

Abstract
Rosiglitazone is an FDA-approved oral antidiabetic agent for the treatment of type 2 diabetes. This compound improves insulin sensitivity through the activation of the nuclear receptor, peroxisome proliferator-activated receptor-γ (PPAR-γ). In addition to sensitizing cells to insulin, the PPAR-γ2 isoform appears to be critical for the regulation of osteoblast and adipocyte differentiation from common mesenchymal bone marrow progenitors. We have demonstrated previously that PPAR-γ2 activated with rosiglitazone acts as a dominant inhibitor of osteoblastogenesis in murine bone marrow in vitro. Here, we show that in vivo, rosiglitazone administration results in significant bone loss. When rosiglitazone (20 μg/g body weight/d) was given to 6-month-old, nondiabetic C57BL/6 mice for 7 wk, a significant decrease in total body bone mineral density was observed. Analysis of bone microarchitecture, using micro-computed tomography, demonstrated a decrease in bone volume, trabecular width, and trabecular number and an increase in trabecular spacing. Histomorphometric analysis showed a decrease in bone formation rate, with a simultaneous increase in fat content in the bone marrow. Changes in bone morphology and structure were accompanied by changes in the expression of osteoblast- and adipocyte-specific marker genes; the expression of the osteoblast-specific genes Runx2/Cbfa1, Dlx5, and α1(I)collagen were decreased, whereas the expression of the adipocyte-specific fatty acid binding protein aP2, was increased. These in vivo data suggest that rosiglitazone therapy may pose a significant risk of adverse skeletal effects in humans.
Glitazones compose a new class of oral antidiabetic agents (1). Two of them, rosiglitazone and pioglitazone, are commonly used for the treatment of type 2 diabetes (2). The primary pharmacological actions of glitazones are an improvement in muscle and adipose insulin sensitivity and the inhibition of hepatic gluconeogenesis (3). In patients with type 2 diabetes, rosiglitazone reduces levels of fasting plasma glucose, glycosylated hemoglobin, insulin, and C-peptide. Moreover, rosiglitazone may potentially sustain and improve β-cell function (1). Recent studies also indicate that glitazones, including rosiglitazone, may be beneficial for the treatment of atherosclerosis, cancer, and brain inflammation that accompany Alzheimer’s disease (4–7).
Rosiglitazone is a high-affinity ligand and activator of the peroxisome proliferator-activated receptor-γ (PPAR-γ), and most of its effects are mediated via this transcription factor (8–10). PPAR-γ is a member of the nuclear receptor superfamily of transcription factors, a large and diverse group of proteins that mediate ligand-dependent transcriptional activation and repression (10). PPAR-γ exists in two isoforms, PPAR-γ1 and PPAR-γ2, as a result of alternative promoter usage and alternative splicing (11). The PPAR-γ1 isoform is expressed in many cell types, including adipocytes, osteoblasts, muscle cells, and macrophages, whereas PPAR-γ2 expression is restricted primarily to adipose cells and is absolutely necessary for fat development in mice (12).
Osteoblasts, or bone-forming cells, share a common mesenchymal precursor in the bone marrow with adipocytes (13, 14). Osteoblast development is controlled by several phenotype-specific transcription factors, among them Runx2/Cbfa1 and Dlx5, whereas formation of terminally differentiated adipocytes in murine marrow requires the activity of PPAR-γ2 (15–19). We demonstrated previously that in the presence of PPAR-γ2, rosiglitazone converts cells of the osteoblast lineage to terminally differentiated adipocytes and simultaneously and irreversibly suppresses the osteoblast phenotype (17, 18). PPAR-γ2 inhibited the ability of osteoblasts to mineralize the extracellular matrix, a hallmark of their activity, and suppressed the expression of Runx2/Cbfa1 and other osteoblast-specific genes such as γ1(I)collagen, osteopontin, alkaline phosphatase, and osteocalcin (17).
To date, there are limited reports of the effects of glitazone administration on human bone. An analysis of the data from the Health, Aging, and Body Composition cohort studies revealed that intake of glitazones (rosiglitazone or pioglitazone) for longer than 24 months by older (age 70–79 yr) diabetic patients decreased bone mineral density (BMD) in the femoral neck and hip (20). Although analysis was performed on only a small group of patients, these results suggest a strong correlation between decreases in BMD, glitazones, and the duration of therapy.
The potential for the broad clinical application of the glitazones for treatment of a variety of pathologies demands detailed analysis of the possible effects of these compounds on target organs that are known to express PPAR-γ. Given that PPAR-γ is expressed in bone, particularly in mesenchymal stem cells, together with our in vitro evidence that rosiglitazone-activated PPAR-γ2 functions as a dominant negative regulator of osteoblast differentiation (17), we tested the effect of in vivo administration of rosiglitazone on murine bone.
Materials and Methods
Animals and treatment regime
Nondiabetic 6-month-old male C57BL/6 mice were obtained from the colony maintained by National Institute of Aging under contractual agreement with Harlan Sprague Dawley, Inc. (Indianapolis, IN). Animals, identified by clipped toes, were housed (four per cage) with free access to water and were maintained at a constant temperature on a 12-h light-dark cycle. The animal treatment and care protocols conformed to National Institutes of Health guidelines and were performed using a University of Arkansas for Medical Sciences Institutional Animal Care and Use Committees-approved protocol.
