

Abstract
Retrotransposons constitute over 40% of the human genome and play important roles in the evolution of the genome. Since certain types of retrotransposons, particularly members of the Alu, L1, and SVA families, are still active, their recent and ongoing propagation generates a unique and important class of human genomic diversity/polymorphism (for the presence and absence of an insertion) with some elements known to cause genetic diseases. So far, over 2,300, 500, and 80 Alu, L1, and SVA insertions, respectively, have been reported to be polymorphic and many more are yet to be discovered. We present here the Database of Retrotransposon Insertion Polymorphisms (dbRIP; http://falcon.roswellpark.org:9090), a highly integrated and interactive database of human retrotransposon insertion polymorphisms (RIPs). dbRIP currently contains a nonredundant list of 1,625, 407, and 63 polymorphic Alu, L1, and SVA elements, respectively, or a total of 2,095 RIPs. In dbRIP, we deploy the utilities and annotated data of the genome browser developed at the University of California at Santa Cruz (UCSC) for user-friendly queries and integrative browsing of RIPs along with all other genome annotation information. Users can query the database by a variety of means and have access to the detailed information related to a RIP, including detailed insertion sequences and genotype data. dbRIP represents the first database providing comprehensive, integrative, and interactive compilation of RIP data, and it will be a useful resource for researchers working in the area of human genetics.
Keywords: retrotransposition, polymorphism, mutation, L1, Alu, SVA, database
INTRODUCTION
The human genome, like those of most other eukaryotic organisms, is highly rich in repetitive elements derived from retrotransposons that amplify themselves in the genome through an RNA-mediated retrotransposition process. Most common among them are Alu elements, which are a class of short interspersed elements (SINEs), and LINE-1s (or L1s), which belong to the long interspersed elements (LINEs). Alu elements have over 1 million members covering ~11% of the human genome, while L1 elements account for ~21% of the genome with ~500,000 members; collectively, they constitute approximately one-third of the human genome [Lander et al., 2001]. While most of the retrotransposable elements in the human genome are considered to be dead fossils from past waves of mobile element amplification, some members belonging to young subfamilies of these elements are still actively mobilizing and continue to make new copies in the genome. The first of such active groups are L1 elements, the only active autonomous retrotransposons in the human genome, which are also responsible for the mobilization of nonautonomous retrotransposons [Boissinot et al., 2000, 2004; Brouha et al., 2003; Dewannieux et al., 2003]. Among the active nonautonomous groups are Alu elements and SVA elements, a composite type of retrotransposon formed of SINE-R, VNTR, and Alu [Boeke, 1997; Boeke and Chapman, 1991; Deininger and Batzer, 2002; Dewannieux et al., 2003; Ostertag et al., 2000, 2002, 2003; Sassaman et al., 1997; Shen et al., 1994; Wang et al., 2005; Wang et al., 2006]. Many of the insertions derived from these active retrotransposons occurred so recently that they are polymorphic with respect to the presence or absence of the insertion in different human populations, families, or even individuals [Batzer et al., 1994; Batzer and Deininger, 1991, 2002; Boissinot et al., 2000, 2004; Badge et al., 2003; Bamshad et al., 2003; Myers et al., 2002; Perna et al., 1992; Sheen et al., 2000; Watkins et al., 2001, 2003]. Newly integrated repetitive elements can cause genetic diseases by interrupting exons, generating aberrant RNA splicing, or altering gene expression [Deininger and Batzer, 1999; Miki, 1998; Ostertag and Kazazian, 2001; Wallace et al., 1991]. Due to their lack of homoplasy and known ancestral state, retrotransposon insertion polymorphisms (RIPs) also serve as excellent genetic markers for studies of human population genetics [Batzer et al., 1991, 1994; Batzer and Deininger, 1991, 2002; Bamshad et al., 2003; Perna et al., 1992; Salem et al., 2005a; Stoneking et al., 1997; Watkins et al., 2001, 2003].
