

Abstract
Proteins evolved through the shuffling of functional domains, and therefore, the same domain can be found in different proteins and species. Interactions between such conserved domains often involve specific, well-determined binding surfaces reflecting their important biological role in a cell. To find biologically relevant interactions we developed a method of systematically comparing and classifying protein domain interactions from the structural data. As a result, a set of conserved binding modes (CBMs) was created using the atomic detail of structure alignment data and the protein domain classification of the Conserved Domain Database. A conserved binding mode is inferred when different members of interacting domain families dock in the same way, such that their structural complexes superimpose well. Such domain interactions with recurring structural themes have greater significance to be biologically relevant, unlike spurious crystal packing interactions. Consequently, this study gives lower and upper bounds on the number of different types of interacting domain pairs in the structure database on the order of 1000–2000. We use CBMs to create domain interaction networks, which highlight functionally significant connections by avoiding many infrequent links between highly connected nodes. The CBMs also constitute a library of docking templates that may be used in molecular modeling to infer the characteristics of an unknown binding surface, just as conserved domains may be used to infer the structure of an unknown protein. The method’s ability to sort through and classify large numbers of putative interacting domain pairs is demonstrated on the oligomeric interactions of globins.
Keywords: protein–protein interactions, conserved binding modes, homology modeling, protein structure, protein domain families
With the advance of numerous experimental techniques in recent years, many protein–protein interactions have been detected enabling construction of protein interaction maps. The understanding of these maps holds promise for a greater understanding of the cell; however, one issue that has arisen so far is the reproducibility of the interactions. Several large analyses of yeast two-hybrid data reveal overlaps of <15% in the types of interactions found (Uetz et al. 2000; Hazbun and Fields 2001; Ito et al. 2001). While some of these discrepancies may be attributed to novel interactions detected in each study, the challenges associated with such high-throughput studies result in significant numbers of false positives and negatives. Verification of interactions is essential to avoid further propagation of errors based on faulty data.
Complementing the large number of putative interactions found in yeast two-hybrid data, protein structure data provide the most detailed interaction information at the molecular level. Structures help sort through thousands of detected protein interactions and decide which are meaningful by identifying detail beyond which two proteins interact. As the rate of structures solved continues to increase, the number of interactions available from the structure data becomes significant compared to verifiable sets of two-hybrid data. Because of this detail and reliability, we use structure data as the basis for our analysis of protein interactions. Unfortunately, annotation is missing from the structure data to specify which regions form biological interactions. The challenge is to decide which interaction surfaces are useful for our understanding of protein interactions and which can be neglected as uninformative or inconclusive (Bahadur et al. 2004).
To distinguish between biological and nonbiological interactions coming from crystal packing, several approaches have been developed. Some of them are based on the observation that biological contacts are larger and more conserved than nonbiological ones (Janin and Rodier 1995; Carugo and Argos 1997; Dasgupta et al. 1997; Janin 1997). Others use knowledge-based pair potentials to estimate the propensities of two proteins to form a contact (Robert and Janin 1998; Ponstingl et al. 2000; Elcock and McCammon 2001). More detailed studies of protein–protein interactions explored how the protein/domain orientations vary among homologs (Bashton and Chothia 2002). It has been shown, for example, that the same interacting domain pair conserves its interaction pattern for close homologs (30% to 40% identity or higher), although for distant homologs the interaction pattern can vary (Aloy et al. 2003; Keskin et al. 2004). Indeed, it has been reported earlier that the prediction of transient protein–protein binding sites has a limited success (Nooren and Thornton 2003; Panchenko et al. 2004). Moreover, domains with several interaction partners tend to use more than one surface area for interaction (Keskin et al. 2004; Littler and Hubbard 2005), which complicates the homology modeling of domain interactions. In these cases, the detailed classification and analysis of different interaction patterns or modes within the broad variety of interaction partners would be of particular use.
Proteins have evolved through the shuffling of functional domains, and as a result, the same domain can be found in different proteins and species. In the course of evolution these conserved domains have developed specific interaction surfaces that can be isolated through the mapping of the conserved domains or their multiple alignments to all existing protein structures. In our study, domains are mapped on the structures using the alignments from the Conserved Domain Database (CDD). This collection is well-suited for this purpose as it contains an expanding set of accurate domain alignments which are curated to conserve the structural and sequence features of a given family together with SMART, PFAM, and COG domain alignments for families not yet curated.