Animals (n = 8 per group) were fed 5 g of food per day with pelleted chow (F05072, Bio-Serv, Frenchtown, NJ) supplemented with rosiglitazone maleate (Avandia, GlaxoSmithKline, King of Prussia, PA) at the concentration of 0.14 mg/g of chow. The control group was fed the same amount of nonsupplemented chow. Animals were fed every other day for 7 wk, and food intake per cage and body weights of individual animals were monitored. At the end of the experiment the average intake of rosiglitazone per gram body weight per cage was calculated and resulted in the dose of 20 μg/g · d. To measure bone formation rates, mice were injected ip with 30 μg/g body weight of tetracycline 7 and 2 d before they were killed.
Blood collection and plasma glucose measurements
Plasma glucose levels were determined for each animal at the beginning and end of the experiment. Animals were fasted for 18 h before blood (from the tail vein) was collected. Plasma glucose levels were determined based on the enzymatic reaction of β-D-glucose with oxygen using Beckman glucose analyzer 2 (Beckman Coulter Inc., Fullerton, CA).
Liver histology
Livers were collected immediately after the mice were killed, snap-frozen, and sectioned. Sectioned livers of representative animals were fixed with formalin (37%), stained with Oil Red O for fat, and counter-stained with methyl green, as described (18).
BMD measurements
BMD was determined using the small-animal dual-energy x-ray absorptiometry (DXA) instrument Piximus (GE Lunar, Madison, WI) and software version 1.46 (21, 22). Mice were anesthetized and scanned before the onset of rosiglitazone treatment, at 4 wk and after killing at 7 wk. Total body BMD (g/cm2), excluding the head region, was obtained from each scan. The percent change in BMD was determined by inserting the values collected for each time point into the calculation: [(Vposttreatment – Vpretreatment)/Vposttreatment] × 100. Internal variations of repeated measures of total murine body BMD have been determined to be 1.7–2.0%.
Micro-computed tomography (micro-CT) analysis
After mice were killed, the right tibia of each animal was dissected and fixed in 4 C Millonig’s phosphate-buffered 10% formalin, pH 7.4. After 24 h, the tibia was dehydrated successively in 70%, 95%, and 100% ethanol and then measured without further sample preparation in a micro-CT 40 (Scanco Medical, Bassersdorf, Switzerland). Micro-CT scans were performed on 256 successively measured slices (12 μm each) to total 3.07 mm of the proximal metaphysis. The three-dimensional information was obtained by stacking the measured slices on top of each other. The volume of interest for subsequent morphometric analysis in three dimensions was the entire secondary spongiosa after extracting automatically the surrounding cortical bone (length of 3.07 mm of the radial axis) and avoiding inclusion of boundaries of the volume of interest (23, 24). The complete secondary spongiosa of the proximal tibia was evaluated to avoid sampling errors incurred by random deviations within a single section. Careful contouring of this region in the secondary spongiosa yields volumetric information of bone volume (BV), total volume (TV), and calculated ratio of BV/TV, as well as trabecular thickness (TbTh), trabecular number (TbN), and trabecular spacing (TbSp).
Estimation of the plate-rod characteristics of the specimen was achieved using the structure model index (SMI). For an ideal plate and rod structure, the SMI values are 0 and 3, respectively. For a mixed structure, the values are between 0 and 3, depending on the volume ratio between rods and plates. The geometrical degree of anisotropy (DA) is defined as the ratio between the maximal and the minimal radius of the mean intercept length ellipsoid. The connectivity density (ConnD) is calculated using the Euler method. The computation of both DA and ConnD are described in detail elsewhere (23).
Bone histomorphometry
After micro-CT analysis, undecalcified tibiae were embedded in methyl methacrylate, sectioned on an automatic, retractable Microtom 355 with a D-profile, tungsten carbide steel knife at 4 μm. Adjacent sections were stained with Masson trichrome, Goldner trichrome, and Von Kossa, and one was left unstained for tetracycline labeling evaluation (25, 26). The histomorphometric examination was performed using an OsteoMeasure system, which includes a Nikon microscope with motorized stage, interfaced with a computer and digitizer tablet (OsteoMetrics Inc., Atlanta, GA). All cancellous measurements were two-dimensional, confined to the secondary spongiosa, and made using a × 40 objective (numerical aperture 0.75). The terminology and units used were those recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research (27). Static measurements and dynamic lengths of single- and double-labeled bone surfaces were obtained as described previously (25, 26). Static measurements were performed on six representative fields per bone sample and included fat volume [fat volume per total volume (BV/TV, percent)] and adipocyte number per high-power field. Histodynamic parameters of mineralized surface per bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate per bone surface (BFR/BS) were measured on the same specimens as above, using six representative fields per bone sample, in epifluorescent light.
