

SUMMARY
The PTEN tumor-suppressor is frequently affected in cancer, and inherited PTEN mutation causes cancer-susceptibility conditions such as Cowden Syndrome. PTEN acts as a plasma-membrane lipid-phosphatase antagonizing the PI-3-Kinase/AKT pathway. However, PTEN is also found in cell nuclei, but mechanism, function and relevance of nuclear localization have remained unclear. Here we show that nuclear PTEN is essential for tumor-suppression and that import is mediated by its mono-ubiquitination. We show that a lysine-mutant of PTEN, which retains catalytic activity yet causes Cowden Syndrome, fails to accumulate in nuclei of patient tissue due to an import defect. We identify this and another lysine-residue as major mono-ubiquitination sites essential for PTEN import. Poly-ubiquitination in contrast, leads to PTEN degradation in the cytoplasm, while nuclear PTEN is stable, antagonizes AKT and causes apoptosis. We thus identify the first cancer-associated mutations of PTEN that target post-translational modification and demonstrate how a discrete molecular mechanism dictates tumor-progression by differentiating between degradation and protection of PTEN.
INTRODUCTION
Since the original identification of the tumor suppressor PTEN (Phosphatase and TENsin homolog on chromosome TEN) (Li et al., 1997; Steck et al., 1997) numerous reports have described PTEN-loss or mutation in cancer specimens, cancer cell lines and inherited cancer predisposing syndromes (Bonneau and Longy, 2000; Li et al., 1997), rendering this gene one of the most frequently affected tumor suppressors of the post-p53 era.
To better understand the causal nature of PTEN-loss in human cancer we and others have genetically modeled the effects of Pten-loss in mouse and shown that such animals faithfully recapitulate the tumor spectrum observed in PTEN-deficient patients (Di Cristofano et al., 1998; Podsypanina et al., 1999; Suzuki et al., 1998; Trotman et al., 2003). These studies have firmly placed Pten-loss to the stage of tumor initiation in epithelial cancer. Most notably, we have demonstrated PTEN haploinsufficiency (reviewed in (Sherr, 2004), as a single copy becomes unable to prevent prostate cancer either in the context of loss of certain other tumor suppressor genes, (Di Cristofano et al., 2001; Ma et al., 2005; Trotman et al., 2006) or, alternatively, in the context of genetically engineered reduction of Pten expression just below heterozygosity (Trotman et al., 2003). In addition, we have shown recently that complete Pten-loss even provokes the p53-dependent cellular senescence response (Chen et al., 2005) that antagonizes prostate tumorigenesis redefining the previously assumed interplay of these two major tumor suppressor genes (Trotman and Pandolfi, 2003). This finding further underscored the importance of a “non-Knudsonian” single-hit working model for loss of PTEN in tumor initiation and clarifies why indeed a majority of human tumors are retaining a wildtype copy of PTEN (Sansal and Sellers, 2004). Thus, understanding the mechanisms of PTEN regulation that govern this “heterozygous tumor progression” is imperative for progress in the field.
PTEN catalyzes the conversion of the membrane lipid second messenger PIP3 to PIP2 (Maehama and Dixon, 1998), and therefore is the key antagonist of various PIP3-dependent proto-oncogenic kinases, chiefly among them AKT/PKB (Stambolic et al., 1998). This firmly positions PTEN action to the cell’s plasma membrane (Cantley, 2002; Di Cristofano and Pandolfi, 2000). However, it has long been observed that in several tissues and cultured cells PTEN is also found in cell nuclei (reviewed in (Lian and Di Cristofano, 2005). While it remains unclear what function can be attributed to this nuclear pool of PTEN specifically, positive correlation between nuclear PTEN and suppression of tumor progression has been described for both cutaneous melanoma and colorectal cancer (Whiteman et al., 2002; Zhou et al., 2002). But in spite of such correlative studies, to date no proof for an essential role of nuclear PTEN in tumor suppression has been accrued. Furthermore, although these data suggested that nuclear PTEN could retain some activity, it still has remained unclear how PTEN subcellular localization would be regulated.
Nuclear protein import is generally mediated by distinct and, importantly, transferable Nuclear Localization Signal (NLS) sequences such as the SV-40 T-antigen-like cluster of positively charged amino acids. Such sequences confer binding to import receptors, which mediate uni- or bidirectional transport across the Nuclear Pore Complexes (NPC’s, see (Chook and Blobel, 2001; Gorlich and Kutay, 1999)). Yet, no functional NLS sequence satisfying the essential criteria has been found in PTEN, suggesting that PTEN either binds to another NLS-bearing protein to be transported in a “piggy-back” fashion or that an altogether novel mechanism could regulate its nuclear import.