After mapping these domains onto protein structures, we check residue distances between domain regions to confirm they come into contact sufficiently to be considered interacting domain pairs. Counting all interacting domain pairs does not reveal much about how domains interact and how likely it is that they are found together. To address these issues we track a more detailed level of geometric information from the structure data that describes the interface between domain pairs. Using structural alignments of different members of interacting domain families, modes of binding are inferred to group unique interface geometries. When two or more nonredundant members show similar spatial interface locations, they constitute a conserved binding mode (CBM). These similarly interacting members, therefore, make it much less likely the domains occur near each other by chance and more likely their proximity is meaningful. While most families interact in a unique conserved manner, some are found to interact in different conserved modes. Such conserved binding modes with recurring structural themes allow us to differentiate biologically relevant from crystal packing interactions, to analyze interaction network topology, and to consider applications of homology modeling to protein–protein interactions.
Results and Discussion
The distribution of conserved binding modes in interacting domain pairs
Table 1 gives an overview of the total interacting domain pairs found from the structure data. The first row shows the number of interacting domain pairs with at least one CBM found, the second row lists the total number of different kinds of interacting domain pairs, the third row shows the number of CBMs among all interacting domains, and the fourth row lists the overall number of possible modes (including nonconserved) among all interacting domains. The above-mentioned counts are categorized further into intra- and inter-chain interacting domain pairs (see columns 2 and 3). It should be noted that these counts include homodimer as well as heterodimer domain pairs. As can be seen from this table, 6250 binding modes are found among 1798 interacting domain pairs, although only 23% of binding modes are regarded as conserved (1416 CBMs). Using CBMs to qualify interacting pairs, only 46% unique conserved pairs remain (833). From these estimates we can get an upper (1798) and lower (833) bound on the number of domain–domain interactions using structural data currently available. This is consistent with the results by Aloy and Russell (2004), who used protein interaction data from different sources and estimated the number of different interaction types as being 735 in Helicobacter pylori, 2000 in yeast, and ~3000 in the worm or fly.
Table 1.
Numbers of interacting pairs and binding modes
| All | Interchain | Intrachain | |
| No. of interaction types with >1 CBM | 833 | 652 | 241 |
| No. of interaction types | 1798 | 1563 | 418 |
| No. of CBMs | 1416 | 1117 | 309 |
| No. of binding modes | 6250 | 5579 | 693 |
The definition of CBM is critical in estimating the overall number of interactions. In this study we require a conserved mode to contain multiple instances from the nonredundant structures. This definition describes biological interactions in structures, but it could be modified, for example, by requiring observations from diverse phyla, thereby searching for “old” interactions. Both of these alternatives have been looked into and could be useful as a triage system in handling interactions with increasing amounts of evidence. In the latter case, the number of domain pairs with at least one CBM drops from 833 to 509, showing that many structural studies are done in a limited number of species.
Figure 1 shows the distribution of the number of binding modes (open boxes) and conserved binding modes (gray boxes) over the interacting domain pairs. It is obvious from the figure that the majority of interacting domain pairs have just one CBM per pair, implying that the same surface and interaction pattern between a pair of similar domains is being reused in similar proteins in the course of evolution. At the same time, some interacting domain pairs, interestingly enough, exhibit a large number of different CBMs (up to 24 CBMs per interacting domain pair), showing that the same domains can interact in different manners using different surface regions and different orientations.
Figure 1.

Binding mode distribution among interacting pairs. Two histograms show the distribution of conserved binding modes (gray) and all binding modes (white) per interacting domain pair. Note that the largest number of CBMs found per interacting pair is 24.
Annotated set of conserved domain interactions can be used to study an evolution of protein–protein interactions. It has been suggested earlier that interface between two different interacting proteins/domains is developed in evolution while they are covalently bonded to each other and interact within one protein (Marcotte et al. 1999). Thus, the hypothesis states that domains from different proteins would most likely interact between each other if there are other examples of their occurrences within one protein chain. To check this, we searched for the same type of interacting domains found both on the same and different protein chains. Interestingly, we found 183 different types of interacting domain pairs (out of 1798) (Table 1) containing both inter- and intra-chain examples, and only 22 of these interactions are found within the same CBM. Furthermore, upon examination a few of these cases turned out to be the result of a designed linker mutation in a complex naturally existing as separate chains. Among the examples there is the case of two transketolase binding domains for which inter- and intra-chain examples are found in the same binding mode across disparate organisms, suggesting an ancient origin of this type of interaction. In contrast to many specialized globin binding modes, which we are going to discuss later in the paper, enzymes of various functions can maintain the same interface regardless of the chain arrangements.
