

Abstract
A facile synthesis of 1-fluoro-1-deoxy-Δ8-THC analogs with side-chains seven carbons in length, in the alkane/ene/yne-series (6, 5 and 4), was achieved from 1-fluoro-3,5-dimethoxybenzene (1). In vitro studies show that substitution by a fluorine has a significant detrimental effect on CB1 binding which is supported by in vivo testing. The implications of these results on the SAR of classical cannabinoids is discussed.
Keywords: Fluorine substitution, tetrahydrocannabinols, CB1 binding affinity
Fluorine’s high electronegativity and small size are among the special properties that contribute to its importance in medicinal chemistry.1 The effects of fluorine substitution on the biological behaviour of biologically active molecules have been used effectively in drug design, especially after the successful use in steroids and the anticancer drug 5-fluorouracil.1 As a result, the presence of fluorine in drugs is now quite common. We were therefore interested in examining the role of fluorine substitution in classical cannabinoids. It is well known in the SAR of classical cannabinoids that substitutions in the C-1, C-3 and C-9 positions play an important role2, 3 in the interaction with CB1 cannabinoid receptors. Hydrogen bonding interactions of the hydroxyl group at C-1 and the presence of a hydroxymethyl at C-9 are of particular interest in this respect. The effect of substituting fluorine for a hydroxyl is especially interesting since the fluorine can only accept hydrogen bonds, whereas hydroxyl groups can both accept and donate hydrogen bonds. Our molecular modeling studies4 suggested that the phenolic hydroxyl of THC corresponded to the terminal hydroxyl of anandamide. Another reason which prompted us to carry out this study was the finding in our laboratory5, 6 that substituting a fluorine atom for the 2’-hydroxyl(O-585) in anandamide (AEA) increased its CB1 binding affinity 10-fold. A similar increase was found in the 2-methyl-2’-F-AEA (O-689) compared to 2-methyl-AEA (O-680). Earlier studies by Charalambous et al.,7 and Martin et al.,8, 9 had shown that substitution of fluorine for hydrogen on C-5’ (or C-5”) of the pentyl side chain had relatively little effect on the pharmacological activity of the THCs (tetrahydrocannabinols) or CBD (cannabidiol). This was attributed to the fact that both fluorine and hydrogen atoms occupy comparable volumes although their electrostatic properties are different. Tius and co-workers10 reported on C-9 difluoro and monofluoro-THC analogs and found them to have marginal anti-inflammatory activity. They also introduced fluorine probes in the aromatic ring of nabilone and found diminished affinity11a for the CB1 receptor thus confirming the hypothesis that the phenolic hydroxyl group is involved in a hydrogen bonding interaction with the receptor. A (−)-5’-18F- Δ8-THC analog was studied12 for brain distribution in a primate, using positron emission tomography (PET) technique, but the results were inconclusive due to low binding affinity of the ligand.