We describe here the discovery of a cancer-associated mutation that targets PTEN post-translational modification leading to an aberrant nuclear exclusion phenotype in a family of patients with Cowden Syndrome (CS). CS is one of three closely related autosomal dominant cancer syndromes, caused by germ line inactivating mutations of PTEN and is characterized by high cancer susceptibility (Eng, 2003). By identifying the biophysical, biochemical and cell-biological deficiencies of this mutant we here establish the first causal relationship between PTEN post-translational modification, nuclear import and tumor initiation.
RESULTS
Nuclear exclusion of PTENK289E in Cowden patient polyps
We have previously solved the PTEN crystal structure (Lee et al., 1999) revealing an N-terminal phosphatase and a C-terminal C2 domain, which stabilizes the catalytic domain and is essential for proper positioning of PTEN at the cell membrane (Georgescu et al., 2000), see also Figure 1C). While most known PTEN missense mutations target the phosphatase domain, such as the catalytic cysteine 124 to arginine mutant (C124R, see abbreviations (Nelen et al., 1997)), some disrupt PTEN function by indirectly affecting catalytic domain architecture (Lee et al., 1999). Yet, cancer associated missense mutations have also been identified in regions of no apparent relevance to PTEN enzymatic function or stability. Prominent among them is a mutation found in a stretch of 32 amino acids within the C2 domain, that we term the C2-loop. This loop of PTEN is highly conserved among vertebrates including Zebrafish (Figure 1A). As previously described, the C2-loop (similar to N-and C-terminal tails of PTEN) interferes with proper crystal formation (Figure 1B) and can be specifically digested by subtilisin, consistent with a random and accessible conformation protruding from the framework of the C2-domain (Figure 1C). Importantly, C2-loop-deficient PTEN retains wildtype (wt) activity in vitro and in vivo (Lee et al., 1999). Yet intriguingly, a missense mutation targeting a conserved residue of the C2-loop, (K289E), has been described (see Figure 1A). In agreement with the above observations PTENK289E displays no significant defects in activity or membrane association (Georgescu et al., 2000) as K289 faces away from the plasma membrane (Figure 1C). Thus, taken together, the C2-loop has no positive structural function, its loss does not impinge on PTEN activity and the C2-loop K289E mutant retains its enzymatic activity in vivo. Yet, the K289E allele of PTEN is found inherited in a family in which carriers with one mutant allele display Cowden Syndrome (Chi et al., 1998), thus demonstrating genetically that the K289E mutant is indeed defective in tumor suppression.
Figure 1. The PTENK289E Cowden mutant shows nuclear exclusion in patient samples.
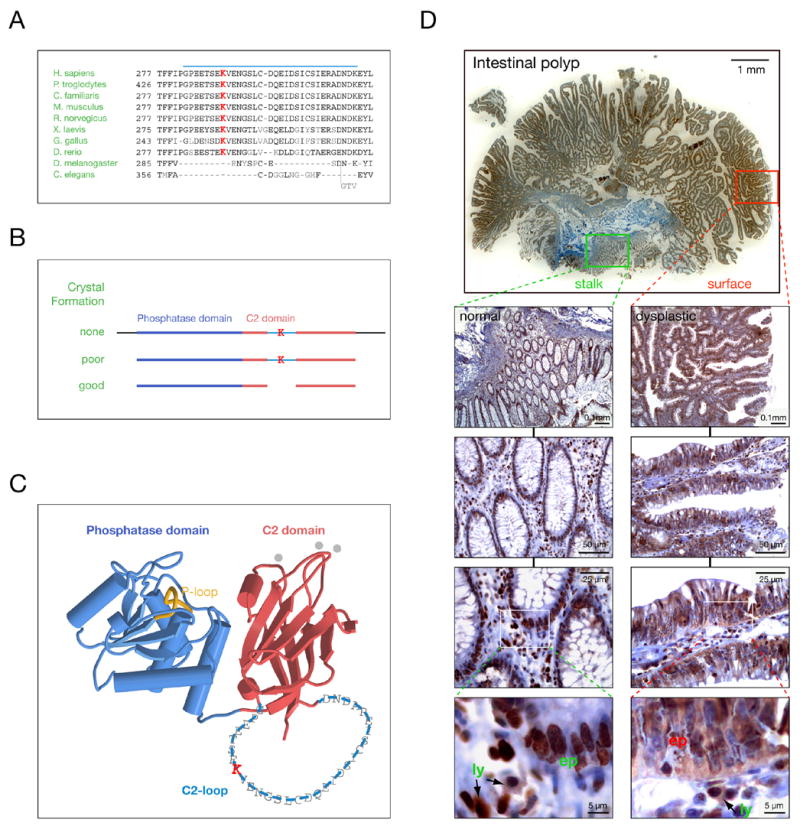
(A) Sequence alignment of the PTEN C2-loop (blue bar) from various species shows K289 conservation in vertebrates (highlighted in red).