Automatic discrimination of biological from nonbiological interactions
Here we illustrate how CBMs can be used to discriminate between biologically relevant interactions and interactions arising as a result of crystal packing. For the first example we focus on one structure (1HA3) of the elongation factor Tu (EF-Tu), which is involved in protein biosynthesis (Vogeley et al. 2001). This structure includes two chains with three domains on each chain. By looking at domain pairs with appreciable interacting surfaces, seven putative pairs are found: four on the same chain and three between the two chains. CBM analysis shows, however, that only two unique intra-chain pairs are found. Grouped in these two CBMs are interactions from another structure (1TTT) from the same species in which tRNA occupies the place of one of the EF-Tu monomers when aligned to 1HA3. EF-Tu structures exist with one, two, and three monomers per unit cell, but orientations between them are not conserved. In contrast, despite the flexibility of the three intra-chain domains, their interactions group neatly into a few CBMs. This suggests that the inter-chain domain interactions probably arise from nonbiological crystal packing arrangements. Obtained results can be compared with two other domain interaction resources. Pibase, for example, computes interaction surfaces within a PDB file and reports five nonredundant inter-chain interactions for EF-Tu (Davis and Sali 2005); similarly, Keskin et al. (2004) include 1HA3 in their list of inter-chain interactions. Thus, for the elongation factor, CBM analysis is crucial for distinguishing between biological domain interfaces and probable inter-chain packing interactions.
The second example comprises globin domain (cd01040), one of the most studied protein families. Besides the well-known monomeric myoglobin and heterotetrameric hemoglobin structures, the globin domain exists in dimers, homotetramers, hexamers, and dodecamers across all three major superkingdoms of life. One contribution to this adaptability is the variety of quaternary states which globin can assume, creating various levels of allosteric influence on the cooperative binding involved at the heme. We look at this diverse structural data to see what can be learned from the analysis of conserved binding modes. There are 630 interacting domain pairs between globins from 196 structures according to our definition. Obviously, an all-against-all comparison of 196 structures by hand is an overwhelming job, and instead, we group these interacting pairs by conserved binding modes as seen in Table 2. The most populated modes, CBM 1 and CBM 2, group the intra- and interheterodimer interfaces, respectively, of the classic hemoglobin tetramer across all jawed vertebrate species without exception.
Table 2.
Globin–globin conserved binding modes
| CBM | No. of structuresa | No. of total structures | No. of species | Taxonomy description |
| 1 | 60 | 154 | 18 | jawed_vertebrates |
| 2 | 42 | 112 | 13 | jawed_vertebrates |
| 3 | 5 | 17 | 2 | clam, earthworm |
| 4 | 3 | 4 | 2 | river and sea lamprey |
| 5 | 3 | 4 | 1 | Vitreoscilla_stercoraria |
| 6 | 2 | 2 | 2 | rice and soybeans |
| 7 | 2 | 2 | 1 | human |
| 8 | 2 | 2 | 2 | river and sea lamprey |
a The number of nonredundant structures with respect to unique cell constants.
As can be seen from Table 2, there are six more conserved modes among globins in addition to CBMs 1 and 2. This means that, unlike most binding partners, which tend to share a common surface (Fig. 1), our mode analysis makes it clear that the globin oligomeric orientations are highly variable. For example, CBM 3 includes protein structures from the earthworm and the clam, both belonging to the protostomia group, with a dodecamer found in the earthworm and both dimeric and tetrameric structures found in the clam. In the case of the lamprey hemoglobin, we find a very complicated network of oligomeric interactions with one structure showing 12 subunits forming 19 interacting globin pairs. From these interactions, CBM 4 isolates all the dimer interfaces, but CBM 8 corresponds to interdimer interacting pairs within one tightly packed hexamer of each structure. CBM 8 is reproduced in both species of lamprey, qualifying it as a conserved interaction. This agrees with an earlier suggestion that the hexamers stabilized by CBM 8 interaction types might occur in vivo (Heaslet and Royer 2001), and a mechanism was offered for oligomeric interactions for unligated globins (Wyman 1948; Royer et al. 2001). Our conserved binding modes also suggest that the lampreys may have developed a conserved hexameric interaction to increase ligand binding cooperativity. We have compared our binding modes to a hand-curated analysis of oligomeric hemoglobins by Royer et al. (2001), who found eight unique oligomers. Most of these structures are accounted for by conserved binding modes, which highlight their ability to find generalized modeling templates corresponding to domain interaction adaptations. Note that the CBM analysis was also recently applied to histones, another well-studied oligomeric family, to help organize the binding surfaces and better understand the key interacting residues (Marino-Ramirez et al. 2005).