Over the last two decades many studies have been carried out to detect genomic variation derived from retrotransposons using various approaches (see http://falcon.roswellpark.org:9090/dbRI-P_ref.html for a complete list of the studies). Earlier studies using genomic library screening or direct DNA sequencing of the suspected mutant gene alleles identified a number of individual Alu and L1 insertions, some of which are disease-related [Arcot et al., 1995; Batzer et al., 1995; Kazazian et al., 1988; Miki et al., 1992, 1996; Wallace et al., 1991]. The recent availability of the human genome draft sequence, and additional diverse human genomic sequence data, have accelerated the process of identifying newly integrated retrotransposons. One strategy that has been very successfully used is to identify members belonging to the young subfamilies of retrotransposons from the reference genome sequence via computational analysis followed by determining their insertion polymorphism status through screening of DNA samples from diverse human populations [Callinan et al., 2003; Carroll et al., 2001; Carter et al., 2004; Myers et al., 2002; Otieno et al., 2004; Salem et al., 2003; 2005b; Sheen et al., 2000; Vincent et al., 2003; Xing et al., 2003]. Furthermore, two recent studies, both employing in silico computational strategies that utilize the sequence data from sources representing different human individuals, have also shown great success. A total of 505 polymorphic Alu, 65 L1, and 39 SVA elements were recovered by comparing the trace sequences derived from different library sources [Bennett et al., 2004]. By comparing the two versions of the human genome sequences (public vs. Celera www.celera.com), we have recently identified over 800 polymorphic Alu elements and 150 L1 elements [Wang et al., 2006; authors’ unpublished data].
Collectively, all these studies have reported, as of this writing, a total of over 3,000 (with redundancy) RIPs. Nevertheless, a database with systematically documented information about retrotransposon derived genomic variation is not in existence, although part of this information has been scattered in several databases, including GenBank [Arcot et al., 1995; Batzer et al., 1995; Holmes et al., 1994; Kazazian et al., 1988; Miki et al., 1992, 1996; Narita et al., 1993; Schwahn et al., 1998; Wallace et al., 1991] and the SNP database (www.ncbi.nlm.nih.gov/SNP) [Bennett et al., 2004]. Here we report a new database called the Database of Retrotransposon Insertion Polymorphisms (dbRIP; http://falcon.roswellpark.org:9090/) to provide a comprehensive compilation of human genome variations derived from retrotransposon insertions.
DATA SOURCES, COLLECTION, AND CURATION
Data for polymorphic Alu, L1, and SVA elements was collected from all available published papers (see a complete list of cited original reports at http://falcon.roswellpark.org:9090/dbRIP_ref.html) and was compiled into XML files. For each reported RIP entry, we collected and compiled the following data items: original identifications (IDs), type of retrotransposon, family and subfamily designation, association with disease, DNA sequences of the elements, target site duplications (TSDs), 400 bp of flanking sequence regions, oligonucleotide primers and PCR conditions, expected PCR product sizes for both filled and empty alleles, ascertainment method(s), source of genome sequences, genotypic data, chromosome position, cytoband position, size of the insertion, and reference(s). The mapping of the exact location for each RIP in the current version of the human genome reference sequences (currently University of California at Santa Cruz [UCSC] hg17 assembly based on National Center for Biotechnology Information [NCBI] Human Genome Build 35) was based on the available sequence information for one or more items among the flanking regions, the primers, TSDs, and the retrotransposon. For each RIP, the raw genotypic data was summed and reported as three genotypes (insertion +/+, +/−, and −/−) for each examined human group.
DATABASE DESIGN AND STRUCTURE
In choosing a design for dbRIP, we reasoned that creating a standalone database with only the RIP data would impose a limitation on its use. By contrast, users would be able to get most out of the RIP data if we could integrate the data into a system such that the RIPs are displayed along with other genome information. For this reason, it was natural for us to choose a system that can display the RIP data in a genome browser style. Among the existing genome browsers that are portable, we found that the UCSC genome browser created by the Genome Bioinformatics Group of University of California at Santa Cruz best fit our purposes [Hsu et al., 2004; Kent et al., 2002]. The UCSC genome browser (hereafter referred to as “the browser”) has gained wide use because of its user-friendly web interface and comprehensive data. In addition, its code and web interface have been stabilized over the past few years, hence it requires very little effort to maintain and update.
To deploy the UCSC genome browser for our use, we first installed the UCSC mirror package and the latest version of the human genome database (hg17). We then added new codes and modified some existing codes for the browser to accommodate dbRIP data as its standard tracks. The dbRIP data is displayed as three separate tracks in the browser with each for polymorphic Alu, L1, and SVA elements, respectively; they are grouped under the title of “Retrotransposon Insertion Polymorphisms in Humans” in the browser and listed within the “Variation and Repeats” category on the display options panel of the browser. Two dbRIP-specific tables are created to contain RIP data: one for genotypic data and the other for all remaining RIP data. In addition, a third table was created to host the start and end genomic positions of all promoter, exon, intron, and intergenic regions in the entire genome based on the current version of NCBI RefGenes, and this table is used to query RIPs by their physical relationship to genes.