(B) PTEN cleavage requirements for crystallization reveal that the C2-loop does not contribute to structural integrity (K denotes K289).
(C) Localization and size of the C2-loop within the PTEN structure (single amino acid code over backbone). Note that the C2-loop faces away from the cell membrane (top) and membrane-interacting loops of the C2-domain (grey dots).
(D) Intestinal polyp from a PTENwt/K289E Cowden patient shows nuclear and cytoplasmic PTEN in normal (PTENwt/K289E) mucosa (left panel magnification series, green) and only cytoplasmic PTENK289E in dysplastic (PTENΔ/K289E) regions (right panel magnification series, red). “Ly” denotes circulating lymphocytes (PTENwt/K289E) that do not show PTEN LOH and “ep” denotes epithelial cells.
To investigate the paradox of how an enzymatically functional PTEN mutant can cause CS we sought to study tumor samples of a family member who inherited the K289E mutation. Since these patients routinely undergo colonoscopy for removal of intestinal polyps we analyzed this tissue (Figure 1D). Dysplastic intestinal polyps have large areas of poorly differentiated glandular structures at their surface (red rectangle, top panel) and this polyps’ stalk featured also a small area of normal colonic mucosa (green rectangle, top panel). As previously shown (Chi et al., 1998), the bulk of these polyps lose the wt PTEN allele while retaining the mutant and thus are expressing only the PTENK289E protein which we sought to study by immunohistochemistry. As depicted, the dysplastic surface area retains PTEN expression (consistent with loss of the wt PTEN copy and demonstrating PTENK289E stability), yet magnification reveals that surprisingly, PTEN is excluded from cell nuclei of epithelial cells (Figure 1D, right panel series). Remarkably, interstitial lymphocytes, which retain both wt and K289E mutant protein (Chi et al., 1998), in contrast also retain nuclear PTEN staining, serving as internal control for the staining procedure (right and left panel series, lowest panels, ly vs. ep). Intriguingly, epithelial cells in the normal areas of the polyp’s stalk, areas that typically retain both mutant and wt PTEN copies, display strong nuclear and cytoplasmic PTEN staining (left panel series), very similar to interstitial lymphocytes (high magnification, lowest panel). Identical results were obtained using two different antibodies for staining (see Experimental Procedures). Thus, the K289E mutant protein appears to display an aberrant subcellular localization, reminiscent of a nuclear exclusion phenotype.
Intrinsic nuclear import defect of PTENK289E
Since the above-shown mislocalization could be either an intrinsic property of the K289E mutation or a polyp specific effect on PTENK289E, we analyzed the mutants’ subcellular distribution in vitro. By immunofluorescence (IF) endogenous PTEN is found in both cell nuclei and cytoplasm of most common human and murine cell lines and primary murine cells (data not shown) similar to the localization shown in the PTEN heterozygous prostate cancer cell line DU-145 (Figure 2A). To test mutant PTEN localization, PTEN-deficient prostate cancer cells (PC3) were transiently transfected. When over-expressed as gfp-fusion, wt PTEN efficiently equilibrated between nucleus and cytoplasm within 10–12 hours after transfection (Figure 2B). Of note, PTEN localization was unaffected by catalytic activity, as revealed by the distribution of the enzyme-dead PTENC124S mutant (lower left panel), demonstrating that AKT activity (which is high in PC3 cells and only lowered upon wt PTEN transfection) does not visibly affect PTEN localization. Strikingly, at this time point the PTENK289E mutant showed dominant cytoplasmic accumulation in most cells, which is also unaffected by loss of enzymatic activity in the PTENK289E, C124S double mutant (Figure 2B, right panels). The same results were obtained by visualizing non gfp-tagged PTEN and the corresponding mutants (Supplemental Figure S1A, upper panels). Notably, nuclear staining of the K289E mutant was found to increase by 24 and 48 hrs after transfection (not shown). Since steady state cytoplasmic accumulation can in principle result from reduced nuclear import or enhanced nuclear export of PTENK289E (indicative of a loss or gain of function mutation, respectively) we next tested the nuclear and cytoplasmic accumulation rates of the PTEN mutant quantitatively. The Fluorescence Recovery After Photo-bleaching (FRAP) technique produces distinguishable (yet chemically identical) cytoplasmic and nuclear PTEN populations by bleaching gfp-fluorescence in either the cell nucleus or cytoplasm. Then, the compartmental equilibration, which occurs via nuclear import and export can be measured by time-lapse fluorescence microscopy (see Methods).
Figure 2. The PTENK289E Cowden mutant has an intrinsic nuclear import defect.