With this background, we synthesized 1-Fluoro-1-deoxy- Δ8-THC analogs and examined their pharmacological activity. We prepared the alkane/ene/yne series of 1-fluoro-THCs (4, 5 and 6) with side-chains seven carbons in length, which previous SAR studies indicated to be near optimum length for cannabinoid activity. The synthesis13a is shown in Scheme 1. Commercially available 1-fluoro-3,5-dimethoxybenzene (1) was demethylated with boron tribromide (2.5 equivalents, CH2Cl2, −78°C for 10 min, then warmed to room temperature for 1 h) to afford 5-fluororesorcinol which was condensed (TsOH catalytic amount, benzene, reflux Dean-Stark trap, 2 h) with cis-p-menth-2-en-1,8-diol to afford a mixture from which two THC isomers were isolated (silica gel chromatography) in approximately equal amounts (~10% each). These isomers differed in the relative substitution pattern on the aromatic THC ring, one isomer being the desired 1-fluoro-3-hydroxy-Δ8-THC (2), the other being the undesired 3-fluoro-1-hydroxy-Δ8-THC (3). The structures were assigned on the basis of shifts found for the aromatic protons (H-2 and H-4) when their NMR’s were taken in CDCl3 and C6D6, as reported by Arnone, A. et al.13b The desired isomer was then activated as a triflate before palladium (0)-catalyzed coupling14 to 1-heptyne to afford 1-fluoro-3-(1-heptynyl)-Δ8-THC (4). This in turn was reduced to both the cis-alkene (5, 1 atm H2, Lindlar’s catalyst, 1 drop of quinoline, alcohol)15 and the alkane (6, 1 atm H2, 5% Pd-C, alcohol) in both cases without reducing the Δ8-double bond. The target compounds were all characterized16 on the basis of their 1HNMR, High resolution mass spectrum, TLC and GLC analyses. In vitro binding assays for CB1 and CB2 receptors were determined and their in vivo activity was examined in the tetrad tests.17 For tail-flick (TF) test, we also tested them by intrathecal (i.t.) route18 in order to investigate if there were any potential differences in receptor interaction or activation based on different routes of administration. The results are shown in Table 1, and for comparison purposes we have also included the activity of the parent 1-hydroxy-THCs, (O-964) for compound 4, and (O-1317) for compound 5 respectively. It is quite clear from Table 1 that substitution of a fluorine at C-1 in THCs has a significant detrimental effect on the CB1 binding affinity. The alkane analog 6 has 38-fold less binding affinity than Δ9-THC whereas the alkene analog 5 has 7-fold less than Δ9-THC and 331-fold less than its parent analog (O-1317). The yne analog 4 is 245-fold and 277-fold less to both Δ9-THC and the parent analog (O-964). Similar findings were reflected in the tetrad tests and the TF (i.t.) tests. Based on these results and the previous literature studies related to the effect of fluorine substitution in the C-5’, C-9 and C-11 positions, it can be concluded that substitution by a fluorine, especially at C-1 position, has a detrimental effect on CB1 binding which is supported by in vivo testing. This is in contrast to our findings in the anandamide analogs (see above).
Scheme 1.
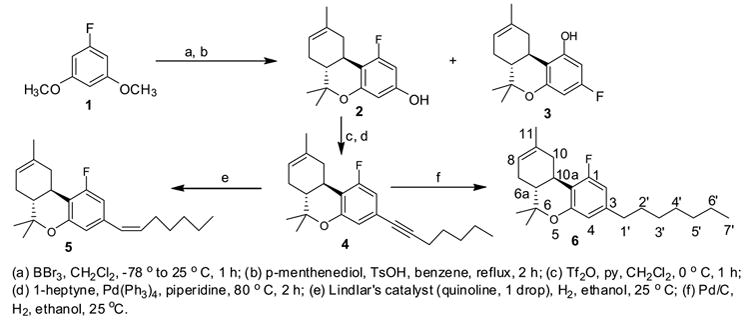
Table 1.
Binding affinity and Tetrad Tests of 1-Fluoro-1-deoxy-THCs
| Compound | structure | CB1 (nM) | CB2 (nM) | Tetrad Tests (ED50, mg/kg) | TF (i.t.) | ||
|---|---|---|---|---|---|---|---|
| SA | TF | RT | μg/mouse | ||||
| Δ9-THC | 40.7 ± 1.7 a | 36.4 ± 10 b | 1.0 | 1.4 | 1.4 | 29 (24–36) c | |
| 4 | >10,000 | >4550 | 8% @ 30 mg | 4% @ 30 mg | −2.2°@ 30 mg | 156.9 (114.1–216) | |
| (O-964) |
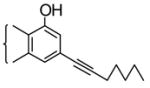
|
36 ± 0.8 | - | 3.68 | 3.24 | 2.96 | - |
| 5 | 285 ± 34 | 40 ± 8 | 0.51 | 5.8 | 4.1 | 99.5 (62.5–158) | |
| (O-1317) |
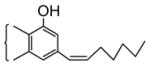
|
0.86 ± 0.09 | - | 0.09 | 0.09 | 0.13 | - |
| 6 | 1557 ± 203 | 1508 | 31.1 | 21.5 | 6.9 | 36% @ 100μ g | |
Behavioral Evaluation (tetrad tests); SA (spontaneous activity), TF (tail-flick), RT (rectal temperature), RI (ring immobility) were carried out in mice. The ED50 data is given in mg/kg. For details see references 17 and 8. RI test was not carried out f or any of the compounds and is therefore not given.