RNA isolation and quantitative real-time RT-PCR analysis
Total RNA was isolated from the left tibia of each animal, and gene expression in each bone was analyzed using quantitative real-time RT-PCR (28). Immediately after an animal was killed, the tibia bone was cleaned of any remaining soft tissue, cut into pieces, and homogenized in the presence of TRIzol reagent followed by RNA isolation as described by the manufacturer (Invitrogen, Carlsbad, CA). The gene-specific primer sequences were selected using the TaqMan probe and primer design function of the Primer Express version 1.5 software (Applied Biosystems, Forest City, CA). RT reactions were carried out using 2 μg RNA, subjected previously to DNase digestion, and a TaqMan reverse transcription reagents (Applied Biosystems), followed by PCR in real time using a SYBR Green PCR master mix (Applied Biosystems) and an ABI Prism 7700 sequence detection system (Applied Biosystems). The reactions were performed in the following cycling conditions: 95 C for 10 min and then 40 cycles of 95 C for 15 sec followed by 60 C for 1 min. The optimal concentrations of primers and templates that were used in each reaction were established based on the standard curve created before the reaction and corresponded to the nearly 100% efficiency of the reaction. The results were then normalized to the expression of 18 S rRNA in the same samples. Gene expression was analyzed using the following pairs of primers: aP2 (forward, GCGTGGAATTCGAT-GAAATCA; reverse, CCCGCCATCTAGGG), Runx2/Cbfa1 (forward, GGGCACAAGTTCTATCTGGAAAA; reverse, CGGTGTCACTGC-GCTGAA), Dlx5 (forward, TGACAGGCGTGTTTGACAGAAGAG; reverse, CGGGAACGGAGCTTGGA), osteocalcin (forward, CGGC-CCTGAGTCTGACAAA; reverse, GCCGGAGTCTGTTCACTACCTT), α1(I)collagen (forward, ACTGTCCCAACCCCCAAAG; reverse, CGT-ATTCTTCCGGGCAGAAA), and 18 S rRNA (forward, TTCGA-ACGTCTGCCCTATCAA; reverse, ATGGTAGGCACGGCGACTA).
Statistical analysis
Statistically significant differences between groups were detected using one-way ANOVA followed by post hoc analysis by Student-Neuman-Keuls within the SigmaStat software (SPSS, Inc., Chicago, IL) after establishing the homogeneity of variances and normal distribution of the data. In all cases, P < 0.05 was considered significant.
Results and Discussion
We examined the in vivo effect of rosiglitazone administration on the skeletons of adult (6 months old) male non-diabetic C57BL/6 mice. The dose of daily administration of rosiglitazone (20 μg/g body weight) was chosen based on previous studies that demonstrated its effectiveness in lowering blood glucose levels in diabetic KK-Ay mice similarly to the pharmacological doses used in humans (1, 29). This dose was also demonstrated to be effective in prevention of atherosclerotic lesions in low-density lipoprotein receptor-deficient mice (4). During 7 wk of rosiglitazone administration no differences were observed between groups with respect to their eating behaviors and animal motor activities. Fasting (18 h) plasma glucose levels measured at the end of the experiment did not differ between groups (rosiglitazone, 232.7 ± 54.9 mg/dl; control, 246.0 ± 34.9 mg/dl), which was consistent with previous reports (4). Nor did rosiglitazone administration have an effect on animal body weights (Fig. 1A).
Fig. 1.
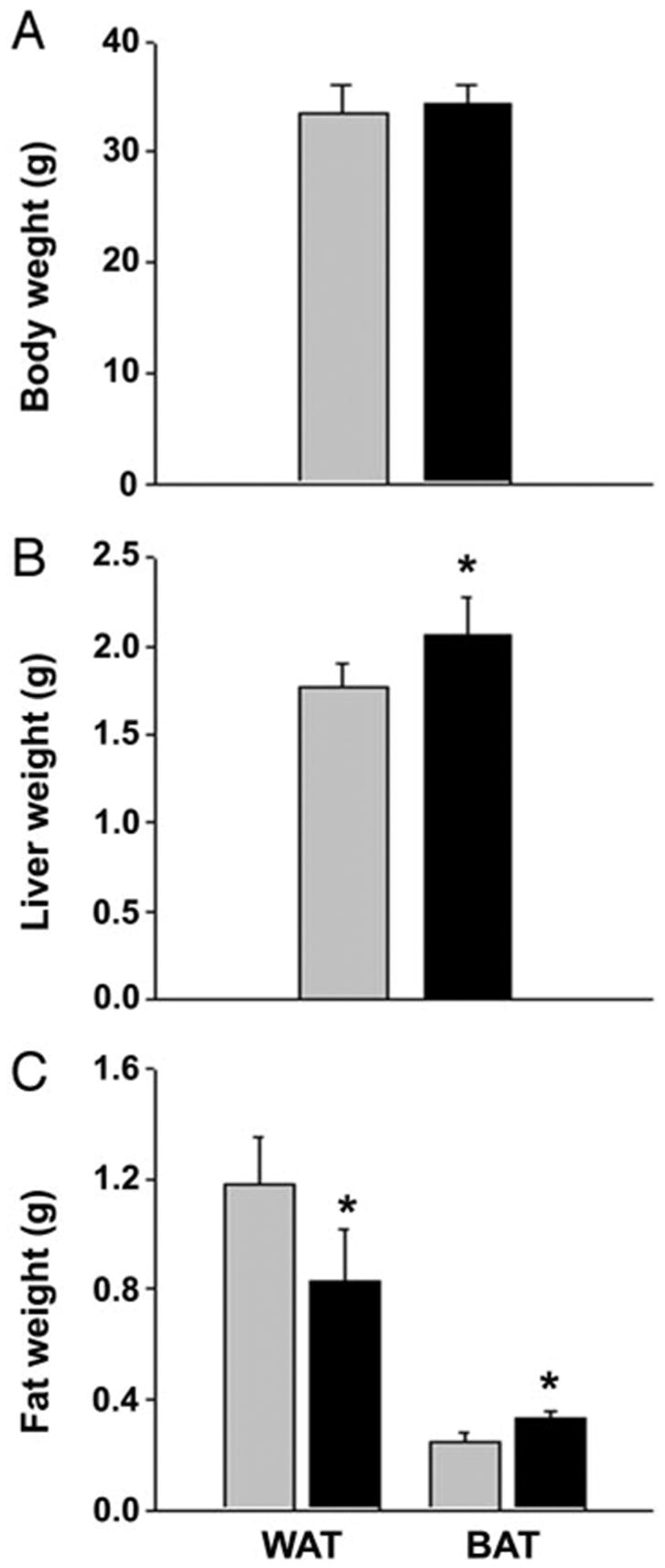
Body and organ weights of animals fed for 7 wk with either nonsupplemented (gray bar) or rosiglitazone-supplemented (black bar) diet. Each bar represents a mean weight (± SD) of eight animals or tissues derived from eight animals. *, Significant difference (P < 0.05) between control and rosiglitazone-fed group. A, Body weights; B, wet weight of livers; C, epididymal WAT and interscapular BAT.