To accommodate queries unique to dbRIP, as well as to increase the speed of some commonly used query functions available from the browser, we implemented a special query interface called SeachdbRIP. SearchdbRIP is implemented as a PERL-based CGI program powered by the MySQL relational database engine (http://dev.mysql.com/). We integrated the SearchdbRIP function into the browser such that it is conveniently accessible within most of the browsing windows via a standard button in the browser. The relationship and data flow among dbRIP tables, human genome annotation tables, SearchdbRIP, and the browser is shown in Figure 1. The database is hosted on a Compaq ProLiant DL360 server machine (Palo Alto, CA; www.hp.com) running the RedHat Linux 8.0 operating system and Apache web server (www.apache.org), and is available freely at http://falcon.roswellpark.org:9090/ and http://batzerlab.lsu.edu/.
FIGURE 1.
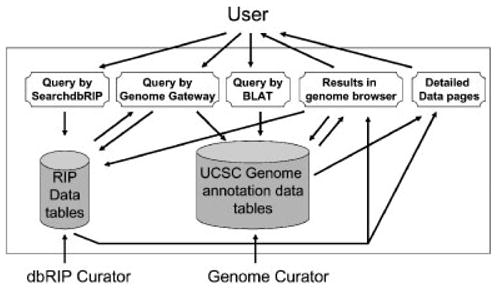
Overall design of dbRIP database. The schematic shows the relationship between dbRIP and the UCSC Genome Browser as well as the data Low and interactions among users, curators, browser, and SearchdbRIP. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DATABASE FUNCTIONS
Utility of SearchdbRIP
The query utilities of SearchdbRIP are divided into the “Quick Search” and “Advanced Search” sections. “Quick Search” allows users to search by IDs, including dbRIP ID and the original IDs, or by chromosome positions. These queries are also available from the Genome Gateway within the browser, but are much quicker in SearchdbRIP, as SearchdbRIP limits the search to the three dbRIP tables. Utilities under “Advanced Search” are largely designed for dbRIP. The available search criteria here include chromosome number, relationship to genes (gene context), genomic source containing the insertion, subfamily of the retrotransposon, the presence or absence within a diverse human group (from a predefined list based on the availability), allele frequency ranges, association with a disease or phenotype, and author name. Users can use these criteria individually or combine two or more criteria. For example, one can query all RIPs that locate inside exons and are known to associate with a disease phenotype and with insertion absent from hg17 by selecting “exon” in the gene context slot and “non-hg17” in the genomic source slot and inputting “all” in the disease slot. The gene context query is made available for users who are more interested in querying RIPs based on the potential effect on gene function, such as those locating inside exons or promoter regions. It is based on the physical relationship of retrotransposon insertions with the boundaries of annotated genes. When a RIP is overlapped with more than one region, an arbitrary priority order of “exon>promoter>intron>intergenic region” is used.
Detailed RIP Information in dbRIP Data Page
In dbRIP, it is our goal to provide all available related information in a detailed manner. All RIP-related data is displayed in the “RIP data page” that is accessible from the output of SearchdbRIP and from the browser, and it represents the most valuable and core part of dbRIP data. As the example in Figure 2 shows, for each RIP we provide 19 data items grouped into the following 10 categories: identification, classification, associated diseases/phenotype, detailed sequence data, PCR-related information, ascertainment methods, sources of RIP, genotypic data, genomic location information, and references. Below, we provide descriptions for a few of these categories and encourage users to explore the database website for the rest.
FIGURE 2.

