

Abstract
Chromosomal translocations are a hallmark of hematopoietic malignancies. The initial molecular events or pathways that lead to translocations in hematopoietic cells are largely unknown, particularly in the stem cell-enriched population postulated to be the initial target for these events. We used in vitro differentiation of mouse embryonic stem (ES) cells combined with an I-SceI endonuclease double-strand break (DSB) repair assay to determine the relative susceptibility of isogenic hematopoietic subpopulations to DSB-induced translocations and the mechanisms that generate them. DSB-induced reciprocal translocations were frequently observed in multipotent progenitors but significantly suppressed in actively proliferating myeloid cells. Most reciprocal translocations in multipotent progenitors resulted from repair consistent with single-strand annealing followed by gap repair. Overexpression of Rad51, a protein central to DNA strand exchange and recombination, did not further increase the frequency of recovered translocations but did increase the frequency of long-tract gene conversion events associated with loss of heterozygosity and tandem duplications. These data directly demonstrate that hematopoietic multipotent progenitor cells are particularly susceptible to the formation of chromosomal rearrangements analogous to those observed in human hematopoietic malignancies. This particular subpopulation apparently represents a window of opportunity for the initiation of potentially oncogenic events following DNA damage.
Keywords: Double-strand break, homologous recombination, hematopoietic stem cell, chromosomal translocation, genome stability, Rad51
Genome integrity relies on the faithful repair of DNA damage such as double-strand breaks (DSBs) (van Gent et al. 2001). Chromosomal DSBs are formed following exposure to irradiation, alkylating agents, and topoisomerase II (topo II) inhibitors (Ward 1988; Osheroff et al. 1994; Gatto et al. 1999). Aberrant repair of DSBs is expected to result in chromosomal rearrangements, including translocations that are a hallmark of hematopoietic malignancies and some soft-tissue sarcomas (Tycko and Sklar 1990; Rowley 2000; Elliott and Jasin 2002).
DSBs are potent inducers of recombination that increase homologous recombination (HR), nonconservative homology-directed single-strand annealing (SSA), and nonhomologous end-joining (EJ) events by several orders of magnitude (Liang et al. 1998; Richardson et al. 1998; Richardson and Jasin 2000b; Elliott et al. 2005), with the relative activity of the pathways varying with the stage of the cell cycle (Liang et al. 1998; Rothkamm et al. 2003; Weinstock et al. 2006). Sequence analysis of recurrent chromosomal translocation breakpoints in human tumors and those generated in mouse and human cell systems suggests chromosomal rearrangements can be generated through multiple DSB repair pathways (Reichel et al. 1998; Lovett et al. 2001; Elliott and Jasin 2002; Kolomietz et al. 2002; Langer et al. 2003; Rassool 2003). These pathways differ based on their requirement for a donor DNA template with significant sequence homology with the site of DNA damage (Rouet et al. 1994; Liang et al. 1998).
Treatment with topo II-inhibiting chemotherapeutic agents such as etoposide is known to promote and stabilize DNA DSBs (Osheroff et al. 1994; Aplan et al. 1996). One class of therapy-related acute myelogenous leukemia (t-AML) is characterized by rearrangements within the mixed lineage leukemia (MLL) locus on chromosome band 11q23 and is associated with prior treatment with etoposide (Super et al. 1993; Lovett et al. 2001; Felix et al. 2006). Since leukemias characterized by MLL rearrangements can involve cells of both myeloid and lymphoid lineages, it is thought that the initiating tumor cell is a stem cell. Leukemia-initiating cells from AML patients are restricted within the stem cell-enriched CD34+ fraction (Sutherland et al. 1996; Bonnet and Dick 1997). Cancer stem cells may be a general paradigm for tumor initiation, and have been identified in multiple tumor types (Al-Hajj et al. 2003; G.D. Richardson et al. 2004; Singh et al. 2004; Jordan 2006; Jordan et al. 2006). However, it remains unclear if the genome rearrangements characteristic of leukemia and lymphoma arise as part of normal DSB repair processes or if they require additional cellular mutations or stimulation of signaling pathways specific to DNA damaging agents.
Use of the I-SceI endonuclease genetic system has demonstrated that two specific DSBs in murine embryonic stem (ES) cells are sufficient to promote chromosomal translocations by mechanisms involving HR between repeat sequences on heterologous chromosomes (Richardson and Jasin 2000b). ES cells may not reflect normal DSB repair in somatic cell types that may lose the potential for HR as part of the normal differentiation program. In addition, DSBs per se may not be sufficient to promote recombinogenic events in otherwise wild-type hematopoietic cell populations. We adapted the I-SceI genetic system to directly determine the potential for DSBs to promote chromosomal translocations and genome rearrangements in isogenic hematopoietic cell populations and provide direct evidence for this type of damage to lead to rearrangements observed in a clinical setting. This study revealed that hematopoietic cells at both early and late stages of differentiation can repair DNA DSBs by interchromosomal HR. Significantly, the induction of DSBs in the early hematopoietic multipotent progenitor subpopulation produced a class of reciprocal translocation repair products consistent with a combination of SSA and gap repair. This outcome was not detected during DSB repair in isogenic ES cells, nor in more differentiated myeloid lineage cells that had a notable absence of any detectable DSB-induced genome rearrangements. In cells engineered to stably overexpress Rad51, central to HR in mammalian cells and the mammalian homolog of the Escherichia coli RecA strand exchange protein (Benson et al. 1994; Baumann et al. 1996), the spectrum of DSB repair events recovered indicated that elevated Rad51 expression significantly increased the frequency of HR events that lead to loss of heterozygosity (LOH) and partial tandem duplications (PTDs). The data presented in this study demonstrate that multipotent stem cell-enriched hematopoietic progenitors can repair DNA DSBs by interchromosomal HR in a manner that frequently leads to reciprocal chromosomal translocations, and that elevated Rad51 expression in these cells increases the proportion of specific repair events that can be mutagenic.
Results
In the genetically engineered murine ES cell line C2FN2, loci on chromosomes 14 and 17 (termed N2 and F, respectively) were each targeted with a defective neomycin phosphotransferase gene (neo) containing the cleavage site for the rare-cutting I-SceI endonuclease (Fig. 1; Richardson and Jasin 2000b). Due to its 18-base-pair (bp) nonpalindromic recognition site, I-SceI endonuclease is expected to cleave only the targeted neo genes (Colleaux et al. 1988). Following I-SceI endonuclease expression and induction of DSBs at one or both I-SceI sites, repair via a homologous recombination mechanism between the two different chromosomes at the neo loci may restore a functional neo gene that confers G418 resistance. Molecular analysis of individual repair structures identified multiple genome rearrangements including deletions, insertions, duplications, and translocations (Richardson and Jasin 2000b).
Figure 1.

Parental targeted alleles for assay of interchromsomal DSB repair with relative location of PCR primers used for analysis or repair products. (F allele) Defective neo S2neo contains an I-SceI site replacing an NcoI site. This was targeted to the pim-1 locus on chr.17 in murine ES cells. (N2 allele) The other defective neo N2neo contains an I-SceI site replacing an EagI site. This was targeted to the Rb locus on chr.14 in murine ES cells (Richardson and Jasin 2000b). Staggering of the I-SceI sites in the two neo sequences results in a 526-bp overlap of neo sequence between the two alleles. Relative primer sites used for PCR classification of repair clone structures are noted. Primers 5′ to neo were on the sense strand, and primers 3′ of neo are on the antisense strand. Encircled number and letter notations of primers are used in figures to show expected band sizes.