After animals were killed, the wet weights of spleen, liver, and depots of white epididymal (white adipose tissue, WAT) and brown interscapular (brown adipose tissue, BAT) fat were measured. Rosiglitazone administration had no effect on spleen weight (not shown) but increased liver weight compared with control (Fig. 1B). Furthermore, histological examination revealed that rosiglitazone administration significantly increased fat liver content compared with the control animals (Fig. 2). Liver steatosis in response to rosiglitazone intake was previously observed in lipoatrophic, A-ZIP/F-1, and obese diabetic, KK-Ay and ob/ob, mice but not in nondiabetic, lean animals (30 –33). C57BL/6 mice are considered a diabetic-prone strain (34, 35); therefore it is possible that their genetic background predisposed them to develop liver steatosis in response to rosiglitazone treatment.
Fig. 2.
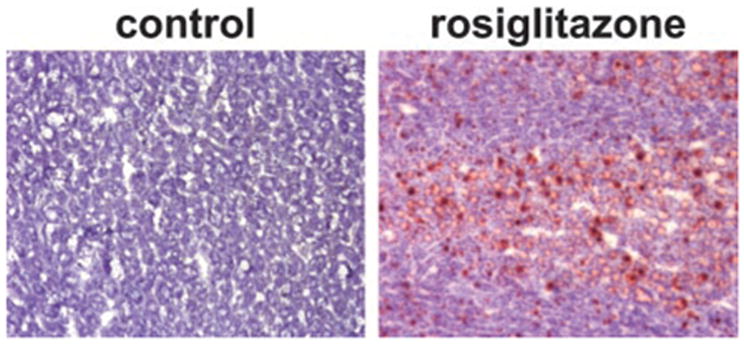
Liver histological cross-sections representative for each group. Snap-frozen specimens were sectioned, fixed with formalin, and stained with Oil Red O for fat and counterstained with methyl green. Magnification, ×40.
An analysis of epididymal WAT and interscapular BAT showed inverse changes in the weight of these tissues in rosiglitazone-fed vs. control animals (Fig. 1C). The weight of epididymal WAT was decreased by 30%, whereas the weight of interscapular BAT was increased by 25% in the rosiglitazone-fed group. A stimulatory effect of rosiglitazone on BAT, an adipose tissue involved in rapid energy dissipation and thermogenesis, and a suppressive effect on WAT, a tissue that is responsible for energy storage, indicates that rosiglitazone effects fatty acid metabolism. These data are consistent with previous reports of glitazone effects on murine adipose tissues (36) and demonstrate the efficacy of our treatment schedule and dose.
DXA analysis of total BMD demonstrated a significant decrease in the BMD of animals fed for 7 wk rosiglitazone-supplemented diet compared with the control mice (0.0462 ± 0.0001 vs. 0.0493 ± 0.0016 g/cm2; P < 0.001) (Fig. 3A). No differences in BMD were observed at 4 wk (data not shown). Longitudinal measurements of the same animals, at the beginning and end of the experiment, demonstrated almost a 10% decrease in BMD of rosiglitazone-fed animals (P < 0.001) (Fig. 3B).
Fig. 3.