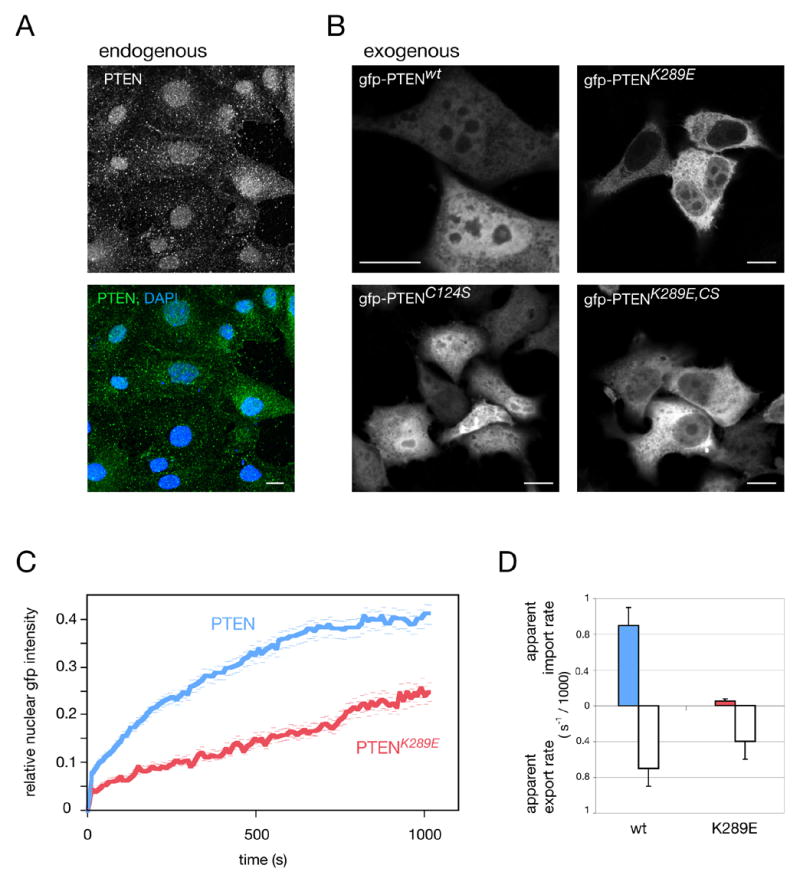
(A) Immunofluorescence of endogenous PTEN in prostate cancer derived PTEN+/− DU-145 cells. Scale bar, 10 μm.
(B) Localization of gfp-PTEN fusions reveals nuclear exclusion of the K289E mutant at 12 hrs post transfection. Scale bars, 10 μm.
(C) FRAP analysis of wt and PTENK289E shows nuclear accumulation after nuclear photo-bleaching. Error bars show S. D.
(D) Quantification of (C) and quantification of nuclear export FRAP results for wt and mutant proteins. Error bars are S.D.
We found that wt PTEN fluorescence re-accumulated inside the nucleus, reaching equilibrium by roughly 15 minutes post nuclear bleaching (Figure 2C). The PTENK289E mutant in contrast, showed clearly reduced nuclear accumulation and no equilibration within the same time. Calculation of the nuclear accumulation rates (i.e. apparent nuclear import rates) revealed an eighteen-fold reduction in nuclear net import for the K289E mutation (Figure 2D). Notably, dynamic analysis revealed that even when steady state nuclear levels of wt and mutant PTEN appear to be similar (by static analysis), the mutant still shows a strongly reduced rate of nuclear accumulation. Importantly, the apparent export rate of the mutant protein was also decreased (Figure 2D), excluding a gain of export function through the K289E mutation. This result also excludes the possibility that wt nuclear localization is due to nuclear retention via K289 (which would result in higher K289E export rates). To exclude charge-conversion effects associated with the K289E mutation, the K289R mutant was measured and also revealed PTEN import-deficiency (Supplemental Figure S1B). Also of note, intra-nuclear and intra-cytoplasmic diffusion coefficients (as measured by FRAP) were not affected by the mutation (not shown, see Experimental Procedures). Therefore, taken together, our experiments revealed that PTENK289E has a nuclear import/export defect, and thus represents a loss of function mutant, defective in nucleo-cytoplasmic shuttling.
Lysine 289 is a major site for PTEN ubiquitination
Given its exposure and accessibility we sought to determine if K289 represented a PTEN ubiquitination site. Ubiquitination is catalyzed by the sequential action of an E1 enzyme that transfers activated Ubiquitin to an E2 ligase. This, in concert with an E3 ligase mediates ubiquitination of target proteins, usually on several variable target lysines, thus defining a system of utmost importance to cancer biology (Hershko and Ciechanover, 1998; Hoeller et al., 2006). We have identified NEDD4-1 as the major E3 ubiquitin ligase of PTEN (Wang et al., submitted) and therefore tested if NEDD4-1 ubiquitinates K289 in an in vitro assay. We used purified PTEN as a substrate and lysine-free ubiquitin (KØ-Ub) to prevent poly-Ub chain formation and promote ubiquitination of all available sites on PTEN, visible as discrete bands in PTEN western blotting. As shown in Figure 3A (left lanes, wt), incubation of PTEN with the E2 ligase alone showed 3 specific adducts, likely representing single-, dual- and triple-mono-ubiquitinated forms of PTEN, while addition of NEDD4-1 revealed at least 7 discrete adduct forms of PTEN, consistent with the role of NEDD4-1 as E3 ligase. When carrying out this assay with the purified PTENK289E mutant we found that at least one adduct band was missing, strongly suggesting that K289 is a target-lysine for PTEN ubiquitination (Figure 3A, right lanes K289E).