see reference 9.
see reference 6.
The results are presented as ED50 (95% confidence limits in parenthesis); see reference 18.
These findings have several implications in connection with the SAR of THCs particularly in connection with the role of the C-1 hydroxyl group of THC with the CB1 receptor and the determination of the common pharmacophore in the THC/AEA molecular modeling overlaying studies. With regard to the former, previous studies19 have attempted to determine the nature of the hydrogen-bonding by preparing and testing specific analogs of Δ9- and Δ8-THCs. However, the interpretation of the data obtained is controversial. So far the studies which support the hypothesis that the phenolic hydroxyl group is involved in a hydrogen-bonding interaction with the CB1 receptor is by Tius et al.,11a and Song and Bonner.11b The latter authors prepared a mutant CB1 receptor in which lysine 192 was replaced by an alanine and examined the binding affinity of various CB1 agonists. Based on this study they arrived at the same conclusions as Tius et al.11a Our results suggest that the hydroxyl group of THC is functioning as a hydrogen-bond donor in its interaction with the CB1 receptor and not solely as a hydrogen-bond acceptor.11c,d In our molecular modeling studies4 of overlaying THC with AEA, we had used the pharmacophore fit involving the pyran oxygen of Δ9-THC with the carbonyl oxygen of AEA thereby leaving the phenolic hydroxyl of THC as the counterpart to the terminal hydroxyl of anandamide. Using this alignment, reasonably good COMFA correlations were obtained for THC and AEA analogs. However, because of our findings in the AEA series, we had expected enhanced binding affinity for the C-1-fluoro-substituted-THCs. Our results do not support the pharmacophore fit we used and lends support to the Tong model20 which uses the superposition of the hydroxyl of the AEA to the cyclohexyl at C-9 of 9-nor-9β-OH-hexahydrocannabinol (HHC). The difference in the CB1 binding affinity9 between 11-F-Δ8-THC and its parent 11-OH-Δ8-THC was marginal, Ki = 107 nM versus 55 nM respectively. It is interesting to note that the alkene analog 5 bound to CB1 receptors significantly better than the alkane and alkyne analogs. A similar pattern was observed in the 11-hydroxy-THC series by Makriyannis and co-workers.21 The alkene analog also showed some CB2 selectivity in binding (7-fold) as found in the 1-deoxy- Δ8-THC-DMH series.22, 23 Moreover, the loss of CB2 affinity in analog 6 was unexpected given the observation that 1-deoxy-THC analogs retain CB2 affinity.22 These findings suggest that electrostatic properties at C-1 are crucial for CB2 receptor affinity.
This study presents some interesting conclusions: (a) substitution of 1-hydroxyl group in Δ8-THC by a fluorine results in a significant decrease in its interaction with the CB1 receptor, (b) the 1-hydroxyl in Δ8-THC is functioning as a hydrogen-bond donor in its interaction with the CB1 receptor, (c) the results support the molecular modeling overlay studies proposed in the Tong model, (d) some CB2 selectivity (7-fold) is observed in the alkene analog 5, and (e) it seems unlikely that any advantage will be gained by substituting fluorine in the template of classical cannabinoids.
Acknowledgments
The authors are grateful to NIDA Grants DA-05488, and DA-03672 for the support of this work.