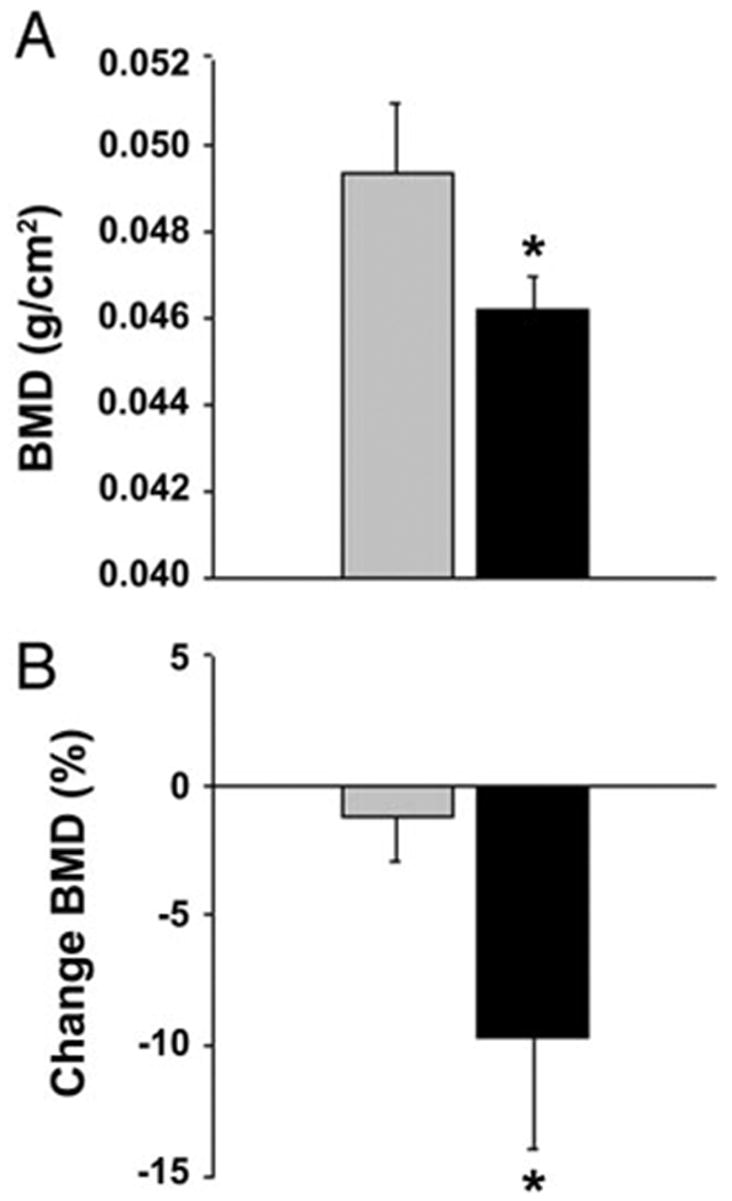
DXA of total-body BMD. Values represent a mean of eight animals per group (±SD) *, Significant differences (P < 0.05) between control (gray bar) and rosiglitazone-fed (black bar) groups. A, Total BMD, measured at the end of the experiment, of animals fed with either nonsupplemented or rosiglitazone-supplemented diet. B, Percent change in BMD within groups based on the measurements at the beginning and end of experiment and calculated as described in Materials and Methods.
To determine the effects of rosiglitazone on three-dimensional bone microarchitecture and morphometry, we performed micro-CT measurements of the proximal tibia. Micro-CT of the tibia demonstrated a significant loss of bone and changes in several morphometric parameters of bone microarchitecture in rosiglitazone-treated animals (Fig. 4 and Table 1). Bone content, as measured by the ratio BV/TV, was decreased by 24.1%. This dramatic decrease was accompanied by significant decreases in TbN (−11.2%) and TbTh (−10.6%) and an increase in TbSp (17.4%).
Fig. 4.
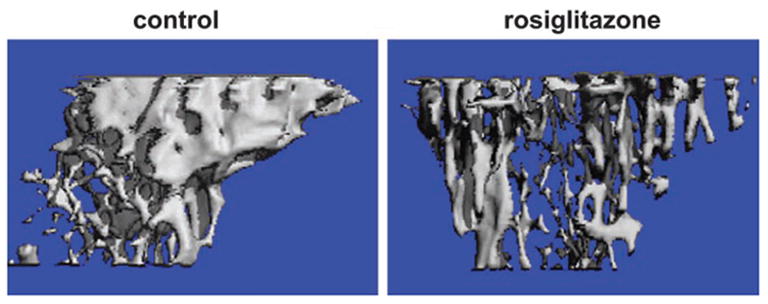
Micro-CT representative renderings of proximal tibia from control and rosiglitazone-treated animals were generated as described in Materials and Methods.
TABLE 1.
Microtomographic measurements of BV and trabecular architecture in tibia distal metaphysis in control and rosiglitazone-fed mice
| Treatment | BV/TV (%) | TbTh (μm) | TbN (mm−1) | TbSp (mm) | ConnD (mm−3) | SMI | DA |
|---|---|---|---|---|---|---|---|
| Control | 15.4 ± 1.7 | 57.6 ± 1.4 | 4.20 ± 0.22 | 0.23 ± 0.05 | 55.78 ± 16.98 | 2.08 ± 0.28 | 2.05 ± 0.10 |
| Rosiglitazone | 11.7 ± 1.6a | 51.5 ± 3.2a | 3.73 ± 0.29a | 0.27 ± 0.02a | 42.21 ± 5.18 | 2.07 ± 0.36 | 2.14 ± 0.18 |
| P (rosiglitazone vs. control) | 0.008 | 0.008 | 0.02 | 0.02 | NS | NS | NS |
Data are given as mean ± SD; n = 5 animals per group.