To analyze how well CBMs can discriminate between biological and nonbiological interactions we have manually divided all globin interacting pairs into biological and nonbiological categories. The details of this study are given in the Supplemental Material section. Although the size of the interaction interface (number of interface contacts) could also be used to select interactions, we show that this measure is not always accurate. As can be seen from the top panel of Figure 2, the properly chosen threshold on the size of the interaction interface does eliminate many nonbiological interacting pairs, but it does not allow for their total removal (two histograms overlap). For example, one nonbiological interaction surface contains 44 contacts, making it larger than many biological interfaces. On the other hand, the bottom panel of Figure 2 shows that CBM analysis correctly identifies a majority of biological interactions (90%) with no false positives, meaning that all pairs of interacting globin structures that exhibit CBMs are biologically significant without exception. CBM analysis can be similarly used to identify nonbiological interfaces as those lacking conserved binding modes. For example, CBM reported 113 nonbiological interacting pairs out of all globin–globin interfaces, with 45 of them being correctly predicted. From the binomial distribution we can calculate the probability of finding that many (45) or more “true” nonbiological interacting pairs by chance, which is very low and equal to 6.77 × 10e-15. Thus, based on the extensive manual analysis of globin oligomers, CBMs are found to reliably predict biological interactions.
Figure 2.

Analysis of globin CBMs by manual curation. The top panel shows two histograms of the interface sizes for all globin–globin interacting pairs. The biological interfaces (light gray) and nonbiological interfaces (dark gray) have been identified by manual curation (see Materials and Methods). Note that the largest number of interface contacts for a nonbiological interface is 44. The bottom panel shows a histogram of the interface sizes for globin–globin pairs with at least one CBM. CBMs have been found only for biological interfaces, and nonbiological interfaces with CBMs are not reported.
Domain interaction network
A map of all conserved domain interactions can be made by clustering the interactions as shown in Figure 3. The edge between two domains is drawn if they have at least one CBM, and only clusters with three or more domains are shown (44 clusters in total). Each cluster is distinguished by color with self-interactions shown by closed loops. Such clustering gives us an idea about the association of domains based on the binding partners they share. As can be seen from this figure, many separable clusters form, which give some general functional groupings. In contrast, when all binding modes are considered (see figure in supplemental data) a single dominant cluster forms from more than half of the interactions, which has Ig as its most connected node but includes many diverse functional networks. Clearly, while being biological, these interactions do not elucidate the overall picture fully given the ability of the IG family to interact with many components of the cell. When we consider only CBMs (Fig. 3), this large cluster is split into several smaller ones. One of these clusters includes the serine protease, kazal, EGF, and clectin domains, which are all related to protease inhibition and activation. Another network includes domains all related to RNA polymerase and transcription. A third, separable network centers around the TPP enzyme and ferredoxin binding domain involved in energy production. We also compared our interaction map with the interaction map obtained by Park and colleagues (Park et al. 2001). The latter analyzed the interacting domain networks of SCOP domains that occur near each other in space in PDB structures, and near each other in sequence from yeast two-hybrid data. Although the number of structures used in their study was almost twice as small as in our test set, the overall network topology is surprisingly similar. The CBMs help prevent the expanding number of interactions from obscuring meaningful associations with a few densely connected nodes and inseparable clusters.
Figure 3.

Clusters of conserved domain interactions. The network of domain interactions is shown, which only includes interactions containing conserved binding modes and gives a minimalist picture of verified interactions. Oligomeric interactions are shown with closed loops. Clusters of domains connected by single linkage clustering are distinguished from other clusters by color. Only clusters of three different domains or more are shown. The largest cluster is encircled with a black line.
Homology modeling of protein–protein interactions
One of the advantages to having interaction data at the molecular level organized by conserved domains and conserved geometries is that domain interactions could be predicted or modeled for domain pairs not observed in the structural database. Based on the previous observation that the interaction interface is conserved among close homologs (Aloy et al. 2003) we make an assumption that proteins clustered together on a phylogenetic tree should exhibit one or a few characteristic CBMs that should vary between the clusters. If this assumption holds true, then an unknown interaction can be inferred by the interaction pattern (i.e., CBM) of a representative structure from the same phylogenetic cluster as an unknown protein. Here we show one example of such analysis performed for the globin family.
Globin’s evolution is directly tied to the local structural changes impacting the heme binding site, but its stereochemistry can also be affected by distal regions including various configurations of oligomeric interfaces. To better understand the relevance of binding modes to homology modeling, we explore the role of quaternary globin structure. Sequences chosen by manual curation to best represent the diversity of the globin family were clustered using the neighbor-joining method (Saitou and Nei 1987; Fig. 4). It is apparent from this figure that most clusters have structure representatives, although some of them have missing interactions because of the monomeric forms of proteins.
Figure 4.