A typical RIP data page. The data page shows the detailed information of locus RIP_Alu_chr1_165_01. A: Information from database ID to genome source. B: Information from genotypic data to reference. This locus was originally identified in two studies as shown by the number of original IDs and the references. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
For RIP IDs, we provide a database ID and the original ID(s). A database ID contains three sections to indicate the type of RIP, the chromosome and position, and the number of the same type of RIPs in the region. For example, “RIP_Alu_chr7_003_01” indicates an Alu RIP on chromosome 7 in the third 3-Mb region starting from the telomere of the short arm. Database IDs are unique and thus represent a nonredundant list of RIPs, making it possible to keep track of the exact number of all compiled RIP loci and to compare the coverage of RIPs derived from different ascertainment methods. Conversely, we kept all original IDs used in the primary literature to facilitate identification of RIPs identified from multiple studies and query of RIPs by any of the original IDs. For classification, we provide the class (i.e., SINE, LINE, etc.), type (such as Alu, L1, and SVA), family, and subfamily of the retrotransposon based on an updated list of consensus sequences and standard nomenclature [Batzer et al., 1996]. For example, we added several newly designated AluY subfamilies, including Yb11, Ya4b, Yf, Yg, Yh, Yj, and Yx [Garber et al., 2005; Salem et al., 2005b; Xing et al., 2003; Wang et al., 2006]. To track all disease-related RIPs, we provide the name of the disease or phenotype associated with each RIP. In the sequence data field, we provide detailed sequence information related to each RIP, which includes the insertion sequence, TSDs, and 400-bp flanking region on each side of the element. Each sequence section is clearly highlighted by a differently-colored font for easy recognition. For a small number of RIPs, we are unable to obtain the complete sequence information based on the data available from the original literature and/or the related GenBank entries. We use “nnnnn” to indicate unknown TSDs and “NNNNNNNNNN” to denote the unknown sequence of the entire or partial section (usually the polyA-tail) of the retrotransposon. Whenever available, we also provide detailed genotypic data for all screened geographic groups or sample sets, as well as the calculated allele frequencies, unbiased heterozygosity, and average allele frequencies. At this moment, due to the database design used, we are unable to include genotypic data for a small number of loci, for which the detailed genotypes were not reported. We encourage users to contact us if they have RIP genotype data not covered by dbRIP.
Browsing RIPs in Genome Context With the UCSC Browser
Since dbRIP is integrated as a part of the browser, the RIP data can be queried using the utilities available from the browser. Being able to browse RIPs alongside other available genome information offers a significant advantage by providing the users a graphic visualization of the genomic context of each RIP. For instance, as shown by the example in Figure 3, the user can easily visualize the fact that this breast cancer–related de novo Alu insertion [Miki et al., 1996] is located inside an exon of the BRCA2 gene and it is flanked by several known SNPs. Users are advised to visit the UCSC genome website for detailed descriptions and tutorials of browser’s utilities at http://genome.ucsc.edu [Kent, 2002; Kent et al., 2002], here we elaborate on a few important functions related to dbRIP. To examine the RIPs within a gene or a genomic region of particular interest, one can search dbRIP using the Genome Gateway by providing a genetic ID, such as a gene ID or an accession ID, or by specifying genomic positions. All RIPs that exist within the region will be displayed in the RIP tracks. Once a RIP is located, one can determine if the RIP contributes to an alternatively-spliced exon by expanding the Expressed Sequence Tag (EST) tracks. Using BLAST-like alignment tool (BLAT) with either DNA or protein sequences, the user can quickly find out if the related genomic region contains any known RIPs. Similarly, one can use BLAT to find out whether or not a newly identified sequence carrying a polymorphic retrotransposon represents a novel RIP.
FIGURE 3.
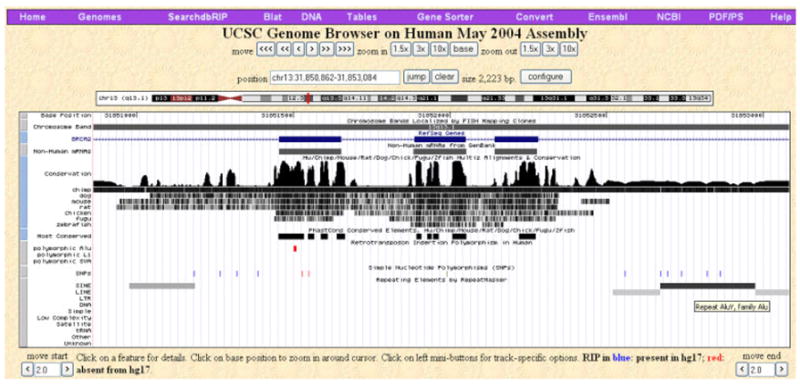
Integrative display of a RIP in the UCSC Genome Browser. In the example shows a RIP that is located within one of the exons of BRCA2. This RIP is absent from the reference genome as indicated by the red color of the RIP tick, as well as the small size of the bar representing the insertion.Within this 2.2-kb genomic region (window), there are multiple SNPs, an Alu repeat, and several other repetitive elements. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Other Accessory Utilities
In addition to the main utilities described above, we also provide several other related resources on the dbRIP main page. These include a complete list of references used in dbRIP, genome-wide plots for all types and individual types of RIPs, and downloadable data in flat files, all of which are accessed via the menu bars at the top of the main page. Although most of the data for individual RIPs is available for downloading through the utilities of the browser, making the entire dbRIP data set available allows advanced users to perform systematic analysis of the large scale RIP data. On the “Help” page, we provide some special instructions for using dbRIP, which may not be so obvious to users. On the main page, a list of examples for utilizing the data in dbRIP is also provided.