NS, not significant (P > 0.05);
P < 0.05.
Adipocytes and osteoblasts originate from common mesenchymal progenitor cells (13, 14, 37, 38). Lineage-specific commitment is preceded by a phase of multilineage activation that is characterized by their plastic ability to interconvert between phenotypes (13, 14, 37, 38). We had demonstrated previously in vitro that rosiglitazone activates murine adipocyte differentiation and inhibits osteoblastic differentiation and that this process occurs only in cells that express the PPAR-α2 transcription factor (17). Therefore, we reasoned that in vivo, bone loss that occurred in response to rosiglitazone administration was due to its action on the osteoblast progenitors that express PPAR-α2. Thus, activation of PPAR-α2 should result in the inhibition of osteoblast differentiation and stimulation of adipocyte differentiation in the bone of treated animals.
Indeed, histomorphometric analysis of the trabecular bone of proximal tibia revealed that rosiglitazone administration increased both fat content and the number of adipocytes by 3-fold (Fig. 5A). This was accompanied by a decrease in the osteoblast surface and/or their activity. The MS/BS (control, 18%; rosiglitazone, 6%) and MAR decreased 3-fold (Fig. 5B), and the rate of BFR/BS decreased by approximately 10-fold (control, 0.7 μm3/μm2 · d; rosiglitazone, 0.06 μm3/μm2 · d).
Fig. 5.
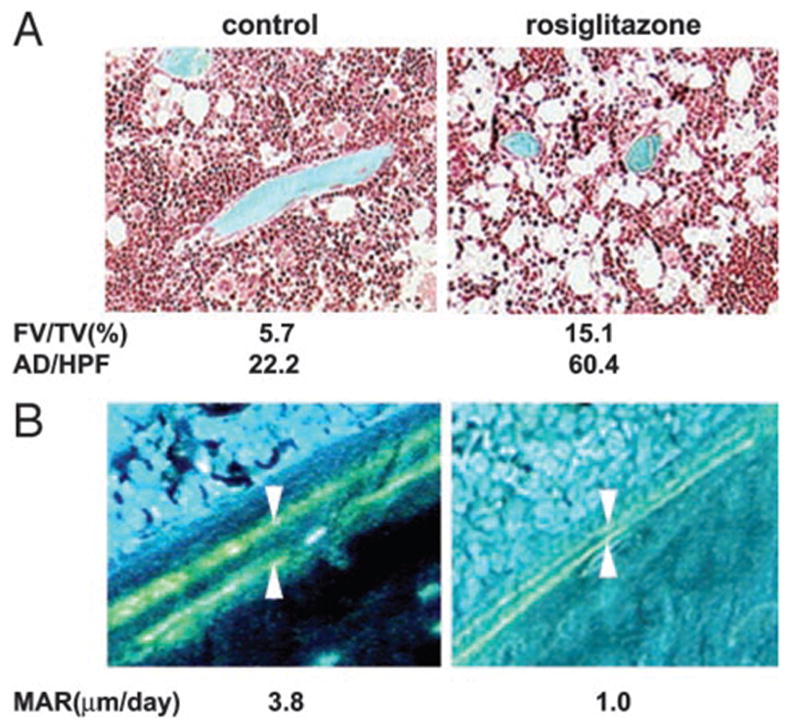
Representative photomicrographs of cancellous tibia. Numbers represent average of six representative fields examined from the same bone. A, Bone sections stained with Goldner trichrome stain. Mineralized bone tissue is stained blue, whereas the unstained area in the bone marrow represents empty spaces previously occupied by adipocytes. Photomicrographs were obtained on the Osteometrics system using a ×10 objective. B, Tetracycline-labeled section of the proximal tibia. The distance between two layers of tetracycline labels (arrows) visualized by epifluorescence represents bone formation that occurred during the 5-d period between tetracycline injections. Photomicrographs were obtained on the Osteometrics system using a ×40 objective. AD/HFP, Number of adipocytes per high-power field.
The observed bone loss and changes in the phenotype of bone marrow mesenchymal cells were reflected by changes in the expression of osteoblast- and adipocyte-specific marker genes measured in the whole tibia. Treatment with rosiglitazone decreased the abundance of mRNAs for two osteoblast-specific transcription factors, Dlx5 and Runx2/Cbfa1, and an osteoblast-specific marker, α1(I)collagen. In contrast, the abundance of mRNA for the adipocyte-specific marker, fatty acids binding protein aP2, was increased (Table 2).
TABLE 2.
Relative expression of phenotype-specific gene markers in the whole tibia normalized to 18 S rRNA
| Treatment | Runx2/Cbfa1 | Dlx5 | Osteocalcin | α1 (I) collagen | aP2 |
|---|---|---|---|---|---|
| Control | 9.3 ± 1.8 | 9.0 ± 1.9 | 7.6 ± 3.7 | 10.3 ± 0.7 | 3.2 ± 0.2 |
| Rosiglitazone | 6.4 ± 0.1a | 6.4 ± 0.7a | 7.4 ± 0.8 | 5.5 ± 1.1a | 13.5 ± 1.9a |
| Ratio (rosiglitazone/vehicle) | 0.6 | 0.7 | 1.0 | 0.6 | 3.4 |
Data are given as mean ± SD; n = 4 animals per group.