To obtain multiple isogenic hematopoietic cell populations that contain the DSB reporter sequences, we employed an in vitro differentiation scheme originally developed to study the hematopoietic stem cell-enriched population (Keller et al. 1993; Helgason 2000). Using specific growth factor combinations, ES cells differentiated out of their totipotent state into embryoid bodies (EBs), then bursting EBs that contain early multipotent hematopoietic progenitors (EB/HPs), then progenitor hematopoietic colonies of myeloid and erythroid lineages, and then terminally differentiated myeloid colonies (see Materials and Methods). Hematopoietic colonies were initially classified based on their morphology (Fig. 2A–F). RT–PCR on individual colonies confirmed appropriate expression of genes within each stage of the in vitro hematopoietic differentiation (Fig. 2G). On the basis of morphology and hematopoietic-specific gene expression, cells within the EB/HP population and differentiated myeloid colonies all demonstrate appropriate gene expression for the stage of hematopoietic differentiation.
Figure 2.

In vitro hematopoietic differentiation and marker expression in differentiated colonies. Morphological analysis of ES cell-derived hematopoietic colonies. (A) Undifferentiated ES cells 200×. (B) Bursting EB/HP 200×. (C) CFU-GEMM 200×. (D) CFU-GM 200×. (E) CFU-Mast 200×. (F) CFU-E 100×. (G) Analysis of expression of hematopoietic stage-specific marker expression during in vitro hematopoietic differentiation by RT–PCR of individual colonies. ES cells, day 14 bursting EB/HP, CFU-GEMM, CFU-GM, CFU-Myeloid, NIH-3T3 mouse fibroblast cell line. OCT-4 restricted to totipotent stem cells. CD45 expressed on the surface of all hematopoietic cells except mature erythrocytes and platelets. TAL/SCL expressed in hematopoietic cells of multipotent and myeloid lineages. β-globin expressed in progenitor and erythroid lineage cells. PU.1 and GATA-2 were restricted to myeloid cells. β-actin as a loading control. NIH3T3 cells negative for all markers except CD45; however, expression of hematopoietic-specific genes in this cell line has been observed by others (Du et al. 2002). Myeloid cells were positive by RT–PCR for additional myeloid-specific markers including c-myb, MAC-1, PR-3, and gp91 (data not shown).
To induce DSBs at the targeted loci in the isogenic hematopoietic subpopulations, the I-SceI expression vector pCBASce (Richardson et al. 1998) was transfected into cells at multiple stages of differentiation. Following induction of DSBs in cells at each stage of differentiation, cells were plated in methylcellulose containing G418 and hematopoietic-specific growth factors. Individual repair events resulting in G418-resistant (G418R) hematopoietic colonies were readily obtained following DSBs in all subpopulations. The average frequency of recombination was 38.3 × 10−5 ± 3.1 in ES cells, consistent with previous studies (Richardson and Jasin 2000b), 3.7 × 10−5 ± 1.2 in EB/HP cells, and 2.5 × 10−5 ± 1.0 in myeloid cells (n = 4). Recombination frequencies were corrected for electroporation efficiency by transfection of the green fluorescent protein (GFP) expression vector pCBAGFP into parallel cohorts of cells (see Materials and Methods). Thus, both multipotent and terminally differentiated cell types retain the potential to repair DSBs with homologous sequence on a heterologous chromosome, although the absolute frequency of recombination between chromosomes in the hematopoietic cells is ∼10- to 15-fold lower than that in undifferentiated ES cells. Furthermore, the potential for interaction between sequences on heterologous chromosomes in wild-type cells is not eliminated by epigenetic factors or chromatin remodeling associated with hematopoietic differentiation programs.
Due to the limited size of individual hematopoietic colonies for analysis (average size 102–103 cells), a nested PCR amplification and diagnostic enzymatic digest strategy of the N2neo (chr.14) and F (chr.17) alleles classified the spectrum of repair products in individual G418R clones (Table 1; Figs. 3, 4). Repair products obtained in parental ES cells were consistent with results reported previously (Richardson and Jasin 2000b; C. Richardson et al. 2004).
Table 1.
Distribution of DSB-induced interchromosomal repair events in differentiating hematopoietic subpopulations as compared with isogenic parental ES cells

aResults from ES cells are the average from control parallel experiments and are similar to those published previously (n = 43) (Richardson and Jasin 2000b).
bp values were determined by Fisher’s exact test comparing the number of events in the total pool of clones from all experiments.
Figure 3.

Classification of DSB-induced gene conversion repair events. (A) DSB repair of parental alleles by gene conversion (STGC). Following DSB at either I-SceI site (S), strand invasion and repair synthesis (denoted by curved arrow) limited to homologous neo sequence results in gene conversion (STGC) and restoration of an NcoI site (N) on the Fneo+ allele or EagI site (E) on the N2neo+ allele. PCR amplification of neo genes in recombinant clones from parental or repaired alleles. The F allele gives a 0.9-kb product using primers 2 and 4, and the N2 allele gives a 1.1-kb product using primers B and D. There is no change in this uncut (U) PCR product size between parental and converted alleles. Conversion leads to loss of the I-SceI site (S) and absence of digestion, and gain of NcoI (N) or EagI (E) site and gain of digestion. (B) DSB repair of parental alleles by gene conversion extending into heterology (LTGC). Following DSB at either I-SceI site (S) on F or N2, strand invasion and repair synthesis (denoted by two-headed arrow) extended beyond homology into downstream heterologous sequence resulting in a neo+ hybrid F/N2 allele. This event is detectable by PCR using primers 1 and E from sequences flanking the DSB sites on opposite alleles (hygR and HPRT) to yield a 3.3-kb product. There is no change in the other allele in these events; the F allele gives a 0.9-kb product using primers 2 and 4, and the N2 allele gives a 1.1-kb product using primers B and D.
Figure 4.

Classification of DSB-induced translocation events. (A) DSB repair leading to translocation by either SSA–gap repair or by SSA–EJ. SSA between the 526 bp of homologous overlap between the neo genes produces an F/N2 allele in all clones (see Fig. 3). In SSA–gap repair, the second hybrid allele is produced by strand invasion and repair synthesis (denoted by curved arrow) limited to a homologous neo sequence resulting in restoration of both the EagI (E) and NcoI (N) sites. The resultant N2/F allele with a full-length neo sequence is detected by PCR using primers B and 4 from sequences flanking the DSB sites on opposite alleles (Rb and pim) to yield a 2.1-kb product. In SSA–EJ, the second hybrid allele is produced by EJ of the broken ends of the nonhomologous extreme 5′ and extreme 3′ portions of neo gene sequences. The resultant N2/F allele is detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (Rb and pim) to yield a 1.6-kb product or smaller depending on processing of ends prior to EJ. (B) PCR detection of the N2/F allele using primers B and 4 in DSB repair colonies from EB/HP cells (see Table 1). Eleven PCR products were 2.1 kb, indicating SSA–gap repair, and four PCR products were 1.6 kb, indicating SSA–EJ with minimal deletion processing of the ends.
The majority of interchromosomal repair products in EB/HP-derived hematopoietic colonies (71% of G418R clones) were consistent with gene conversion limited to the region of homology in the alternate neo sequence (short-tract gene conversion, STGC) (Table 1; Fig. 3A). In this class, the PCR products amplifying the region across the DSB site on the F allele and on the N2 allele were unaltered from the parental configuration (Fig. 3A). DSB repair of either neo sequence eliminated the I-SceI site and reconstituted an NcoI site on the F allele or an EagI site the N2 allele. Of the 42 STGC clones, three underwent EJ at the I-SceI site on the nonconverted chromosome, but none converted both alleles to neo+. PCR using primers from sequences flanking the DSB sites on opposite alleles (e.g., hygR and HPRT to identify hybrid F/N2; Rb and pim to identify hybrid N2/F) did not recover products, confirming that these clones underwent gene conversion without exchange of flanking markers (data not shown).