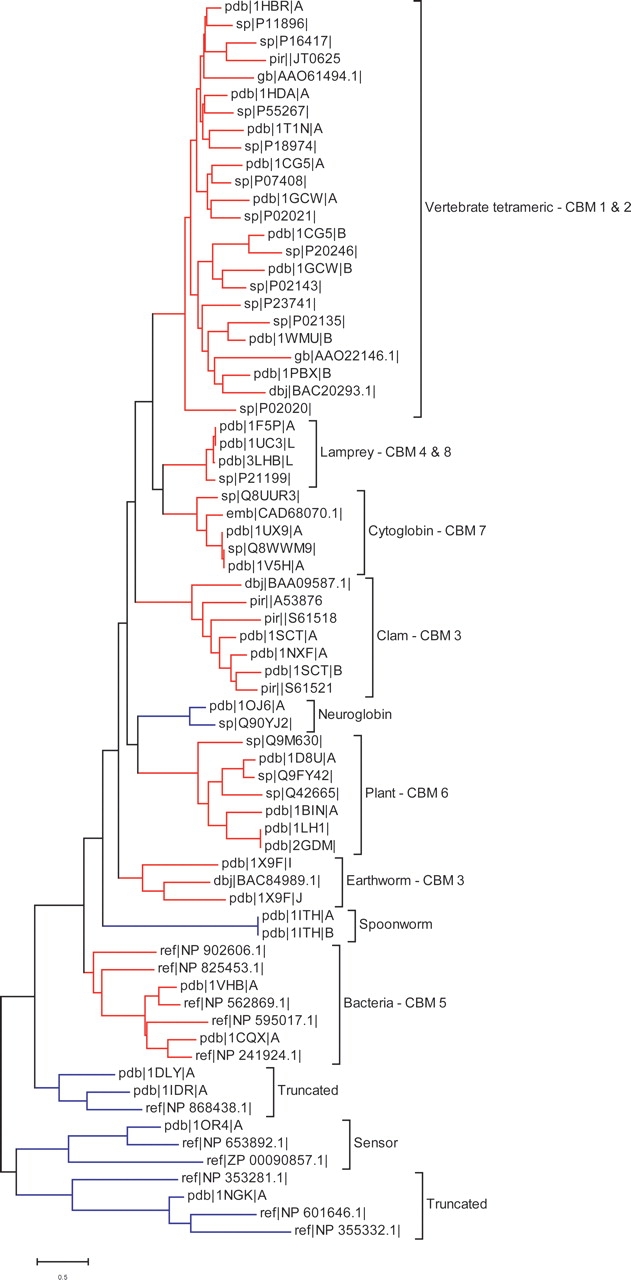
Globin sequence cluster tree. Globin sequences are clustered by the neighbor-joining method using the Jones–Taylor–Thornton distance matrix (Jones et al. 1992). The identity of each conserved mode from Table 2 is listed after each cluster.
We find that proteins with a particular CBM tend to group together on a cluster tree. For example, the vertebrate tetramers comprise a large group of structures from 36 species that are all found to interact in the same way (one CBM). While the conserved binding modes correspond to sequence clusters, they also help distinguish clusters from each other. This is true with one exception: The earthworm and the clam hemoglobin form the same CBM, although they belong to different sequence clusters. A few clusters contain multiple modes that occur together for higher order oligomers. We tried to extend our analysis to more distantly related proteins by relaxing our definition of conserved mode (as explained in the supplemental data), but in this case the correlation between CBM and sequence clusters was shown to be very weak. Thus, it seems promising that conserved binding modes, as a complement to sequence homology, can assist in modeling potential interactions of uncrystallized proteins.
Materials and methods
Benchmark construction
In order to find conserved domain–domain interactions with functional relevance in all protein structures, we first mapped protein structures to the domain models defined by the CDD. Using CDD alignments instead of SCOP let us in many cases avoid alignment problems at the domain boundaries reported earlier (Littler and Hubbard 2005). Our queries (52,439 chains from 25,192 protein structures of the Molecular Modeling Database [MMDB] [Wang et al. 2002]) were searched using RPS-BLAST (Marchler-Bauer and Bryant 2004) with the default parameters (E = 0.01) against the nonredundant CDD version 2.01 (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) (Marchler-Bauer et al. 2002, 2005). This version of protein domain alignments includes curated CDDs and preprocessed domain family alignments imported from SMART, PFAM, and COG: 5282 protein domain families altogether. The redundancy between CDD domain families was checked by using the procedure implemented in the CDART algorithm (Geer et al. 2002), and RPS-Blast search models were derived from CDD multiple sequence alignments using the pseudocount method described previously (Marchler-Bauer and Bryant 2004).