Figure 3. K13 and K289 are major sites of PTEN ubiquitination.
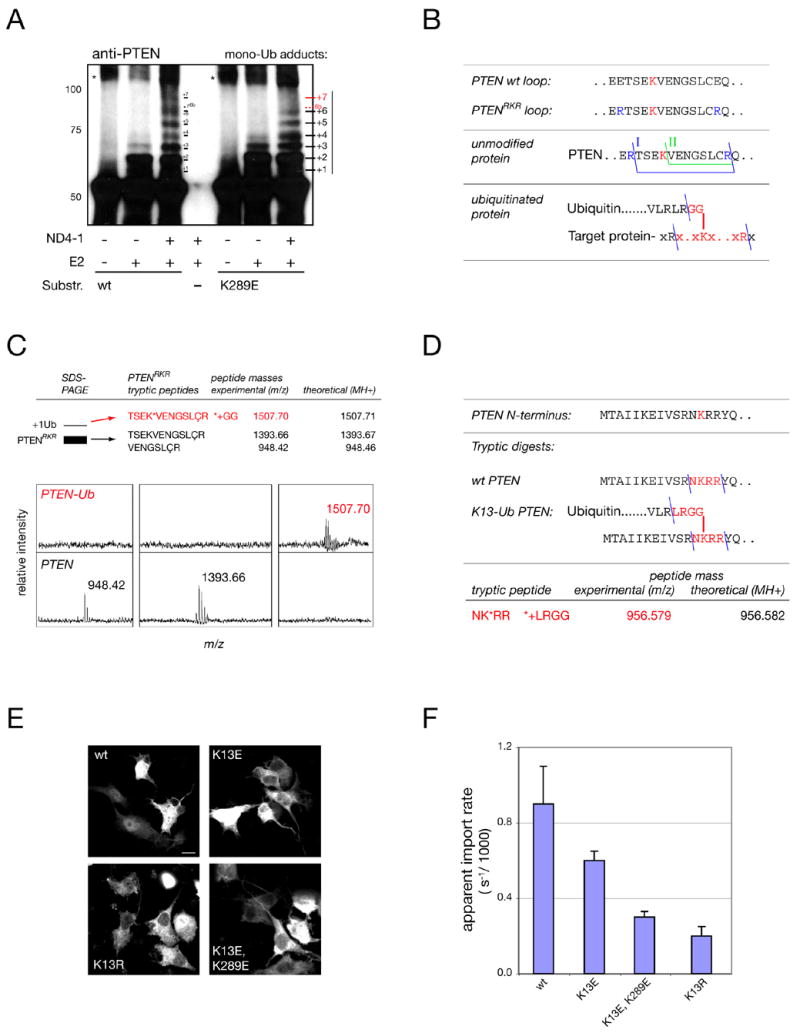
(A) In vitro PTEN mono-ubiquitination assays using NEDD4-1 E3 ligase (ND4-1) reveal several target sites in wt PTEN and loss of at least one corresponding target site(s) (“+7/ 6b”) in the K289E mutant (red). Note that numbering reflects only an estimate of the true adduct-number. Asterisks denote non-Ub specific aggregates. Molecular weights in kD.
(B) Top: location of engineered tryptic sites for mass spectrometry identification in PTENRKR. Middle: the two expected tryptic C2-loop fragments for PTENRKR (I, II). Bottom: expected fragment for K289-ubiquitinated PTENRKR (red).
(C) Mass spectrometry of PTENRKR and the first adduct band reveals the expected fragments for unmodified PTEN and PTEN ubiquitinated at K289 (upper panels, red). Lower panels show spectra of unmodified and modified PTEN with masses. ‘Ç’ denotes cysteine-acryl-amide (see Experimental Procedures).
(D) Mass spectrometry of immunoprecipitated PTEN after overexpression in cells reveals the peptide expected for a tryptic Ub adduct on lysine 13.
(E) Immunofluorescence staining of gfp-tagged K13 mutants. Scale bar, 10 μm.
(F) FRAP and quantification of the apparent import rates of mutants visualized in (E). Error bars show S. D.