Footnotes
Selected spectroscopic data; 1HNMR (100 MHz, CDCl3) (a) compound 2. δ 1.11 (s, 3H), 1.37 (s, 3H), 1.70 (br s, 3H), 2.57–3.07(m, 2H), 4.92 (s, 1H), 5.43 (br d, J = 4 Hz, 1H), 6.04–6.23 (m, 2H); (C6D6) δ 6.09 (dd, J = 12.0, 2.5 Hz, 1H), 6.18 (d, J = 3 Hz, 1H); TLC, Rf 0.4 (1:4 EtOAc-hexanes); (b) compound 4. δ 0.91 (t, J = 6 Hz, 3H), 1.09 (s, 3H), 1.38 (s, 3H), 1.71 (br s, 3H), 2.37 (t, J = 6.6 Hz, 2H), 2.58–3.07 (m, 2H), 5.43 (br d, J = 5 Hz, 1H), 6.52–6.70 (m, 2H); TLC Rf 0.5 (1:19 EtOAc-hexanes); GLC >84%; HRMS (CI) calcd. For C23H30FO (MH+) 341.2281, found 341.2247, Δ = + 3.4 mmu; (c) compound 5. δ 0.89 (t, J = 6 Hz, 3H), 1.12 (s, 3H), 1.39 (s, 3H), 1.72 (br s, 3H), 2.33 (q, J = 6 Hz, 2H), 2.6–3.1 (m, 2H), 5.44 (br d, J = 4 Hz, 1H), 5.62 (dt, J = 12, 7 Hz, 1H), 6.23 (br d, J = 12 Hz, 1H), 6.52(dd, J = 12.2, 1.6 Hz, 1H), 6.54 (br s, 1H); TLC Rf 0.2 (5 x hexanes); GLC 90%; HRMS (CI) calcd. For C23H32FO (MH+) 343.2437, found 343.2444 Δ = −0.7 mmu; (d) compound 6. δ 0.88 (t, J = 6 Hz, 3H), 1.11 (s, 3H), 1.28 (br s, 8H), 1.38 (s, 3H), 1.71 (br s, 3H), 2.49 (t, J = 7 Hz, 2H), 2.6–3.1 (m, 2H), 5.43 (br d, J = 4 Hz, 1H), 6.41 (dd, J = 11.9, 1.8 Hz, 1H), 6.44 (br s, 1H); TLC Rf 0.3 (5 x hexanes); GLC 87%; (HRMS) (CI) calcd. For C23H34FO (MH+) 345.2594, found 345.2603, Δ = −0.9 mmu.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Kirk KL. Curr Topics Med Chem. 2006;6:1447. doi: 10.2174/156802606777951073. [DOI] [PubMed] [Google Scholar]
- 2.Razdan RK. Pharmacol Rev. 1986;38:21. [PubMed] [Google Scholar]
- 3.Rapaka RS, Makriyannis A. National Institute on Drug Abuse, MD. 1987. NIDA Research Monograph 79. [Google Scholar]
- 4.Thomas BF, Adams IB, Mascarella SW, Martin BR, Razdan RK. J Med Chem. 1996;39:471. doi: 10.1021/jm9505167. [DOI] [PubMed] [Google Scholar]
- 5.Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK, Martin BR. J Pharmacol Exp Ther. 1995;273:1172. [PubMed] [Google Scholar]
- 6.Showalter VM, Compton DR, Martin BR, Abood ME. J Pharmacol Exp Ther. 1996;278:989. [PubMed] [Google Scholar]
- 7.Charalambous A, Lin S, Marciniak G, Banijamali A, Friend FL, Compoton DR, Martin BR, Makriyannis A. Pharmacol Biochem Behav. 1991;40:509. doi: 10.1016/0091-3057(91)90355-6. [DOI] [PubMed] [Google Scholar]
- 8.Martin BR, Compton DR, Semus SF, Lin S, Marciniak G, Grzybovska J, Charalambous A, Makriyannis A. Pharmacol Biochem Behav. 1993;46:295. doi: 10.1016/0091-3057(93)90356-x. [DOI] [PubMed] [Google Scholar]