We impose the following requirements on the alignments between queries and the CDD domains: First, we exclude those CDD domains, which consist of several non-overlapping CDD domains (Geer et al. 2002); second, if two or more CDD footprints defined by RPS-BLAST alignments on the query sequence overlap, the longest footprint is used to map the CDD domain to a query sequence. The footprint here is defined as a region on a query sequence between the first and the last residues aligned by RPS-BLAST. As a result of this filtering procedure we end up with a set of 42,278 CDD footprint regions. We note that for the purpose of convenience we will refer hereafter to the CDD footprint regions on the query as “domains,” although one should keep in mind that an RPS-BLAST alignment does not necessarily include the whole CDD domain; therefore, the footprint region defined on a query might be shorter than the actual CDD domain.
Definition of conserved binding modes
Two domains qualify an interacting domain pair to be interacting if there are at least five residue–residue contact pairs made between their residues. Residue contacts are counted between residues of one interacting domain and any other residue of another interacting domain whose Ca–Ca distances are within 8 A °. All residues involved in such contacts constitute the domain interface. For each interacting domain pair there are two sets of interfacial residues coming from two corresponding domains. Altogether, 34,095 interacting domain pairs are found for all the queries, and among them both inter- (27,957) and intra-chain (6138) domain–domain interactions are present. Using a domain as our unit of interaction, some protein chains in close proximity will not qualify as interacting without identifiable domain regions, and likewise, some chains will contain multiple domains. In counting the number of putative domain interactions, one drawback lies in overestimating the number of interacting domains with very small interfaces. Such interactions typically arise from crystal packing, as biological interfaces tendto be more extensive. One could simply use a threshold on the number of residue contacts or on a minimum surface area at the binding interface, but considerable variation exists in the size of domain interfaces, making a single threshold somewhat arbitrary. Therefore, weused the more elaborate criterion, namely, the criterion of conserved binding modes in defining biologically relevant interfaces.
To define the conserved binding modes we first collect all structure queries that correspond to the same interacting domain pair. Then we apply the Vector Alignment Search Tool (VAST) (Gibrat et al. 1996) to obtain the structure—structure alignments between the queries. In the case where a VAST alignment is available for both full-length queries and each domain, the longest alignment has been used. To measure the similarity between interaction interfaces, we first map interfacial residues of two aligned domain structures with respect to each other, and calculate the fraction of equivalently aligned interfacial residues. This fraction is calculated for each of the four interfaces corresponding to each domain in the structure–structure alignment as a ratio between the number of structurally aligned interfacial residues (with a minimum of two) and the overall number of residues in a given interface. We then cluster all interacting domain pairs based on their interface similarity using single linkage clustering. Those with the fraction of equivalently aligned positions >50% for all four interfaces of two interacting domain pairs are joined with an edge and clustered together. To reduce the redundancy between queries we examine the crystallographic cell constant and symmetry group of their structures. Since these quantities depend on crystallization conditions and on the size of the molecules, they can be used to remove redundancy, while retaining structural differences such as the addition of a cofactor. Not more than one query with similar cell constants (within 2%) and the same symmetry group is included in the cluster. At the end, each cluster corresponds to a binding mode, and clusters with more than one nonredundant query are defined as conserved binding modes.
Figure 5 gives an illustration of a conserved binding mode with the example of two interacting CDD families (cd00043 and cd00180). As shown in this figure, there are three structures that map to the corresponding CDD domain pair with an inter-chain domain–domain interaction. Two of the structure queries (1GY3 and 1E9H) can be very well aligned by the VAST algorithm, and the interfacial residues show strong structural conservation between 1GY3 and 1E9H forming the conserved binding mode. This is not the case for the 1E9H and 1OL2 structure queries. Since VAST does not align the domains corresponding to cd00180, the interfacial residues are not equivalently superimposed as well, and this type of interaction cannot manifest the conserved binding mode as defined here.
Figure 5.