DISCUSSION
Database Statistics and Genome Distribution of RIPs
As shown by the summary statistics of dbRIP data in Table 1, dbRIP, as of this writing, covers the data compiled from 2,300 Alu, 520 L1, and 80 SVA elements, respectively, from over 50 original reports. By determining their exact genome locations, we obtained a nonredundant set of 1,625 Alu, 407 L1, and 63 SVA elements or a total of 2,095 RIPs. For a small fraction of reported RIPs, we failed to map them to a genome location due to the lack of sufficient sequence information; therefore, we excluded them from dbRIP. Among the mapped RIPs, over 600 loci have been subjected to genotyping using DNA samples from diverse human groups. It would also be very useful to have the remaining RIPs that were largely identified via in silico comparative genomic approaches confirmed by the gold standard of genotyping a human diversity panel. Interestingly, a total of 531 loci (or 25%) among the compiled RIPs are elements that are absent from the reference genome sequences (hg17 or NCBI Human Genome Build 35). These RIPs were identified using methods that are either independent of the known reference genome sequences [Badge et al., 2003; Buzdin et al., 2003, 2005; Roy et al., 1999; Mamedov et al., 2005; Sheen et al., 2000] or have utilized genomic sequences representing additional human individuals [Bennett et al., 2004; Wang et al., 2006]. The relatively high ratios of RIPs outside of reference genome sequences suggests to us that there are a large number of new RIPs yet to be identified. This notion is further supported by the fact that methods independent of known genomic sequences identify high ratios of novel RIPs, many of which fall into regions that have not been sequenced [Badge et al., 2003; Buzdin et al., 2003, 2005; Roy et al., 1999; Sheen et al., 2000] and the fact that there is very low ratio of overlapping loci identified by different methods. Our data indicates that the minimal polymorphism rates (the number of RIPs vs. the total number of members in the class) for Alu, L1, and SVA are 0.14%, 0.10%, and 2.29%, respectively. The much higher polymorphism rate (greater than one order of magnitude) of SVA elements in comparison with that of the other two retrotransposon families may be indicative of a higher current rate of retrotransposition and/or a much younger evolutionary age for SVA elements.
TABLE 1.
dbRIP Summary Statistics
| RIP type | RIP counts (unique/total) | RIPs outside hg17 | RIP with genotypea | Loci gene context (5 kb promotor/exon/intron/intergenic) | Disease-related loci |
|---|---|---|---|---|---|
| Alu | 1625/2299 | 395 | 528 | 41/18/623/943 | 33 |
| L1 | 407/524 | 132 | 81 | 7/9/130//261 | 15 |
| SVA | 63/74 | 4 | 31 | 7/3/22/31 | 3 |
| Total | 2095/2897 | 531 | 640 | 55/30/775/1235 | 51 |
Excluding those with only calculated allele frequency.
Among the 2,100 RIPs, we have identified 51 cases of disease-related loci that are examples of the involvement of retrotransposon insertions in genetic diseases. The detection of such rare elements has been greatly hindered by the lack of sensitive techniques for identifying individual de novo retrotransposon insertions. We expect that with the advancement of these approaches it will be possible to identify many more such disease-related loci in the future and facilitate a more comprehensive understanding of the contribution that retrotransposons make to human genetic disorders.
Future Development
To increase the utility of dbRIP data, we will contact the UCSC Bioinformatics group to inquire about the possibility of adding a new track for the RIP data in their browser or modifying the existing “RepeatMasker” track to accommodate RIPs. We will also work with other experts in the genetic variation community, such as HGVS, to develop a nomenclature for this type of variation and to make the RIP data available for deposition into other related central databases, such as the ongoing international HUGO Mutation Central Database. Nevertheless, even with the integration of RIP data into central databases, we will continue to maintain dbRIP as an independent platform for purposes of raw data collection and validation, as well as for providing special queries that may not be available from the central databases. We will also implement a data input form for others to deposit their RIP data into dbRIP, and update the database on a regular basis.