Two repair products in EB/HP-derived hematopoietic colonies (3% of G418R clones) were consistent with gene conversion via a replication-based mechanism that extended beyond one neo sequence into downstream heterologous sequence (long-tract gene conversion, LTGC) (Table 1; Fig. 3B; Moynahan and Jasin 1997; Richardson et al. 1998). The frequency of these events in colonies derived from EB/HP cells was significantly lower than detected in parental ES cells (3% vs. 21%; p = 0.003). In both, the converted neo+ hybrid F/N2 allele was detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (hygR and HPRT) (Fig. 3B). By contrast, the donor allele remained unaltered and was amplified by PCR in the parental configuration.
The remaining 26% of G418R EB/HP-derived hematopoietic colonies were reciprocal translocations (Table 1; Fig. 4). This genome rearrangement results from the dual exchange of flanking markers on both alleles resulting in two hybrid alleles, F/N2 and N2/F. We used PCR across the breakpoint junctions and sequencing analysis to provide insight on the repair mechanisms used in individual repair events. As expected based on previous studies, recovered DSB-induced reciprocal translocations from parental ES cells arose exclusively from SSA repair coupled with EJ (SSA–EJ) (Fig. 4A, right column; Richardson and Jasin 2000b). By contrast, only a small number of reciprocal translocations from isogenic EB/HP-derived hematopoietic colonies had structures consistent with this mechanism (7% of G418R clones) (Table 1). The F/N2 allele in all clones was detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (hygR and HPRT) (Fig. 4A, right column). The reciprocal hybrid N2/F allele resulting from EJ of the broken ends of the nonhomologous extreme 5′ and extreme 3′ portions of neo gene sequences was detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (Rb and pim, Fig. 4B). Sequence analysis of the N2/F breakpoint junctions of the four translocations arising from SSA–EJ demonstrated minimal if any deletion (0, 0, 5, and >27 bp, respectively) from the two ends during processing followed by EJ within microhomology (overlapping ATAA, ATAA, AGGG, and C, respectively).
Surprisingly, the majority of reciprocal translocations in EB/HP-derived hematopoietic colonies (11 of 15 translocations, 19% of all G418R clones) had novel repair structures that contained hybrid F/N2 and N2/F alleles both with full-length neo genes (Table 1; Fig. 4A, left column). The F/N2 allele was detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (hygR and HPRT), and the reciprocal N2/F allele was detected by PCR using primers from sequences flanking the DSB sites on opposite alleles (Rb and pim) (Fig. 4B). Recovery of clones with full-length neo+ sequences on both hybrid alleles could occur by either of two distinct mechanisms: (1) gene conversion associated with crossing-over via resolution of two Holliday junctions (Szostak et al. 1983), or (2) SSA and subsequent gap repair (SSA–gap repair) (Fig. 4A, left column; Ferguson and Holloman 1996). These two pathways can be distinguished by sequence analysis of the N2/F hybrid allele. A crossover event would retain staggered I–SceI mutations after Holliday junction resolution and remain neo−, but a SSA–gap repair event would restore a full-length neo+ gene during the gene conversion gap repair event using neo+ sequence from the F/N2 allele as a template (Fig. 4A, left column). Sequencing of three clones confirmed that the neo genes were both intact, favoring SSA–gap repair. Overall, multipotent hematopoietic cells are particularly susceptible to DSB-induced reciprocal translocations and often use a different repair pathway than ES cells.
To determine if the potential for DSBs to promote reciprocal translocations is a characteristic of all hematopoietic cells or is limited to specific subpopulations, we induced DSBs in isogenic myeloid cells. For this, ES-derived hematopoietic myeloid colonies (colony-forming unit (CFU)-Myeloid) were isolated and pooled (see Materials and Methods), transfected with I-SceI to induce DSBs, and then replated in methylcellulose. Individual G418R clones were isolated and the repair products characterized, as described above. G418R clones recovered after induction of DSBs in three independent experiments demonstrated a unique product spectrum. All 43 clones (100%) repaired their DSBs by STGC (Table 1). There was no evidence of additional genome rearrangements in any of the clones (data not shown).
Reciprocal translocation products with two full-length neo genes are not recovered in wild-type ES cells using this genetic system; transient Rad51 overexpression overrides this suppression to promote crossovers and also results in aneuploidy (Richardson et al. 1998; C. Richardson et al. 2004). Since our results indicated that the EB/HP population is proficient at repair that results in reciprocal translocations with two full-length neo sequences at the junctions, we questioned whether Rad51 overexpression in this cell population would further stimulate these events during DSB repair. Parental C2FN2 ES cells were engineered to stably overexpress hsRad51 by transfection with the hsRad51 cDNA expression vector, pCBARad51 (Stark et al. 2002; C. Richardson et al. 2004). A pool of clones (Rad51p) and a single clone (Rad51c) showed an eightfold to 10-fold increase in Rad51 protein expression by Western blotting of whole-cell extracts as compared with control. A four- to fivefold increase in Rad51 protein expression was detected in pooled hematopoietic cells as compared with control (Fig. 5). Constitutive overexpression of Rad51 had no gross effect on the growth characteristics of undifferentiated cells or on the distribution and numbers of ES-derived hematopoietic colonies. Elevated levels of Rad51 protein did not inhibit in vitro differentiation of ES cells into EB/HP cells as the average number of cells per EB/HP colony was similar among groups: 8.6 × 103 for control and 11.3 × 103 for +Rad51 cells. Overexpression of Rad51 resulted in an average fourfold increase in the overall frequency of G418R clones following DSB induction with I-SceI: 16.5 × 10−5 ± 0.9 in +Rad51 cells as compared with 3.7 × 10−5 ± 1.2 in controls.
Figure 5.
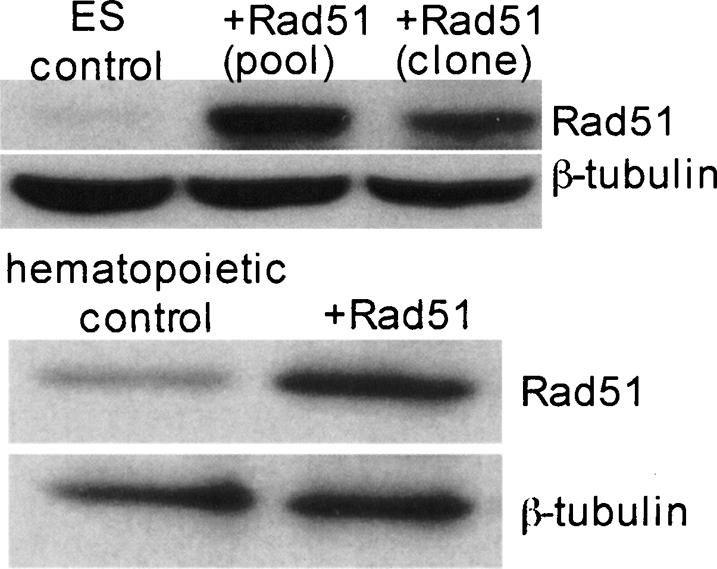
Elevated Rad51 protein levels in ES and hematopoietic cells stably transfected with CBARad51. Control ES cells contain low to undetectable levels of Rad51 protein. Following stable transfection of cells (see Materials and Methods), a pool of +Rad51 clones and a single +Rad51 clone both show increased Rad51. Control differentiated hematopoietic cells contain low levels of Rad51 protein. A pool of stably transfected cells differentiated to hematopoietic cells show elevated Rad51 protein levels. β-Tubulin was used as a loading control.