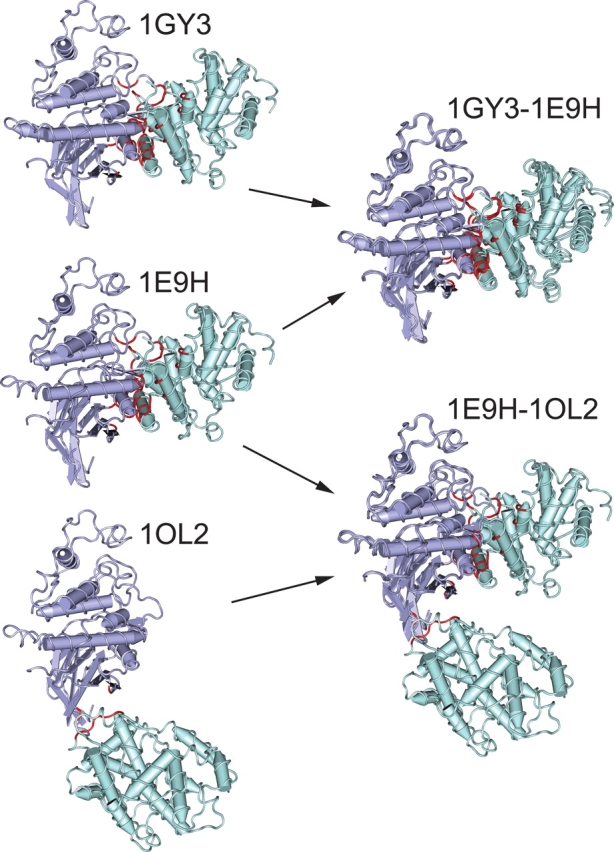
Determining conservation of interaction modes. The cyclin domain (cd00043) is shown in green interacting with a protein kinase domain (cd00180) in purple to illustrate the definition of CBMs. On the left, three different structures are shown containing this interaction, with interacting residues highlighted in red. On the right, structural superpositions are shown between these structures to determine conserved binding modes. In the alignment of 1GY3 to 1E9H, a sufficient fraction of the interfacial residues overlap, and the two structures create a conserved binding mode. The alignment of 1E9H to 1OL2, however, fails the definition of overlap to be grouped in the same binding mode. The 1OL2 interaction is, in fact, due to crystal packing and is isolated as a nonconserved mode.
Conclusion
Complementing the large number of protein interactions found through large-scale experiments, a set of conserved protein–protein interaction patterns or CBMs has been extracted from the protein structure data. These interactions have been isolated as domain interactions required to contain multiple observations of the same docking location. It has been shown that the conserved binding mode analysis helps remove spurious, nonbiological interactions and prioritize binding surfaces that have biological relevance. Moreover, the majority of interacting domain pairs exhibiting the conserved binding patterns are found to have just one CBM per pair, while a substantial number of the interacting domain pairs also show several different CBMs per pair. Observed commonality in interaction patterns between proteins allows us to estimate from the available structural data the number of different types of interactions on the order of 1000 (833 CBMs) and not >2000 (1798 interacting domain pairs).
The globin domain family has been examined in detail to learn how conserved modes classify the interactions between its subunits. It has been shown that the globin interactions can be grouped into eight representative conserved binding modes. Two of these modes contain the majority of globin interactions describing the classic tetrameric hemoglobin. The conserved modes give a good overview of the structural adaptations of the ubiquitous globin interface while avoiding packing interactions. The CBMs for this diverse family were found to correlate very well with the groups on a sequence cluster tree, confirming the prospect of using them as modeling templates. Thus, conserved binding modes introduce a robust resource for studying protein interactions by combining the evolutionary relationships of ancient conserved sequence domains with the structural comparison of interaction geometries.
Acknowledgments
We thank Aron Marchler-Bauer and Maricel Kann for helpful discussions and Lewis Geer for use of the CDart database. This work was supported in whole by the Intramural Research Program of the National Library of Medicine at National Institutes of Health/DHHS.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051760806.
Supplemental material: www.proteinscience.org
References
- Aloy, P. and Russell, R.B. 2004. Ten thousand interactions for the molecular biologist. Nat. Biotechnol. 22: 1317–1321. [DOI] [PubMed] [Google Scholar]
- Aloy, P., Ceulemans, H., Stark, A., and Russell, R.B. 2003. The relationship between sequence and interaction divergence in proteins. J. Mol. Biol. 332: 989–998. [DOI] [PubMed] [Google Scholar]
- Bahadur, R.P., Chakrabarti, P., Rodier, F., and Janin, J. 2004. A dissection of specific and non-specific protein–protein interfaces. J. Mol. Biol. 336: 943–955. [DOI] [PubMed] [Google Scholar]
- Bashton, M. and Chothia, C. 2002. The geometry of domain combination in proteins. J. Mol. Biol. 315: 927–939. [DOI] [PubMed] [Google Scholar]
- Carugo, O. and Argos, P. 1997. Protein–protein crystal-packing contacts. Protein Sci. 6: 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta, S., Iyer, G.H., Bryant, S.H., Lawrence, C.E., and Bell, J.A. 1997. Extent and nature of contacts between protein molecules in crystal lattices and between subunits of protein oligomers. Proteins 28: 494–514. [DOI] [PubMed] [Google Scholar]
- Davis, F.P. and Sali, A. 2005. PIBASE: A comprehensive database of structurally defined protein interfaces. Bioinformatics 21: 1901–1907. [DOI] [PubMed] [Google Scholar]