CONCLUSION
A unique feature of dbRIP is the integration of RIP data with existing genome annotation data in a genome browser. This feature allows the graphic visualization of each RIP in context with all gene annotation in the regions, which can be extremely useful in understanding the potential functional effects of the variation. The utilities of the UCSC genome browser also provide users with many convenient tools for manipulation and retrieval of data. To our knowledge, dbRIP is the only currently available database that offers a comprehensive compilation of human genome variations/mutations related to retrotransposon insertions in a highly integrated and interactive manner. In this sense, dbRIP also represents a good example for utilizing existing genome browsing systems and genome annotation data to develop highly integrative and interactive specialized databases. We believe that dbRIP will be a very useful resource and tool for studying human genome variation, retrotransposition, human genome evolution, and population genetics, as well as forensic genomics.
Acknowledgments
This research was supported in part by a Roswell Park Cancer Institute (RPCI) development fund (to P.L.), by NIH grants, CA101515 (to P.L.), CA16056 (RPCI), and GM59290 (to M.A.B.), NSF grants, BCS-0218338 (to M.A.B.) and EPS-0346411 (to M.A.B.), and by the Louisiana Board of Regents Millennium Trust Health Excellence Fund HEF (2000–05)-01 (to M.A.B.), and the State of Louisiana Board of Regents Support Fund (to M.A.B.). We thank all researchers who have contributed to the RIP data, especially those who posted their full datasets online.
Footnotes
This article is a U.S. Government work and, as such, is in the public domain of the United States of America.
Communicated by Alastair Brown
Grant sponsor: Roswell Park Cancer Institute (RPCI) Development Fund; Grant sponsor: State of Louisiana Board of Regents Support Fund; Grant sponsor: Louisiana Board of Regents Millennium Trust Health Excellence Fund; Grant number: HEF (2000-05)-01; Grant sponsor: NIH; Grant numbers: CA101515; CA16056; and GM59290; Grant sponsor: NSF; Grant numbers: BCS-0218338; and EPS-0346411.
References
- Arcot SS, Fontius JJ, Deininger PL, Batzer MA. Identification and analysis of a “young” polymorphic Alu element. Biochim Biophys Acta. 1995;1263:99–102. doi: 10.1016/0167-4781(95)00080-z. [DOI] [PubMed] [Google Scholar]
- Badge RM, Alisch RS, Moran JV. ATLAS: a system to selectively identify human-specific L1 insertions. Am J Hum Genet. 2003;72:823–838. doi: 10.1086/373939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Wooding S, Watkins WS, Ostler CT, Batzer MA, Jorde LB. Human population genetic structure and inference of group membership. Am J Hum Genet. 2003;72:578–589. doi: 10.1086/368061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. A human-specific subfamily of Alu sequences. Genomics. 1991;9:481–487. doi: 10.1016/0888-7543(91)90414-a. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Gudi VA, Mena JC, Foltz DW, Herrera RJ, Deininger PL. Amplification dynamics of human-specific (HS) Alu family members. Nucleic Acids Res. 1991;19:3619–3623. doi: 10.1093/nar/19.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Rubin CM, Hellmann-Blumberg U, Alegria-Hartman M, Leeflang EP, Stern JD, Bazan HA, Shaikh TH, Deininger PL, Schmid CW. Dispersion and insertion polymorphism in two small subfamilies of recently amplified human Alu repeats. J Mol Biol. 1995;247:418–427. doi: 10.1006/jmbi.1994.0150. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL, Hellmann-Blumberg U, Jurka J, Labuda D, Rubin CM, Schmid CW, Zietkiewicz E, Zuckerkandl E. Standardized nomenclature for Alu repeats. J Mol Evol. 1996;42:3–6. doi: 10.1007/BF00163204. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Bennett EA, Coleman LE, Tsui C, Pittard WS, Devine SE. Natural genetic variation caused by transposable elements in humans. Genetics. 2004;168:933–951. doi: 10.1534/genetics.104.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD. LINEs and Alus—the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Chapman KB. Retrotransposition mechanisms. Curr Opin Cell Biol. 1991;3:502–507. doi: 10.1016/0955-0674(91)90079-e. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol. 2000;17:915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Young L, Munson PJ, Furano AV. The insertional history of an active family of L1 retrotransposons in humans. Genome Res. 2004;14:1221–1231. doi: 10.1101/gr.2326704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzdin A, Ustyugova S, Gogvadze E, Lebedev Y, Hunsmann G, Sverdlov E. Genome-wide targeted search for human specific and polymorphic L1 integrations. Hum Genet. 2003;112:527–533. doi: 10.1007/s00439-002-0904-2. [DOI] [PubMed] [Google Scholar]