The structure and spectrum of G418R repair events was determined using the PCR approach described above (Fig. 1; Table 2). Overexpression of Rad51 in EB/HP cells was associated with a significant decrease in STGC events and concomitant increase in LTGC events, as compared with controls. STGC events declined from 71% to an average of 57% and LTGC events rose from 3% to 16% of +Rad51 EB/HP-derived G418R clones (p = 0.044 and p < 0.05, respectively). A single +Rad51 clone converted both the F and N2 alleles. EJ repair events on the nonconverted allele were detected in an average of 25% of STGC clones from the +Rad51 cells. These results differed significantly from the control clones, in which only 7% demonstrated EJ repair at the nonconverted allele (p ≤ 0.05). There was no shift in the portion of reciprocal translocations in clones that overexpressed Rad51 as compared with controls (Table 2). Twenty-four percent of +Rad51 EB/HP-derived G418R clones were reciprocal translocations, similar to the recovery of reciprocal translocations in 26% of controls. PCR and sequencing across breakpoints indicated no significant shift in the repair mechanisms used to form the translocations. SSA–gap repair events occurred in an average of 66% of +Rad51 and 73% of control clones. SSA–EJ events were detected in an average of 34% of +Rad51 and 27% of control clones. Sequence analysis of the N2/F breakpoint junctions of the SSA–EJ translocations from +Rad51 clones demonstrated minimal deletion from the ends and microhomology (0–4 bp) at the breakpoint junctions. Eleven of these had deletions <10 bp, and another four had deletions of <60 bp. A single clone deleted 1012 bp and inserted GCCT at the junction.
Table 2.
Distribution of DSB-induced interchromosomal repair events in hematopoietic EB/HP cells promoted by overexpression of Rad51

Following transient expression of I-SceI in control EB/HP cells or in EB/HP cells that stably overexpress Rad51, neo+ hematopoietic clones were isolated and analyzed.
aResults from control EB/HP cells are also presented in Table 1 (n = 3).
bp values were determined by Fisher’s exact test comparing the number of events in the total pool of clones from all experiments.
cIn these, one neo+ hybrid allele was recovered without detection of the reciprocal hybrid allele or parental F or N2 alleles.
Seven clones had evidence of a rearrangement and allele loss during DSB repair. Five clones amplified the expected PCR product from a F/N2 allele, but did not amplify a product from a N2/F allele (Table 2). Two clones amplified a N2/F allele but did not amplify a product from a F/N2 allele (Table 2). Parental F and N2 alleles were not detected in these. These results are most consistent with SSA–gap repair occurring in S/G2 with segregation of hybrid alleles to different daughter cells, which could produce and maintain either a neo+ F/N2 allele or a neo+ N2/F allele (see Fig. 4A).
Discussion
The data presented in this study provide evidence that multipotent stem cell-enriched hematopoietic progenitors can repair DNA DSBs by interchromosomal HR in a manner that frequently leads to reciprocal chromosomal translocations. Significantly, the induction of DSBs in the early hematopoietic multipotent progenitor subpopulation produced a large class of reciprocal translocations consistent with a combination of SSA and gap repair. This outcome was unique to this subpopulation and was not detected during DSB repair in isogenic ES cells or in more differentiated myeloid cells that had a notable absence of any detectable DSB-induced genome rearrangements.
Chromosomal translocations are a frequent outcome of DSB repair in multipotent hematopoietic cells: repair by SSA–gap repair
Interchromosomal DSB repair resulted in a significant number of reciprocal translocations in EB/HP cells. Although some reciprocal translocations in EB/HP cells had repair structures similar to those previously observed in ES cells, the significant majority contained hybrid alleles with two full-length neo genes. The presence of full-length neo sequences on both hybrid alleles could occur either by gene conversion associated with crossing-over or by SSA and subsequent gap repair. Staggered placement of the I-SceI mutations favors crossover products that contain one neo+ F/N2 allele and one nonfunctional neo− N2/F allele, while repair by SSA–gap repair may produce F/N2 and N2/F alleles that both have full-length neo+ genes. Other well-characterized mechanisms could generate one functional neo+ F/N2 allele and one nonfunctional mutated neo− N2/F allele, including (1) SSA–EJ with extensive deletion involving at least one PCR primer site, (2) nonreciprocal translocations in which SSA produced a neo+ F/N2 allele while the two remaining unjoined chromosome ends were lost, and (3) repair by SSA–EJ or by gene conversion associated with crossover during S/G2 with segregation of the two repair products to opposing poles. Sequence analysis of repair products here suggests that both neo sequences were intact and is most consistent with repair by SSA followed by gap repair. Further support for a SSA–gap repair mechanism comes from the seven clones recovered that contained a single hybrid neo+ allele, F/N2 or N2/F. If repair occurs in S/G2, then segregation of hybrid alleles to different daughter cells would result in clones that maintain either a neo+ F/N2 allele or a neo+ N2/F allele able to confer G418 resistance. Crossing-over is suppressed during interchromosomal mitotic gene conversion in wild-type mammalian cells, and the repair products recovered here suggest that it is not a significant mechanism in hematopoietic stem cell populations either.
In SSA followed by gap repair, DSBs in the neo genes allow repair by SSA to form a hybrid F/N2 allele. The two remaining free ends would then utilize this F/N2 allele as a homologous template for gap repair to create the other N2/F allele. This mechanism suggests a temporal model of events, and suggests the four chromosome ends remain in close proximity during repair, as opposed to two independent repair events occurring in distinct loci. Consistent with this, immunofluorescence studies show colocalization of DNA repair factors and multiple broken chromosomes in mitotic cells indicative of “repairosomes” (Lisby et al. 2003) that likely include Rad51 protein foci thought to represent active sites of repair (Scully et al. 1997; Raderschall et al. 1999).
Recent analysis of primary leukemic samples has examined translocation breakpoints of both derivative chromosomes to define the mechanisms that lead to these events. In two reports analyzing samples with t(4;11) and t(9;11) translocations, more than half contained one parental sequence present at the breakpoint site on both derivative chromosomes (Reichel et al. 1998; Langer et al. 2003). One explanation for this phenomenon has been initiation of the translocation event by single-strand DNA breaks leaving long single-strand tails. Following annealing of both tails to partner chromosome ends, subsequent fill-in of the single strands would lead to duplication of the parental sequence on both resultant derivative chromosomes. An alternative explanation would be analogous to the events observed here (Fig. 4A, left column). Following DSBs and annealing to produce one derivative chromosome, use of gap repair to repair and ligate the remaining two ends to form the second derivative chromosome would also result in a duplication of one parental sequence at the breakpoint.
Hematopoietic differentiation limits gene conversion tract length
Differences in developmental stage and cell lineage correlated with a shift in the DSB repair products recovered. STGC events remained the majority of gene conversion events at all stages of differentiation. LTGC events progressively and significantly decreased as cells differentiated into hematopoietic subpopulations while the frequency of STGC events increased. The complete lack of LTGC repair events in myeloid cells suggests this population stringently limits the extent of conversion events as well as suppresses other repair mechanisms that would produce neo+ clones.