P < 0.05 vs. control.
In summary, we have demonstrated that in vivo, rosiglitazone administration to nondiabetic C57BL/6 mice results in significant bone loss. When considered with our previous in vitro evidence (17, 18), these data suggest that rosiglitazone-induced bone loss may be due to the suppression of the osteoblast differentiation of multipotential mesenchymal bone marrow progenitors that express PPAR-α2. The observed decrease in bone formation and osteoblast number correlates with increased fat content and increased adipocyte number. These data support the hypothesis that an inverse relationship exists between osteoblast and adipocyte differentiation from a common mesenchymal progenitor (13, 14, 37, 39). Thus, stimulation of adipocyte formation by rosiglitazone appears to occur at the expense of osteoblast formation.
When considered in light of the recent studies by Tornvig et al. (40) using troglitazone and our previous in vitro observation (18), the studies presented here suggest that PPAR-γ agonists differentially regulate the reciprocal relationship between osteoblastic and adipocytic cell differentiation in the bone marrow. Further evaluation of the efficacy and potency of selective PPAR-γ modulators are required to address these important observations.
In conclusion, our results indicate that murine bone marrow is a target for rosiglitazone, an antidiabetic drug and activator of the PPAR-γ 2 adipocyte-specific transcription factor. Furthermore, these results suggest that longitudinal rosiglitazone therapy may pose a significant risk to human bone, and provides the rationale for the development of new, selective and effective antidiabetic PPAR-γ agonists with little or no adverse effects on bone.
Acknowledgments
We thank Kui Teng for performing real-time RT-PCR and Frances Swain for preparing bone sections.
Abbreviations
- BAT
Brown adipose tissue
- BFR/BS
bone formation rate per bone surface
- BMD
bone mineral density
- BS
bone surface
- BV
bone volume
- ConnD
connectivity density
- CT
computed tomography
- DA
degree of anisotropy
- DXA
dual-energy x-ray absorptiometry
- FV/TV
fat volume per total volume
- MAR
mineral apposition rate
- MS
mineral surface
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- SMI
structure model index
- TbN
trabecular number
- TbSp
trabecular spacing
- TbTh
trabecular thickness
- WAT
white adipose tissue
Footnotes
This work was supported by National Institute on Aging Grant R01 AG17482 (to B.L.-C.).
References
- 1.Werner AL, Travaglini MT. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy. 2001;21:1082–1099. doi: 10.1592/phco.21.13.1082.34615. [DOI] [PubMed] [Google Scholar]
- 2.Henney JE. From the Food and Drug Administration. JAMA. 1999;282:932. [PubMed] [Google Scholar]
- 3.Kahn CR, Chen L, Cohen SE. Unraveling the mechanism of action of thiazolidinediones. J Clin Invest. 2000;106:1305–1307. doi: 10.1172/JCI11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li AC, Brown KK, Silvestre MJ, Wilson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Girnun GD, Spiegelman BM. PPAR-γ ligands: taking part in chemo-prevention. Gastroenterology. 2003;124:564 –567. doi: 10.1053/gast.2003.50070. [DOI] [PubMed] [Google Scholar]
- 6.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 7.Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor-γ agonists in Alzheimer’s disease. Neurobiol Aging. 2001;22:937–944. doi: 10.1016/s0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor-γ (PPAR-γ) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 9.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 10.Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Qi C, Korenberg JR, Chen X, Noya D, Rao MS, Reddy JK. Structural organization of mouse peroxisome proliferator-activated receptor-γ (mPPAR-γ) gene: alternative promoter use and different splicing yield two mPPAR-γ isoforms. Proc Natl Acad Sci USA. 1995;92:7921–7925. doi: 10.1073/pnas.92.17.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS. PPARγ knockdown by engineered transcription factors: exogenous PPARγ 2 but not PPARγ 1 reactivates adipogenesis. Genes Dev. 2002;16:27–32. doi: 10.1101/gad.953802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 14.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180 –192. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 15.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Rao Y-H, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 16.Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126:3795–3809. doi: 10.1242/dev.126.17.3795. [DOI] [PubMed] [Google Scholar]
- 17.Lecka-Czernik B, Gubrij I, Moerman EJ, Kajkenova O, Lipschitz DA, Manolagas SC, Jilka RL. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPAR-γ 2. J Cell Biochem. 1999;74:357–371. [PubMed] [Google Scholar]
- 18.Lecka-Czernik B, Moerman EJ, Grant DF, Lehmann JM, Manolagas SC, Jilka RL. Divergent effects of selective peroxisome proliferator-activated receptor-γ 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143:2376 –2384. doi: 10.1210/endo.143.6.8834. [DOI] [PubMed] [Google Scholar]