Since lack of ubiquitination in this system could also be due to an indirect effect of the mutation, we sought to confirm K289 ubiquitination directly by Mass-Spectrometry (MS). However, our attempts of identifying even the unmodified tryptic fragment containing the K289 residue by MS failed since the tryptic loop fragment was too large to be retrieved in sample preparation (see Experimental Procedures). Therefore, we engineered two tryptic target sites flanking K289 into the C2-loop to produce PTENRKR (Figure 3B, top panel), which was indistinguishable from wt PTEN regarding enzymatic activity (Supplemental Figure S1C). GST-fused PTENRKR was ubiquitinated in vitro, enriched and purified by GST-pull down and, after SDS-PAGE/ coomassie staining, the fast migrating unmodified PTENRKR (below referred to as PTEN) and the first shifted band (termed +1) were both excised and processed for MS analysis. As intended, two expected tryptic fragments of the C2-loop (see Figure 3B, middle panel, fragments “I” and “II”) were now clearly detectable by analysis of the PTEN band, demonstrating that the main PTEN coomassie band contained only the unmodified C2 loop (Figure 3C, see upper panels for masses and lower panels for MS-spectra). Analysis of the +1 band in contrast, revealed a higher mass peptide of 1507.70 Daltons, as predicted for the loop-peptide linked by branching to two additional glycine residues, the leftover of a Ub adduct after tryptic digestion (Figure 3C, PTEN-Ub, and Figure 3B, lowest panel). Thus, K289 is a target lysine for PTEN ubiquitination, coherent with its structural exposure in the C2-loop.
A second, N-terminal ubiquitination site is mutated in cancer and regulates PTEN import
To test for the presence of ubiquitin adducts on PTEN-residues in cells, samples obtained from PTEN overexpression/IP in PC3 cells were processed for MS, which resulted in the identification of another UB-adduct site, PTEN lysine-13 (K13, see Experimental Procedures). The molecular weight of the tryptic T-peptide including K13 was measured at 956.579 Dalton (Figure 3D). Similar to the internal C2-loop, the N-terminal 13 residues of PTEN are also unstructured and exposed (Lee et al., 1999) and thus form targets for post-translational modification. Strikingly, K13 of PTEN is also found mutated to glutamic acid (K13E) in spontaneous cancer (Duerr et al., 1998). To test if this mutation also affects PTEN shuttling we analyzed its localization. As shown in Figure 3E, PTENK13E was enriched in the cytoplasm and FRAP analysis revealed a defect in nuclear import as well as export (Figure 3F, not shown). Notably, as shown in Supplemental Figure S1D, K13 is also highly conserved.
Since K13 is part of a positively charged cluster (RNKRR), which could potentially function as an SV-40 type NLS for PTEN, the K13E mutation could simply be disrupting a basic NLS by replacing a positive charge with the negative charge of glutamic acid. We therefore tested whether the charge conserving but ubiquitination abolishing K13R mutation could still impeach import. Also this mutant displayed strong cytoplasmic accumulation due to a nuclear import defect (Figures 3E–F, K13R), demonstrating that the positively charged patch does not function as PTEN-specific NLS but instead that the presence of the lysine residue is required, consistent with requirement for its ubiquitination to regulate import.
Our experiments with the K13E, K289E double mutant revealed that the two lysine residues can cooperate in promoting PTEN import (Figure 3F, K13E,K289), consistent with the notion that they are part of the limited set of ubiquitin target lysines of PTEN (revealed in Figure 3A).
Taken together, we have identified 2 lysine residues, which function as ubiquitination sites, are essential for PTEN shuttling and import, and are implicated in hereditary (K289E) and spontaneous (K13E) cancer.
PTEN ubiquitination in vivo
Next, we asked to what extent PTEN ubiquitination was occurring in vivo. While endogenous PTEN is efficiently mono- and poly-ubiquitinated upon overexpression of His-tagged ubiquitin (Wang et al., submitted), overexpressed PTEN was found similarly ubiquitinated in cells: Western blot analysis of lysates stained for PTEN revealed the appearance of two PTEN-specific slower migrating bands (Figure 4A) which increased over time (using two different antibodies, see Experimental Procedures). In addition, a smear was seen at later time points, as expected for poly-ubiquitination. The discrete bands co-migrated with various tagged forms of PTEN as expected for covalently linked adducts (Figure 4B, see also Supplemental Figure S1E for lower exposure). To test if these adducts where indeed formed by ubiquitin (Ub) we co-expressed both lysine-free (KØ-Ub) and wt Ub plasmids together with gfp-PTEN followed by immunoprecipitation (IP). As shown, over-expression of PTEN and KØ-Ub resulted in several discrete mono-Ub adducts, bands that were even more prominent upon co-expression of unmodified Ub (Figure 4C, left panel, 1–3) due to the nature of that plasmid (see Experimental Procedures, Figure 5C, and Supplemental Figure S1F for expression efficiencies and levels of the Ub plasmids). Ub overexpression also prompted a strong non-resolvable smear of poly-ubiquitinated PTEN (Figure 4C, left panel, poly). Analyzed vis-à-vis the PTEN input, the discrete bands coincided with the shifted bands of PTEN, in a pattern similar to the one shown above, but revealing an additional adduct (termed +1) (Figure 4C, right panel). Our results therefore showed that similar to endogenous PTEN (Wang et al., submitted), overexpressed PTEN forms mono- as well as poly-ubiquitin adducts in cells.