Mechanistically, suppression of LTGCs in more differentiated populations may be due to a reduction in processivity of the D-loop DNA synthesis repair mechanism, or a reduction in nuclease end degradation. Gene conversion tracts in both mammalian and yeast cells tend to be short, with average tract lengths ranging from <100 to 400 bp (Palmer et al. 2003; Paffett et al. 2005; Weinstock et al. 2006). Some gap repair events in yeast require successive rounds of invasion, reinitiation, and synthesis, suggesting that DNA synthesis during DSB repair has low processivity (Paques et al. 1998; McVey et al. 2004). LTGCs may be limited by frequent displacement of the newly synthesized strand to promote annealing within complementary sequence on the broken chromosome (Richardson and Jasin 2000a; McVey et al. 2004). Reduced nuclease degradation of broken chromosome ends in hematopoietic cells would reduce the requirement for strand synthesis and limit LTGC events. In support of this, analysis of the EJ breakpoints from the hybrid N2/F alleles of translocations from EB/HP neo+ clones showed little or no degradation at the junctions, with two out of three junctions having no deletion, and the remaining junction having a 5-bp deletion. In contrast, all N2/F alleles from neo+ clones in ES cells had deletions at the junctions ranging from 13 bp to >1 kb (Richardson and Jasin 2000b; this study).
Alternatively, the reduced overall frequency of neo+ HR events for analysis in the myeloid cells could indicate a shift toward EJ or cell death as a predominant outcome of DNA damage, including cells that repair DSBs to result in LTGCs or rearrangements. The recovery of LTGC events in previous studies of I-SceI-induced DSBs in ES cells led to the model of one-sided invasion and repair via a replication-based mechanism (Richardson et al. 1998; Richardson and Jasin 2000a). Repair in myeloid cells may occur via a two-sided invasion mechanism expected to exclusively produce STGC events. We cannot rule out that only one I-SceI site is cleaved within a single cell in this subpopulation, which has been linked to a strong bias of STGC events (Richardson et al. 1998).
Overexpression of Rad51 in hematopoietic multipotent progenitor cells is sufficient to complement the suppression of LTGC repair events observed during hematopoietic lineage differentiation. This is consistent with the idea that Rad51 increases DNA synthesis processivity or stabilizes pairing events to favor longer conversion tracts and is supported by studies in P element excision-induced gap repair in Drosophila and gap repair in Saccharomyces cerevisiae (Symington et al. 2000; McVey et al. 2004). In addition, elevated Rad51 may be sufficient to form more stable strand invasion intermediates or overcome disruption of Rad51 presynaptic filaments by SWI/SNF-associated Rad54 (Solinger et al. 2002) or by DNA helicases, as shown with Srs2 (Krejci et al. 2003; Veaute et al. 2003), thus favoring the formation of illegitimate intermediate structures.
Multiple immortalized and tumor cell lines overexpress Rad51, and screening of primary tumor samples has identified elevated levels of wild-type Rad51 protein (Maacke et al. 2000a, b; Slupianek et al. 2001, 2002; Raderschall et al. 2002; Skorski 2002). There has been speculation about whether disregulation of Rad51 protein can be an early step in transformation or secondary to mutations in regulators of Rad51. In this report, Rad51 is sufficient to promote certain mutagenic DSB repair pathways associated with LOH and PTDs analogous to those observed in leukemias (Moynahan and Jasin 1997; Strout et al. 1998; Richardson and Jasin 2000a). Hematopoietic cells may be sensitive to Rad51 protein levels to stimulate alternative DSB repair in the same cell subpopulation postulated to be the direct target of the initiating chromosomal translocations that are a hallmark of hematopoietic malignancies.
Stem cells as a target population for chromosomal rearrangements
Clinical parameters and tumor pathology of multiple tumor types have suggested that stem cells are the target of the initiating lesion. Hierarchical models propose that “cancer stem cells” possess some distinct phenotype from the bulk of the population that permit them to act as tumor initiators. Stem cells could act as tumor initiators because they possess a greater capacity for proliferation into multiple lineages even if the initial event is no more likely in any particular cell, or because they are more susceptible than differentiated cell types to the initial acquisition of genome alterations. Our data support the idea that hematopoietic cells do not lose the potential for HR as part of the differentiation program, but DSB repair pathways utilized in multipotent stem cells are distinct to this population and more likely to give rise to genome rearrangements than pathways used by terminally differentiated, but actively proliferating, myeloid cells. Suppression of rearrangements in terminally differentiated myeloid cells could be due to transcriptional changes or epigenetic alterations resulting in reduced activity of certain proteins. Overall, the multipotent progenitor subpopulation apparently represents a “window of opportunity” for the initiation of potentially oncogenic events following exposure to DNA-damaging agents. This suggests that development of targeted anti-cancer therapies should be aimed at this somewhat elusive population that clearly has a distinct reactive profile to DNA damage, repair, and survival.
Materials and methods
Cell lines and tissue culture
Cultures were maintained at 37°C and 5% CO2. The C2FN2 targeted murine ES cell line was maintained as previously described (Richardson and Jasin 2000b). Cells were passaged every 48 h by trypsinization.
In vitro hematopoietic differentiation
ES cells were plated for predifferentiation at 2.85 × 105 cells per 10-cm, gelatinized tissue culture dish in Iscove’s Modified Dulbecco’s Medium (IMDM), 15% ES-Qualified FBS, 1 mM sodium pyruvate, 100 U/mL penicillin-streptomycin, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1000 U/mL ESGRO LIF, and 100 μM monothioglycerol (MTG) for 48 h. Cells were then plated at 500 cells per 3.5-cm, low-adherence tissue culture dish in IMDM, 1% methylcellulose, 15% FBS, 100 U/mL penicillin-streptomycin, 2 mM L-glutamine, 150 μM MTG, and 40 ng/mL murine stem cell factor (mSCF) to form embryoid bodies (EBs). On day 7, EB cultures were given IMDM, 0.5% methylcellulose, 15% FBS, 100 U/mL penicillin-streptomycin, 150 μM MTG, 160 ng/mL mSCF, 30 ng/mL interleukin-3 (mIL-3), 20 ng/mL human interleukin-6 (hIL-6), and 3 U/mL human erythropoietin (hEpo), and cultures were incubated for an additional 7 d. Secondary differentiation: On day 14, bursting EB/HPs were harvested by diluting each culture 1:3 in IMDM and 2% FBS, and were disrupted into a single-cell suspension by treatment with 0.25% collagenase for 60 min at 37°C and passage through a 21-gauge needle three times, then addition of equal volume of IMDM and 5% FBS. Single cells were plated at a density of 1.0 × 105 cells per 3.5-cm, low-adherence tissue culture dish in medium containing 1% methylcellulose, 15% FBS, 100 U/mL penicillin-streptomycin, 2 mM L-glutamine, 150 μM MTG, 1% bovine serum albumin (BSA), 10 μg/mL insulin, 200 μg/mL transferrin, 150 ng/mL mSCF, 30 ng/mL mIL-3, 30 ng/mL hIL-6, and 3 U/mL hEpo for 12–14 d, after which hematopoietic colonies were scored. Mature colonies derived from individual precursors included the CFU granulocyte–erythrocyte–monocyte–megakaryocte (CFU-GEMM), CFU granulocyte–monocyte (CFU-GM), CFU granulocyte (CFU-G), CFU monocyte (CFU-M), CFU erythrocyte (CFU-E), CFU megakaryocyte (CFU-Mk), and CFU mast (CFU-Mast). Myeloid cells: After day 14, CFU-Myeloid colonies were a pooled mix of CFU-G, CFU-M, CFU-Mk, and CFU-Mast colonies that were isolated, pooled, and maintained in IMDM, 1% methylcellulose, 15% FBS, 1% BSA, 10 μg/mL insulin, 200 μg/mL transferrin, 100 μM β-ME, 2 mM L-glutamine, 100 U/mL penicillin-streptomycin, 50 ng/mL mSCF, 10 ng/mL mIL-3, 10 ng/mL hIL-6, and 3 U/mL hEpo until induction of DSBs.