- Buzdin A, Vinogradova T, Lebedev Y, Sverdlov E. Genome-wide experimental identification and functional analysis of human specific retroelements. Cytogenet Genome Res. 2005;110:468–474. doi: 10.1159/000084980. [DOI] [PubMed] [Google Scholar]
- Callinan PA, Hedges DJ, Salem AH, Xing J, Walker JA, Garber RK, Watkins WS, Bamshad MJ, Jorde LB, Batzer MA. Comprehensive analysis of Alu-associated diversity on the human sex chromosomes. Gene. 2003;317:103–110. doi: 10.1016/s0378-1119(03)00662-0. [DOI] [PubMed] [Google Scholar]
- Carroll ML, Roy-Engel AM, Nguyen SV, Salem AH, Vogel E, Vincent B, Myers J, Ahmad Z, Nguyen L, Sammarco M, Watkins WS, Henke J, Makalowski M, Jorde LB, Deininger PL, Batzer MA. Large-scale analysis of the Alu Ya5 and Yb8 subfamilies and their contribution to human genomic diversity. J Mol Biol. 2001;311:17–40. doi: 10.1006/jmbi.2001.4847. [DOI] [PubMed] [Google Scholar]
- Carter AB, Salem AH, Hedges DJ, Keegan CN, Kimball B, Walker JA, Watkins WS, Jorde LB, Batzer MA. Genome-wide analysis of the human Alu Yb-lineage. Hum Genomics. 2004;1:167–178. doi: 10.1186/1479-7364-1-3-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Mammalian retroelements. Genome Res. 2002;12:1455–1465. doi: 10.1101/gr.282402. [DOI] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH., Jr A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet. 1994;7:143–148. doi: 10.1038/ng0694-143. [DOI] [PubMed] [Google Scholar]
- Hsu F, Pringle TH, Kuhn RM, Karolchik D, Diekhans M, Haussler D, Kent WJ. The UCSC Proteome Browser. Nuc Acids Res. 2004;33(Suppl 1):D454–D458. doi: 10.1093/nar/gki100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la BM, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, Szustakowki J, De Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Mamedov IZ, Arzumanyan ES, Amosova AL, Lebedev YB, Sverdlov ED. Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acids Res. 2005;33:e16. doi: 10.1093/nar/gni018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- Miki Y, Katagiri T, Kasumi F, Yoshimoto T, Nakamura Y. Mutation analysis in the BRCA2 gene in primary breast cancers. Nat Genet. 1996;13:245–247. doi: 10.1038/ng0696-245. [DOI] [PubMed] [Google Scholar]
- Miki Y. Retrotransposal integration of mobile genetic elements in human diseases. J Hum Genet. 1998;43:77–84. doi: 10.1007/s100380050045. [DOI] [PubMed] [Google Scholar]
- Myers JS, Vincent BJ, Udall H, Watkins WS, Morrish TA, Kilroy GE, Swergold GD, Henke J, Henke L, Moran JV, Jorde LB, Batzer MA. A comprehensive analysis of recently integrated human Ta L1 elements. Am J Hum Genet. 2002;71:312–326. doi: 10.1086/341718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita N, Nishio H, Kitoh Y, Ishikawa Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M. Insertion of a 5′ truncated L1 element into the 3′ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest. 1993;91:1862–1867. doi: 10.1172/JCI116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000;28:1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Ostertag EM, DeBerardinis RJ, Goodier JL, Zhang Y, Yang N, Gerton GL, Kazazian HH., Jr A mouse model of human L1 retrotransposition. Nat Genet. 2002;32:655–660. doi: 10.1038/ng1022. [DOI] [PubMed] [Google Scholar]
- Ostertag EM, Goodier JL, Zhang Y, Kazazian HH., Jr SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet. 2003;73:1444–1451. doi: 10.1086/380207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otieno AC, Carter AB, Hedges DJ, Walker JA, Ray DA, Garber RK, Anders BA, Stoilova N, Laborde ME, Fowlkes JD, et al. Analysis of the human Alu Ya-lineage. J Mol Biol. 2004;342:109–118. doi: 10.1016/j.jmb.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Perna NT, Batzer MA, Deininger PL, Stoneking M. Alu insertion polymorphism: a new type of marker for human population studies. Hum Biol. 1992;64:641–648. [PubMed] [Google Scholar]