- 19.Gimble JM, Robinson CE, Wu X, Kelly KA, Rodriquez BR, Kliewer SA, Lehmann JM, Morris DC. Peroxisome proliferator-activated receptor-γ activation by thiazolidinediones induces adipogenesis in bone marrow stromal cells. Mol Pharmacol. 1996;50:1087–1094. [PubMed] [Google Scholar]
- 20.Schwartz AV, Sellmeyer DE, Feingold KR, Strotmeyer E, Resnick HE, Carbone L, Beamer BA, Lane NE, Cummings SR. Thiazolidinedione (TZD) use and bone density in older adults with diabetes. Diabetes. 2002;51:A237. (Abstract) [Google Scholar]
- 21.Samuels A, Perry MJ, Gibson R, Tobias JH. Effects of combination therapy with PTH and 17β-estradiol on long bones of female mice. Calcif Tissue Int. 2001;69:164 –170. doi: 10.1007/s002230020026. [DOI] [PubMed] [Google Scholar]
- 22.Iida-Klein A, Lu SS, Yokoyama K, Dempster DW, Nieves JW, Lindsay R. Precision, accuracy, and reproducibility of dual x-ray absorptiometry measurements in mice in vivo. J Clin Densitom. 2003;6:25–33. doi: 10.1385/jcd:6:1:25. [DOI] [PubMed] [Google Scholar]
- 23.Hildebrand T, Ruegsegger P. Quantification of bone microarchitecture with the structure model index. Comput Methods Biomech Biomed Engin. 1997;1:15–23. doi: 10.1080/01495739708936692. [DOI] [PubMed] [Google Scholar]
- 24.Amblard D, Lafage-Proust MH, Laib A, Thierry T, Ruegsegger P, Alexandre C, Vico L. Tail suspension induces bone loss in skeletally mature mice in the C57BL/6J strain but not in the C3H/HeJ strain. J Bone Miner Res. 2003;18:561–569. doi: 10.1359/jbmr.2003.18.3.561. [DOI] [PubMed] [Google Scholar]
- 25.Suva LJ, Seedor JG, Endo N, Quartuccio HA, Thompson DD, Bab I, Rodan GA. Pattern of gene expression following rat tibial marrow ablation. J Bone Miner Res. 1993;8:379 –388. doi: 10.1002/jbmr.5650080315. [DOI] [PubMed] [Google Scholar]
- 26.Jilka RL, Weinstein RS, Takahashi K, Parfitt AM, Manolagas SC. Linkage of decreased bone mass with impaired osteoblastogenesis in a murine model of accelerated senescence. J Clin Invest. 1996;97:1732–1740. doi: 10.1172/JCI118600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 28.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169 –193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 29.Reginato MJ, Bailey ST, Krakow SL, Minami C, Ishii S, Tanaka H, Lazar MA. A potent antidiabetic thiazolidinedione with unique peroxisome proliferator-activated receptor-γ-activating properties. J Biol Chem. 1998;273:32679 –32684. doi: 10.1074/jbc.273.49.32679. [DOI] [PubMed] [Google Scholar]
- 30.Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, Gavrilova O, Reitman ML. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest. 2000;106:1221–1228. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsusue K, Haluzik M, Lambert G, Yim S-H, Gavrilova O, Ward JM, Brewer B, Jr, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu S, Matsusue K, Kashireddy P, Cao W-Q, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem. 2003;278:498 –505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 33.Bedoucha M, Atzpodien E, Boelsterli UA. Diabetic KKAy mice exhibit increased hepatic PPARγ1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J Hepatol. 2001;35:17–23. doi: 10.1016/s0168-8278(01)00066-6. [DOI] [PubMed] [Google Scholar]
- 34.Kaku K, Fiedorek FT, Jr, Province M, Permutt MA. Genetic analysis of glucose tolerance in inbred mouse strains. Evidence for polygenic control. Diabetes. 1988;37:707–713. doi: 10.2337/diab.37.6.707. [DOI] [PubMed] [Google Scholar]
- 35.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 36.Tai TAC, Jennermann C, Brown KK, Oliver BB, MacGinnitie MA, Wilkison WO, Brown HR, Lehmann JM, Kliewer SA, Morris DC, Graves RA. Activation of the nuclear receptor peroxisome proliferator-activated receptor gamma promotes brown adipocyte differentiation. J Biol Chem. 1996;271:29909 –29914. doi: 10.1074/jbc.271.47.29909. [DOI] [PubMed] [Google Scholar]
- 37.Beresford JN, Bennett JH, Devlin C, Leboy PS, Owen ME. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci. 1992;102:341–351. doi: 10.1242/jcs.102.2.341. [DOI] [PubMed] [Google Scholar]
- 38.Nuttall ME, Patton AJ, Olivera DL, Nadeau DP, Gowen M. Human trabecular bone cells are able to express both osteoblastic and adipocytic phenotype: implications for osteopenic disorders. J Bone Miner Res. 1998;13:371–382. doi: 10.1359/jbmr.1998.13.3.371. [DOI] [PubMed] [Google Scholar]
- 39.Nuttall ME, Gimble JM. Is there a therapeutic opportunity to either prevent or treat osteopenic disorders by inhibiting marrow adipogenesis? Bone. 2000;27:177–184. doi: 10.1016/s8756-3282(00)00317-3. [DOI] [PubMed] [Google Scholar]
- 40.Tornvig L, Mosekilde LI, Justesen J, Falk E, Kassem M. Troglitazone treatment increases bone marrow adipose tissue volume but does not affect trabecular bone volume in mice. Calcif Tissue Int. 2001;69:46 –50. doi: 10.1007/s002230020018. [DOI] [PubMed] [Google Scholar]