RNA isolation and RT–PCR
Cells from individual colonies were pelleted by centrifugation. RNA was isolated using Qiagen RNeasy Mini Kit with the on-column Qiagen RNase-Free DNase Set. RNA concentration was determined by A260 spectrophotometric measurement. RT–PCR was performed using 25 ng of template RNA in each 25-μL reaction using the Qiagen OneStep RT–PCR kit per the manufacturer’s protocol. Samples were incubated for 30 min at 50°C; for 15 min at 95°C, followed by 35 cycles of 30 sec at 94°C, 30 sec at 60°C, 1 min at 72°C; and extension for 10 min at 72°C.
Transfection
Electroporations were performed with 0.8 mL of cells at 10 × 106 to 25 × 106 cells per milliliter and 60 μg of circular pCBASce or circular pCBAGFP expression vector (Richardson et al. 1998). Settings were 250 mV and 950 μF (ES) or 220 mV and 1600 μF (EB/HP and myeloid). Twenty-four hours later, the number of surviving cells was determined by hemacytometer. G418 was added at 200 μg/mL for ES cells, 400 μg/mL for EB/HP cells, and 900 μg/mL for myeloid cells. Colonies were scored after 10–14 d in selection. G418R hematopoietic colonies per 1 × 107 cells averaged 925 from ES cells, 39 from EB/HP cells, and six from myeloid cells. Recombination frequencies were calculated as the number of colonies scored divided by the number of cells surviving transfection. Recombination frequencies were corrected for electroporation efficiency by transfection of the GFP expression vector pCBAGFP into parallel cohorts of cells, and 24 h later cells were collected, washed, resuspended in 1 mL of Dulbecco’s phosphate buffered saline, and analyzed by flow cytometry for GFP positivity on FL1. An average of 60% of ES cells, 26% of EB/HP cells, and 6% of myeloid cells were GFP+ as compared with <1% of untransfected cells. For stable overexpression of Rad51, 20 × 106 cells were electroporated with 80 μg of human Rad51 expression vector pCBARad51 (Stark et al. 2002) linearized by SalI restriction digest along with 8 μg of the puromycin phosphotransferase expression vector pGKpuro linearized by ClaI digest at 250 mV and 950 μF (Elliott et al. 1998). Twenty-four hours post-electroporation, cells were selected in 1.5 μg/mL of puromycin for 14 d to yield puroR clones. Five puroR clones were examined for increased expression of Rad51 protein by Western blotting, and a pool of clones (+Rad51 pool) and a single clone (+Rad51 clone) were chosen for further study.
DNA analysis
Genomic DNA was extracted from individual colonies using the Qiagen DNeasy Tissue Kit and amplified using Qiagen Repli-G as per the manufacturer’s protocol. Following amplification, samples were purified with Qiagen DNeasy mini spin columns. DNA concentrations were determined by A260 spectrophotometry.
PCR and sequencing
DNA template (1.0 μg) was used. For nested PCR, 1.0 μg of DNA template was used for the first round of amplification, and 5 μL of the first-round product were used as a template for the second round of amplification. Each 25-μL PCR reaction contained template DNA, 10× reaction buffer, 1.5 mM MgCl2, 200 μM each dNTP, 0.48 μM each primer, and 2.5 U Taq DNA polymerase. Amplification was performed for 5 min at 94°C, followed by 40 cycles of 30 sec at 94°C, 30 sec at 60°C, 2 min at 72°C; and 15 min at 72°C. Three microliters of nested PCR product from amplification of F and N2 alleles were digested with 10 U of NcoI, EagI, or I-Sce I overnight at 37°C. Sequencing: Sequencing of the F and N2 alleles as well as the breakpoint junction of the hybrid N2/F allele was performed using nested PCR products. During second-round amplification, a modified reverse primer that contained the M13 reverse sequence was used. F and N2/F alleles: p1-M13rev, 5′-CAGGAAACAGCTAT GACTTCCAGGCCGGCACCTTG-3′; N2 allele: nR-M13rev, 5′-CAGGAAACAGCTATGACCTCGCTCGATGCGATGTTT C-3′. Sequencing was performed using the M13 reverse sequencing primer.
Protein analysis
Cells (100 × 106) were resuspended in 100 μL of PBS, mixed with 200 μL of M-PER protein extraction buffer (Pierce Biotechnology) containing protease inhibitor cocktail (Roche), and incubated for 30 min on ice. Following purification using centrifugal filtration, samples were mixed with NuPAGE LDS sample buffer and NuPAGE reducing agent (Invitrogen) and separated on 10% SDS-PAGE gels. Samples were transferred to a Polyscreen PVDF membrane (Bio-Rad) and probed with anti-Rad51 IgG rabbit polyclonal antibody (Ab-1; Calbiochem) or rabbit-anti-β-tubulin (H-235; Santa Cruz Biotechnology), then HRP-goat-anti-rabbit secondary antibody (Amersham Biosciences). Immunoblot signals were detected with an ECL kit (Amersham Biosciences).