- Roy AM, Carroll ML, Kass DH, Nguyen SV, Salem AH, Batzer MA, Deininger PL. Recently integrated human Alu repeats: finding needles in the haystack. Genetica. 1999;107:149–161. [PubMed] [Google Scholar]
- Salem AH, Myers JS, Otieno AC, Watkins WS, Jorde LB, Batzer MA. LINE-1 preTa elements in the human genome. J Mol Biol. 2003;326:1127–1146. doi: 10.1016/s0022-2836(03)00032-9. [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Batzer MA. Identity by descent and DNA sequence variation of human SINE and LINE elements. Cytogenet Genome Res. 2005a;108:63–72. doi: 10.1159/000080803. [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Hedges DJ, Jurka J, Batzer MA. Analysis of the human Alu Ye lineage. BMC Evol Biol. 2005b;5:18. doi: 10.1186/1471-2148-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassaman DM, Dombroski BA, Moran JV, Kimberland ML, Naas TP, DeBerardinis RJ, Gabriel A, Swergold GD, Kazazian HH., Jr Many human L1 elements are capable of retrotransposition. Nat Genet. 1997;16:37–43. doi: 10.1038/ng0597-37. [DOI] [PubMed] [Google Scholar]
- Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FP, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- Sheen FM, Sherry ST, Risch GM, Robichaux M, Nasidze I, Stoneking M, Batzer MA, Swergold GD. Reading between the LINEs: human genomic variation induced by LINE-1 retrotransposition. Genome Res. 2000;10:1496–1508. doi: 10.1101/gr.149400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Wu LC, Sanlioglu S, Chen R, Mendoza AR, Dangel AW, Carroll MC, Zipf WB, Yu CY. Structure and genetics of the partially duplicated gene RP located immediately upstream of the complement C4A and the C4B genes in the HLA class III region. Molecular cloning, exon-intron structure, composite retroposon, and breakpoint of gene duplication. J Biol Chem. 1994;269:8466–8476. [PubMed] [Google Scholar]
- Stoneking M, Fontius JJ, Clifford SL, Soodyall H, Arcot SS, Saha N, Jenkins T, Tahir MA, Deininger PL, Batzer MA. Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent BJ, Myers JS, Ho HJ, Kilroy GE, Walker JA, Watkins WS, Jorde LB, Batzer MA. Following the LINEs: an analysis of primate genomic variation at human-specific LINE-1 insertion sites. Mol Biol Evol. 2003;20:1338–1348. doi: 10.1093/molbev/msg146. [DOI] [PubMed] [Google Scholar]
- Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS. A de novo Alu insertion results in neurofibromatosis type 1. Nature. 1991;353:864–866. doi: 10.1038/353864a0. [DOI] [PubMed] [Google Scholar]
- Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, Batzer MA. SVA elements: a hominid-specific retroposon family. J Mol Biol. 2005;354:994–1007. doi: 10.1016/j.jmb.2005.09.085. [DOI] [PubMed] [Google Scholar]
- Wang J, Song L, Gonder MK, Azrak S, Ray DA, Batzer MA, Tishkoff SA, Liang P. Whole genome computational comparative genomics: a fruitful approach for ascertaining Alu insertion polymorphisms. Gene. 2006;365:11–20. doi: 10.1016/j.gene.2005.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WS, Ricker CE, Bamshad MJ, Carroll ML, Nguyen SV, Batzer MA, Harpending HC, Rogers AR, Jorde LB. Patterns of ancestral human diversity: an analysis of Alu-insertion and restriction-site polymorphisms. Am J Hum Genet. 2001;68:738–752. doi: 10.1086/318793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WS, Rogers AR, Ostler CT, Wooding S, Bamshad MJ, Brassington AM, Carroll ML, Nguyen SV, Walker JA, Prasad BV, Reddy PG, Das PK, Batzer MA, Jorde LB. Genetic variation among world populations: inferences from 100 Alu insertion polymorphisms. Genome Res. 2003;13:1607–1618. doi: 10.1101/gr.894603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Salem AH, Hedges DJ, Kilroy GE, Watkins WS, Schienman JE, Stewart CB, Jurka J, Jorde LB, Batzer MA. Comprehensive analysis of two Alu Yd subfamilies. J Mol Evol. 2003;57(Suppl 1):S76–S89. doi: 10.1007/s00239-003-0009-0. [DOI] [PubMed] [Google Scholar]