- Elcock, A.H. and McCammon, J.A. 2001. Identification of protein oligomerization states by analysis of interface conservation. Proc. Natl. Acad. Sci. 98: 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer, L.Y., Domrachev, M., Lipman, D.J., and Bryant, S.H. 2002. CDART: Protein homology by domain architecture. Genome Res. 12: 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat, J.F., Madej, T., and Bryant, S.H. 1996. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6: 377–385. [DOI] [PubMed] [Google Scholar]
- Hazbun, T.R. and Fields, S. 2001. Networking proteins in yeast. Proc. Natl. Acad. Sci. 98: 4277–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslet, H.A. and Royer Jr., W.E. 2001. Crystalline ligand transitions in lamprey hemoglobin. Structural evidence for the regulation of oxygen affinity. J. Biol. Chem. 276: 26230–26236. [DOI] [PubMed] [Google Scholar]
- Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., and Sakaki, Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. 98: 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin, J. 1997. Specific versus non-specific contacts in protein crystals. Nat. Struct. Biol. 4: 973–974. [DOI] [PubMed] [Google Scholar]
- Janin, J. and Rodier, F. 1995. Protein–protein interaction at crystal contacts. Proteins 23: 580–587. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8: 275–282. [DOI] [PubMed] [Google Scholar]
- Keskin, O., Tsai, C.J., Wolfson, H., and Nussinov, R. 2004. A new, structurally nonredundant, diverse data set of protein–protein interfaces and its implications. Protein Sci. 13: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler, S.J. and Hubbard, S.J. 2005. Conservation of orientation and sequence in protein domain–domain interactions. J. Mol. Biol. 345: 1265–1279. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer, A. and Bryant, S.H. 2004. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 32: W327–W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Panchenko, A.R., Shoemaker, B.A., Thiessen, P.A., Geer, L.Y., and Bryant, S.H. 2002. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 30: 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Anderson, J.B., Cherukuri, P.F., DeWeese-Scott, C., Geer, L.Y., Gwadz, M., He, S., Hurwitz, D.I., Jackson, J.D., Ke, Z., et al. 2005. CDD: A Conserved Domain Database for protein classification. Nucleic Acids Res. 33: D192–D196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein–protein interactions from genome sequences. Science 285: 751–753. [DOI] [PubMed] [Google Scholar]
- Marino-Ramirez, L., Kann, M.G., Shoemaker, B.A., and Landsman, D. 2005. Histone structure and nucleosome stability. Expert Rev. Proteomics 2: 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooren, I.M. and Thornton, J.M. 2003. Structural characterisation and functional significance of transient protein–protein interactions. J. Mol. Biol. 325: 991–1018. [DOI] [PubMed] [Google Scholar]
- Panchenko, A.R., Kondrashov, F., and Bryant, S. 2004. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 13: 884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J., Lappe, M., and Teichmann, S.A. 2001. Mapping protein family interactions: Intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J. Mol. Biol. 307: 929–938. [DOI] [PubMed] [Google Scholar]
- Ponstingl, H., Henrick, K., and Thornton, J.M. 2000. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins 41: 47–57. [DOI] [PubMed] [Google Scholar]
- Robert, C.H. and Janin, J. 1998. A soft, mean-field potential derived from crystal contacts for predicting protein–protein interactions. J. Mol. Biol. 283: 1037–1047. [DOI] [PubMed] [Google Scholar]
- Royer Jr., W.E., Knapp, J.E., Strand, K., and Heaslet, H.A. 2001. Cooperative hemoglobins: Conserved fold, diverse quaternary assemblies and allosteric mechanisms. Trends Biochem. Sci. 26: 297–304. [DOI] [PubMed] [Google Scholar]
- Saitou, N. and Nei, M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Uetz, P., Giot, L., Cagney, G., Mansfield, T.A., Judson, R.S., Knight, J.R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. 2000. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627. [DOI] [PubMed] [Google Scholar]
- Vogeley, L., Palm, G.J., Mesters, J.R., and Hilgenfeld, R. 2001. Conformational change of elongation factor Tu (EF-Tu) induced by antibiotic binding. Crystal structure of the complex between EF-Tu.GDP and aurodox. J. Biol. Chem. 276: 17149–17155. [DOI] [PubMed] [Google Scholar]
- Wang, Y., Anderson, J.B., Chen, J., Geer, L.Y., He, S., Hurwitz, D.I., Liebert, C.A., Madej, T., Marchler, G.H., Marchler-Bauer, A., et al. 2002. MMDB: Entrez’s 3D-structure database. Nucleic Acids Res. 30: 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman Jr., J. 1948. Heme proteins. Adv. Protein Chem. 4: 407–531. [DOI] [PubMed] [Google Scholar]