- 9.Compton DR, Rice KC, DeCosta B, Razdan RK, Melvin LS, Johnson MR, Martin BR. J Pharmacol Exp Ther. 1993;265:218. [PubMed] [Google Scholar]
- 10.Tius MA, Kannangara GSK, Kerr MA, Grace KJS. Tetrahedron. 1993;49:3291. [Google Scholar]
- 11.(a) Tius MA, Kawakami JK, Hill WAG, Makriyannis A. Chem Commun. 1996:2085. [Google Scholar]; (b) Song ZH, Bonner TI. Mol Pharmacol. 1996;49:891. [PubMed] [Google Scholar]; (c) Semus SF, Martin BR. Life Sci. 1990;46:1781. doi: 10.1016/0024-3205(90)90142-e. [DOI] [PubMed] [Google Scholar]; (d) Reggio PM, Seltzman HH, Compton DR, Prescott WP, Jr, Martin BR. Mol Pharmacol. 1990;38:854. [PubMed] [Google Scholar]
- 12.Charalambous A, Marciniak G, Shiue CY, Dewey SL, Schlyer DJ, Wolf AP, Makriyannis A. Pharmacol Biochem Behav. 1991;40:503. doi: 10.1016/0091-3057(91)90354-5. [DOI] [PubMed] [Google Scholar]
- 13.(a) Singer M, Ryan WJ, Saha B, Martin BR, Razdan RK. J Med Chem. 1998;41:4400. doi: 10.1021/jm9803875. [DOI] [PubMed] [Google Scholar]; (b) Arnone A, Bernardi R, Merlini L, Servi S. Gazz Chim Ital. 1975;105:9. [Google Scholar]
- 14.Alami M, Ferri F, Linstrumelle G. Tetrahedron Lett. 1993;34:6403. [Google Scholar]
- 15.Crocker PJ, Saha B, Ryan WJ, Wiley JL, Martin BR, Ross RA, Pertwee RG, Razdan RK. Tetrahedron. 1999;55:13907. [Google Scholar]
- 16.See selected spectroscopic data listed after references.
- 17.Martin BR, Jefferson R, Winckler R, Wiley JL, Huffman JW, Crocker PJ, Saha B, Razdan RK. J Pharmacol Exp Ther. 1999;290:1065. [PubMed] [Google Scholar]
- 18.Wiley JL, Patrick GS, Crocker PC, Saha B, Razdan RK, Martin BR. Eu J Pharmacol. 2000;397:319. doi: 10.1016/s0014-2999(00)00259-4. [DOI] [PubMed] [Google Scholar]
- 19.Reggio PH, Seltzman HH, Compton DR, Prescott JWR, Martin BR. Mol Pharmacol. 1990;38:854. [PubMed] [Google Scholar]
- 20.Tong W, Collantes ER, Welsh WJ, Berglund BA, Howlett AC. J Med Chem. 1998;41:4207. doi: 10.1021/jm970239z. [DOI] [PubMed] [Google Scholar]
- 21.Busch-Peterson J, Hill WA, Fan P, Khanolkar A, Xie X, Tius MA, Makriyannis A. J Med Chem. 1996;39:3790. doi: 10.1021/jm950934b. [DOI] [PubMed] [Google Scholar]
- 22.Huffman JW, Yu S, Showalter V, Abood ME, Compton DR, Martin BR, Bramblett RD, Reggio PH. J Med Chem. 1996;39:3875. doi: 10.1021/jm960394y. [DOI] [PubMed] [Google Scholar]
- 23.Gareau Y, Dufresne C, Gallant M, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Weech PK, Metters KM, Labelle M. Bioorg Med Chem Lett. 1996;6:189–194. [Google Scholar]