Figure 4. K13 and K289 are ubiquitinated in vivo.

(A) PTEN shows adduct formation in vivo and a smear (poly) at 36 hours post transfection. Molecular weights in kD. ‘n’ denotes non-specific band and asterisks indicate migration of unmodified (single) and modified PTEN (double).
(B) Discrete adducts co-migrate with various PTEN-fusions. Asterisks as in (A). Molecular weights in kD.
(C) Immunoprecipitation of gfp-PTEN after HA-Ubiquitin co-expression confirms discrete mono- (labeled 1–3) and poly-Ub adducts (labeled poly). Migration of gfp-PTEN is shown. Molecular weights in kD.
(D) Low steady state levels (left panel), strong ubiquitination (middle panel) and PTEN-specific ubiquitination (right panel) of the gfp-PTEN-UbKØ fusion. Molecular weights in kD.
(E) Defects in mono-ubiquitination of the indicated lysine mutants. Left panel shows overview (location of stacking gel and molecular weights are indicated) and right panels show magnification and quantification of ratios for mono-Ub : main PTEN-band intensities.
Figure 5. Mono-ubiquitination regulates PTEN nuclear import and shuttling.
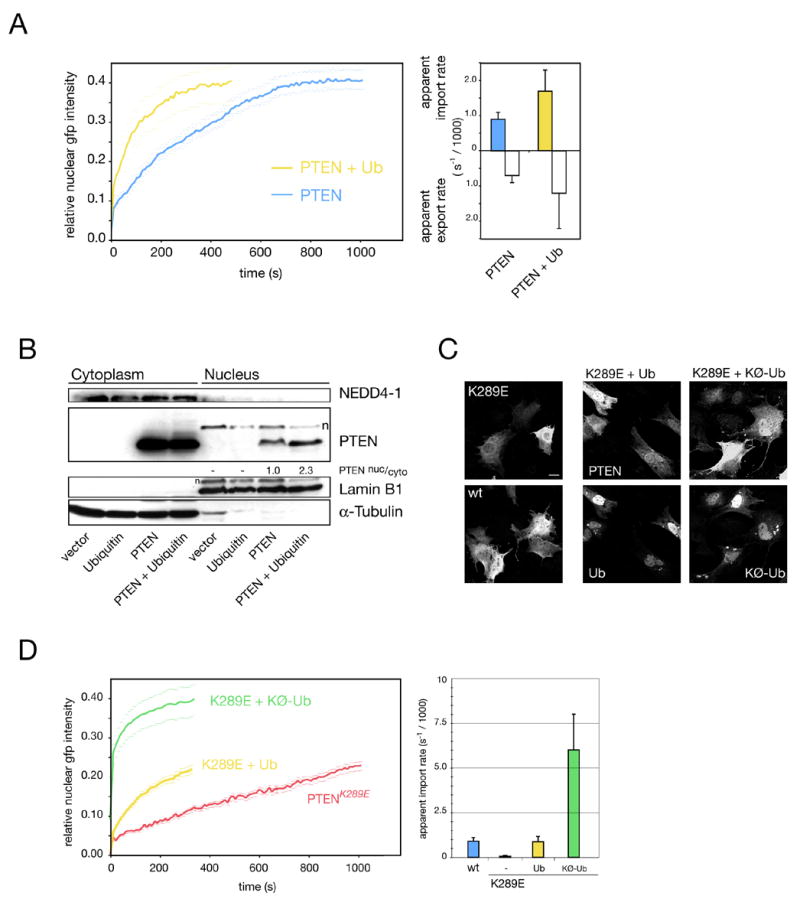
(A) FRAP assay measuring effect of ubiquitin overexpression on PTEN import (left panel) and quantification of nuclear and cytoplasmic PTEN accumulation rates (right panel). Error bars are S. D.
(B) Nuclear/ cytoplasmic fractionation of PTEN after ubiquitin overexpression. Lamin B1 and α-Tubulin serve as nuclear and cytoplasmic markers, respectively. Numbers denote nuclear to cytoplasmic PTEN ratios (relative to Lamin and Tubulin, see Supplemental Data). A non PTEN-specific band is indicated (n).
(C) Immunofluorescence imaging of PTEN and K289E with ubiquitin co-expression. Scale bar, 10 μm.
(D) Ubiquitination rescues K289E nuclear import defect as measured by FRAP (left panel; quantifications: right panel). Note that mono-ubiquitination is most efficient in rescuing. Error bars show S. D.
