowledgments
We thank lab members for discussion, H. Weiner for initial brain tumor samples and D. Trono for lentivectors. This work was supported by a private donation from the Lombard-Odier-Darier-Hentsch bank to NdT and grants from the Louis-Jeantet Foundation, the Leenaards Foundation, the Swiss National Foundation, the Swiss Cancer League and the NIH to ARA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253–1265. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 2.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 3.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 5.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 6.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 7.Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz i Altaba A. The SHH-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 8.Ruiz i Altaba A, editor. HEDGEHOG-GLI Signaling in Human Disease. Landes Bioscience; Plenum Publishers (Georgetown): 2006. [Google Scholar]
- 9.Katayam M, Yoshida K, Ishimori H, Katayama M, Kawase T, Motoyama J, Kamiguchi H. Patched and smoothened mRNA expression in human astrocytic tumors inversely correlates with histological malignancy. J Neurooncol. 2002;59:107–115. doi: 10.1023/a:1019660421216. [DOI] [PubMed] [Google Scholar]
- 10.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Inhibition of prostate cancer proliferation by interference with Hedgehog-GLI1 signaling. Proc Nat Acad Sci USA. 2004;101:12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Nature. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 14.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinzler KW, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 16.Xiao H, Goldthwait DA, Mapstone T. A search for gli expression in tumors of the central nervous system. Pediatr Neurosurg. 1994;20:178–82. doi: 10.1159/000120783. [DOI] [PubMed] [Google Scholar]
- 17.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 18.Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, Vandenberg S, Alvarez-Buylla A. PDGFRa-Positive B cells are neural Stem Cells in the Adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 19.Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 20.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, Depinho RA. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 22.Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61:3826–3836. [PubMed] [Google Scholar]
- 23.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 24.Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–345. doi: 10.1242/dev.00930. [DOI] [PubMed] [Google Scholar]
- 25.Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Ruiz i Altaba A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 27.Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA, Rubin LL, Dudek H, McMahon AP, Fishell G. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech Dev. 2005;122:223–230. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.