Acknowledgments
We thank Jonathan McCafferty and Na Lin for technical assistance, and Vladin Miljkovic for sequencing. This work was supported by NCI (5R01CA100159) and the Stewart Trust Pilot Project for Cancer Research. C.R. is an ACS Research Scholar for Beginning Investigators.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1522807
References
- Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F., Benito-Hernandez A., Morrison S.J., Clarke M.F., Morrison S.J., Clarke M.F., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplan P.D., Chervinsky D.S., Stanulla M., Burhans W.C., Chervinsky D.S., Stanulla M., Burhans W.C., Stanulla M., Burhans W.C., Burhans W.C. Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood. 1996;87:2649–2658. [PubMed] [Google Scholar]
- Baumann P., Benson F.E., West S.C., Benson F.E., West S.C., West S.C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- Benson F.E., Stasiak A., West S.C., Stasiak A., West S.C., West S.C. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D., Dick J.E., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Colleaux L., d’Auriol L., Gailbert F., Dujon B., d’Auriol L., Gailbert F., Dujon B., Gailbert F., Dujon B., Dujon B. Recognition and cleavage site of the intron-encoded omega transposase. Proc. Natl. Acad. Sci. 1988;85:6022–6026. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Campbell J.L., Nalbant D., Youn H., Bass A.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Campbell J.L., Nalbant D., Youn H., Bass A.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Nalbant D., Youn H., Bass A.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Youn H., Bass A.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Bass A.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Cobos E., Tsai S., Keller J.R., Williams S.C., Tsai S., Keller J.R., Williams S.C., Keller J.R., Williams S.C., Williams S.C. Mapping gene expression patterns during myeloid differentiation using the EML hematopoietic progenitor cell line. Exp. Hematol. 2002;30:649–658. doi: 10.1016/s0301-472x(02)00817-2. [DOI] [PubMed] [Google Scholar]
- Elliott B., Jasin M., Jasin M. Double-strand breaks and translocations in cancer. Cell. Mol. Life Sci. 2002;59:373–385. doi: 10.1007/s00018-002-8429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B., Richardson C., Winderbaum J., Nickoloff J.A., Jasin M., Richardson C., Winderbaum J., Nickoloff J.A., Jasin M., Winderbaum J., Nickoloff J.A., Jasin M., Nickoloff J.A., Jasin M., Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell. Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B., Richardson C., Jasin M., Richardson C., Jasin M., Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol. Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Felix C.A., Kolaris C.P., Osheroff N., Kolaris C.P., Osheroff N., Osheroff N. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst.) 2006;5:1093–1108. doi: 10.1016/j.dnarep.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Ferguson D.O., Holloman W.K., Holloman W.K. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc. Natl. Acad. Sci. 1996;93:5419–5424. doi: 10.1073/pnas.93.11.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto B., Capranico G., Palumbo M., Capranico G., Palumbo M., Palumbo M. Drugs acting on DNA topoisomerases: Recent advances and future perspectives. Curr. Pharm. Des. 1999;5:195–215. [PubMed] [Google Scholar]
- Helgason C.D. In vitro hematopoietic differentiation of embryonic stem (ES) cells. StemCell Technologies Inc.; Vancouver, British Columbia: 2000. pp. 1–25. [Google Scholar]
- Jordan C.T. Searching for leukemia stem cells—Not yet the end of the road? Cancer Cell. 2006;10:253–254. doi: 10.1016/j.ccr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Jordan C.T., Guzman M.L., Noble M., Guzman M.L., Noble M., Noble M. Cancer stem cells. N. Engl. J. Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- Keller G., Kennedy M., Papayannopoulou T., Wiles M.V., Kennedy M., Papayannopoulou T., Wiles M.V., Papayannopoulou T., Wiles M.V., Wiles M.V. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol. Cell. Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E., Meyn M.S., Pandita A., Squire J.A., Meyn M.S., Pandita A., Squire J.A., Pandita A., Squire J.A., Squire J.A. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- Krejci L., Van Komen S., Li Y., Villemain J., Reddy M.S., Klein H., Ellenberger T., Sung P., Van Komen S., Li Y., Villemain J., Reddy M.S., Klein H., Ellenberger T., Sung P., Li Y., Villemain J., Reddy M.S., Klein H., Ellenberger T., Sung P., Villemain J., Reddy M.S., Klein H., Ellenberger T., Sung P., Reddy M.S., Klein H., Ellenberger T., Sung P., Klein H., Ellenberger T., Sung P., Ellenberger T., Sung P., Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- Langer T., Metzler M., Reinhardt D., Viehmann S., Borkhardt A., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Metzler M., Reinhardt D., Viehmann S., Borkhardt A., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Reinhardt D., Viehmann S., Borkhardt A., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Viehmann S., Borkhardt A., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Borkhardt A., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Reichel M., Stanulla M., Schrappe M., Creutzig U., Ritter J., Stanulla M., Schrappe M., Creutzig U., Ritter J., Schrappe M., Creutzig U., Ritter J., Creutzig U., Ritter J., Ritter J., et al. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: almost identical MLL breakpoints in therapy-related AML after treatment without etoposides. Genes Chromosomes Cancer. 2003;36:393–401. doi: 10.1002/gcc.10167. [DOI] [PubMed] [Google Scholar]
- Liang F., Han M., Romanienko P.J., Jasin M., Han M., Romanienko P.J., Jasin M., Romanienko P.J., Jasin M., Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Mortensen U.H., Rothstein R., Mortensen U.H., Rothstein R., Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- Lovett B.D., Lo Nigro L., Rappaport E.F., Blair I.A., Osheroff N., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Lo Nigro L., Rappaport E.F., Blair I.A., Osheroff N., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Rappaport E.F., Blair I.A., Osheroff N., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Blair I.A., Osheroff N., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Osheroff N., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Zheng N., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Megonigal M.D., Williams W.R., Nowell P.C., Felix C.A., Williams W.R., Nowell P.C., Felix C.A., Nowell P.C., Felix C.A., Felix C.A. Near-precise interchromosomal recombination and functional DNA topoisomerase II cleavage sites at MLL and AF-4 genomic breakpoints in treatment-related acute lymphoblastic leukemia with t(4;11) translocation. Proc. Natl. Acad. Sci. 2001;98:9802–9807. doi: 10.1073/pnas.171309898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maacke H., Jost K., Opitz S., Miska S., Yuan Y., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Jost K., Opitz S., Miska S., Yuan Y., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Opitz S., Miska S., Yuan Y., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Miska S., Yuan Y., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Yuan Y., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Hasselbach L., Luttges J., Kalthoff H., Sturzbecher H.W., Luttges J., Kalthoff H., Sturzbecher H.W., Kalthoff H., Sturzbecher H.W., Sturzbecher H.W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000a;19:2791–2795. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- Maacke H., Opitz S., Jost K., Hamdorf W., Henning W., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Opitz S., Jost K., Hamdorf W., Henning W., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Jost K., Hamdorf W., Henning W., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Hamdorf W., Henning W., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Henning W., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Kruger S., Feller A.C., Lopens A., Diedrich K., Schwinger E., Feller A.C., Lopens A., Diedrich K., Schwinger E., Lopens A., Diedrich K., Schwinger E., Diedrich K., Schwinger E., Schwinger E., et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer. 2000b;88:907–913. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- McVey M., Adams M., Staeva-Vieira E., Sekelsky J.J., Adams M., Staeva-Vieira E., Sekelsky J.J., Staeva-Vieira E., Sekelsky J.J., Sekelsky J.J. Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics. 2004;167:699–705. doi: 10.1534/genetics.103.025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Jasin M., Jasin M. Loss of heterozygosity induced by a chromosomal double-strand break. Proc. Natl. Acad. Sci. 1997;94:8988–8993. doi: 10.1073/pnas.94.17.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff N., Corbett A., Robinson M., Corbett A., Robinson M., Robinson M. Mechanism of action of topoisomerase II-targeted antineoplastic drugs. Adv. Pharmacol. 1994;29B:105–126. doi: 10.1016/s1054-3589(08)61134-5. [DOI] [PubMed] [Google Scholar]
- Paffett K.S., Clikeman J.A., Palmer S., Nickoloff J.A., Clikeman J.A., Palmer S., Nickoloff J.A., Palmer S., Nickoloff J.A., Nickoloff J.A. Overexpression of Rad51 inhibits double-strand break-induced homologous recombination but does not affect gene conversion tract lengths. DNA Repair (Amst.) 2005;4:687–698. doi: 10.1016/j.dnarep.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Palmer S., Schildkraut E., Lazarin R., Nguyen J., Nickoloff J.A., Schildkraut E., Lazarin R., Nguyen J., Nickoloff J.A., Lazarin R., Nguyen J., Nickoloff J.A., Nguyen J., Nickoloff J.A., Nickoloff J.A. Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res. 2003;31:1164–1173. doi: 10.1093/nar/gkg219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F., Leung W., Haber J., Leung W., Haber J., Haber J. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell. Biol. 1998;18:2045–2054. doi: 10.1128/mcb.18.4.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E., Golub E.I., Haaf T., Golub E.I., Haaf T., Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E., Stout K., Freier S., Suckow V., Schweiger S., Haaf T., Stout K., Freier S., Suckow V., Schweiger S., Haaf T., Freier S., Suckow V., Schweiger S., Haaf T., Suckow V., Schweiger S., Haaf T., Schweiger S., Haaf T., Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- Rassool F.V. DNA double strand breaks (DSB) and non-homologous end joining (NHEJ) pathways in human leukemia. Cancer Lett. 2003;193:1–9. doi: 10.1016/s0304-3835(02)00692-4. [DOI] [PubMed] [Google Scholar]
- Reichel M., Gilert E., Nilson I., Siegler G., Greil J., Fey G.H., Marschalek R., Gilert E., Nilson I., Siegler G., Greil J., Fey G.H., Marschalek R., Nilson I., Siegler G., Greil J., Fey G.H., Marschalek R., Siegler G., Greil J., Fey G.H., Marschalek R., Greil J., Fey G.H., Marschalek R., Fey G.H., Marschalek R., Marschalek R. Fine structure of translocations breakpoints in leukemic blasts with chromosomal translocation t(4;11): The DNA damage-repair model of translocation. Oncogene. 1998;17:3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- Richardson C., Jasin M., Jasin M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol. Cell. Biol. 2000a;20:9068–9075. doi: 10.1128/mcb.20.23.9068-9075.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C., Jasin M., Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000b;405:697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- Richardson C., Moynahan M.E., Jasin M., Moynahan M.E., Jasin M., Jasin M. Double-strand break repair by interchromosomal recombination: Suppression of chromosomal translocations. Genes & Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C., Stark J.M., Ommundsen M., Jasin M., Stark J.M., Ommundsen M., Jasin M., Ommundsen M., Jasin M., Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–553. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- Richardson G.D., Robson C.N., Lang S.H., Neal D.E., Maitland N.J., Collins A.T., Robson C.N., Lang S.H., Neal D.E., Maitland N.J., Collins A.T., Lang S.H., Neal D.E., Maitland N.J., Collins A.T., Neal D.E., Maitland N.J., Collins A.T., Maitland N.J., Collins A.T., Collins A.T. CD133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 2004;117:3539–3545. doi: 10.1242/jcs.01222. [DOI] [PubMed] [Google Scholar]
- Rothkamm K., Kruger I., Thompson L.H., Lobrich M., Kruger I., Thompson L.H., Lobrich M., Thompson L.H., Lobrich M., Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P., Smih F., Jasin M., Smih F., Jasin M., Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J.D. Cytogenetic analysis in leukemia and lymphoma: An introduction. Semin. Hematol. 2000;37:315–319. doi: 10.1016/s0037-1963(00)90012-x. [DOI] [PubMed] [Google Scholar]
- Scully R., Chen J., Ochs R.L., Keegan K., Hoekstra M., Feunteun J., Livingston D.M., Chen J., Ochs R.L., Keegan K., Hoekstra M., Feunteun J., Livingston D.M., Ochs R.L., Keegan K., Hoekstra M., Feunteun J., Livingston D.M., Keegan K., Hoekstra M., Feunteun J., Livingston D.M., Hoekstra M., Feunteun J., Livingston D.M., Feunteun J., Livingston D.M., Livingston D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B., Henkelman R.M., Cusimano M.D., Dirks P.B., Cusimano M.D., Dirks P.B., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Skorski T. Oncogenic tyrosine kinases and the DNA-damage response. Nat. Rev. Cancer. 2002;2:351–360. doi: 10.1038/nrc799. [DOI] [PubMed] [Google Scholar]
- Slupianek A., Schmutte C., Tombline G., Nieborowska-Skorska M., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Schmutte C., Tombline G., Nieborowska-Skorska M., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Tombline G., Nieborowska-Skorska M., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Nieborowska-Skorska M., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Hoser G., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Nowicki M.O., Pierce A.J., Fishel R., Skorski T., Pierce A.J., Fishel R., Skorski T., Fishel R., Skorski T., Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- Slupianek A., Hoser G., Majsterek I., Bronisz A., Malecki M., Blasiak J., Fishel R., Skorski T., Hoser G., Majsterek I., Bronisz A., Malecki M., Blasiak J., Fishel R., Skorski T., Majsterek I., Bronisz A., Malecki M., Blasiak J., Fishel R., Skorski T., Bronisz A., Malecki M., Blasiak J., Fishel R., Skorski T., Malecki M., Blasiak J., Fishel R., Skorski T., Blasiak J., Fishel R., Skorski T., Fishel R., Skorski T., Skorski T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol. Cell. Biol. 2002;22:4189–4201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger J.A., Kiianitsa K., Heyer W.D., Kiianitsa K., Heyer W.D., Heyer W.D. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- Stark J.M., Hu P., Pierce A.J., Moynahan M.E., Ellis N., Jasin M., Hu P., Pierce A.J., Moynahan M.E., Ellis N., Jasin M., Pierce A.J., Moynahan M.E., Ellis N., Jasin M., Moynahan M.E., Ellis N., Jasin M., Ellis N., Jasin M., Jasin M. ATP-hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J. Biol. Chem. 2002;28:28. doi: 10.1074/jbc.M112132200. [DOI] [PubMed] [Google Scholar]
- Strout M.P., Marcucci G., Bloomfield C.D., Caligiuri M.A., Marcucci G., Bloomfield C.D., Caligiuri M.A., Bloomfield C.D., Caligiuri M.A., Caligiuri M.A. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. 1998;95:2390–2395. doi: 10.1073/pnas.95.5.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Super H.J., McCabe N.R., Thirman M.J., Larson R.A., Le Beau M.M., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., McCabe N.R., Thirman M.J., Larson R.A., Le Beau M.M., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., Thirman M.J., Larson R.A., Le Beau M.M., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., Larson R.A., Le Beau M.M., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., Le Beau M.M., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., Pedersen-Bjergaard J., Philip P., Diaz M.O., Rowley J.D., Philip P., Diaz M.O., Rowley J.D., Diaz M.O., Rowley J.D., Rowley J.D. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood. 1993;82:3705–3711. [PubMed] [Google Scholar]
- Sutherland H.J., Blair A., Zapf R.W., Blair A., Zapf R.W., Zapf R.W. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood. 1996;87:4754–4761. [PubMed] [Google Scholar]
- Symington L.S., Kang L.E., Moreau S., Kang L.E., Moreau S., Moreau S. Alteration of gene conversion tract length and associated crossing over during plasmid gap repair in nuclease-deficient strains of Saccharomyces cerevisiae. Nucleic Acids Res. 2000;28:4649–4656. doi: 10.1093/nar/28.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak J.W., Orr-Weaver T.L., Rothstein R.J., Stahl F.W., Orr-Weaver T.L., Rothstein R.J., Stahl F.W., Rothstein R.J., Stahl F.W., Stahl F.W. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Tycko B., Sklar J., Sklar J. Chromosomal translocations in lymphoid neoplasia: A reappraisal of the recombinase model. Cancer Cells. 1990;2:1–8. [PubMed] [Google Scholar]
- van Gent D.C., Hoeijmakers J.H., Kanaar R., Hoeijmakers J.H., Kanaar R., Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- Veaute X., Jeusset J., Soustelle C., Kowalczykowski S.C., Le Cam E., Fabre F., Jeusset J., Soustelle C., Kowalczykowski S.C., Le Cam E., Fabre F., Soustelle C., Kowalczykowski S.C., Le Cam E., Fabre F., Kowalczykowski S.C., Le Cam E., Fabre F., Le Cam E., Fabre F., Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Ward J. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- Weinstock D.M., Richardson C.A., Elliott B., Jasin M., Richardson C.A., Elliott B., Jasin M., Elliott B., Jasin M., Jasin M. Modeling oncogenic translocations: Distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair (Amst.) 2006;5:1065–1074. doi: 10.1016/j.dnarep.2006.05.028. [DOI] [PubMed] [Google Scholar]