Next, we sought to determine if the lysine mutants show a ubiquitination defect. To this end we made use of a gfp-PTEN-KØ-ubiquitin (gfp-PTEN-UbKØ) fusion protein (see Experimental Procedures). As shown in Figure 4D, this PTEN construct is present at lower levels in cells due to massive ubiquitination (for unknown reasons) resulting in its degradation, yet shows no effect on general ubiquitination (right panel). This tool allowed us to study PTEN-ubiquitination directly by enhancing ubiquitination of PTEN exclusively, in contrast to Ub-overexpression or proteasome inhibition, which enhance ubiquitination of all cellular Ub target proteins.
To test the contribution of K289 and K13 to PTEN ubiquitination cells were transfected with the gfp-PTEN-UbKØ-K289E and gfp-PTEN-UbKØ-K13E mutants, respectively. As shown (Figure 4E), the wt fusion protein displayed a distinct mono-Ub band as well as a poly-Ub smear (note molecular weights), which characteristically extended into the stacking gel. Notably, the mono-Ub specific shift (Figure 4E, mono-Ub) was clearly reduced in both the K13 and K289 mutants (see magnification and quantification in Figure 4E, right panels) confirming the function of these lysines as important targets for PTEN mono-ubiquitination in vivo. Note that forced Ub overexpression in contrast always rescued PTEN mutant ubiquitination, as expected given the number of possible target sites (not shown, see also Figures 5C,D below).
Mono-ubiquitination dictates PTEN nuclear import
Ubiquitin has previously been linked to protein transport (Li et al., 2003; Lohrum et al., 2001; Massoumi et al., 2006; Plafker et al., 2004) and we sought to determine if it could affect PTEN shuttling by FRAP. Ub co-expression resulted in a significant acceleration of PTEN nuclear import (Figure 5A, left panel). Quantification revealed a greater than 50% increase in apparent import rates and export rates (Figure 5A, right panel), suggesting that PTEN-ubiquitination may enhance shuttling. Moreover, we found that the nuclear resident fraction of PTEN was also increased upon Ub over-expression (Figure 5B, compare PTEN and PTEN + Ubiquitin in nuclear fractions). Notably, nuclear PTEN was not quantitatively mono-ubiquitinated, suggesting that PTEN is de-ubiquitinated to remain nuclear or else re-exported. In sum, our results demonstrated that ubiquitination positively affects PTEN shuttling, complementing the finding of reduced ubiquitination and transport of the PTEN mutants.
To test if the nuclear import defect of the K289E mutant could be overcome by forced ubiquitination, we co-expressed PTENK289E with ubiquitin and assessed PTEN localization by IF at 12 hours post transfection. As shown in Figure 5C, PTENK289E was enriched in the cytoplasm while wt PTEN had equilibrated into the nucleus at this time point. Ubiquitin co-expression in contrast, resulted in nuclear accumulation of the PTENK289E mutant (Figure 5C, K289E + Ub). Since the Ub plasmid promotes both PTEN mono-and poly-Ub formation (see Figure 4C), we next tested if the KØ-Ub construct, which only supports mono-ubiquitination, was also able to rescue import. Indeed, cells co-transfected with the K289E mutant and KØ-Ub displayed very clear nuclear PTENK289E staining, strongly suggesting that mono-Ub suffices to rescue the import defect. To confirm and quantify these results, we performed rescue experiments using FRAP. Figure 5D shows that co-expression of PTENK289E and the Ub-plasmid similarly enhanced the overall import of the mutant (Figure 5D, left panel, K289E + Ub). But most importantly, co-expression of the KØ-Ub mutant accelerated PTENK289E import far more efficiently (K289E + KØ-Ub). Quantification of this rescue series showed that while Ub reset the K289E defect to approximately wt PTEN import levels, co-expression of KØ-Ub was seven-fold more powerful (right panel, K289E + KØ-Ub).
Next, we sought to determine if endogenous PTEN localization could be affected by lack of ubiquitination. To this end we used the temperature sensitive cell line ts20 in which ubiquitination is strongly reduced due to E1-ligase disruption at the restrictive temperature (Chowdary et al., 1994). As shown (Figure 6A), Pten localization at the permissive temperature was strongly nuclear in these NIH 3T3-derived cells, similar to the corrected control sub-line H35 before or after temperature shift and similar to endogenous Pten localization in mouse embryonic fibroblasts (MEFs, Figure 6B). But already 18hrs after the temperature shift Pten started to accumulate in the cytoplasm leading to a strong cytoplasmic stain by 36 and 60 hrs, consistent with an import defect of newly synthesized Pten. As expected, the cells also revealed an aberrant Ub-localization pattern at 36 and 60 hrs (Figure 6A, right panels).
Figure 6. NEDD4-1 dictates PTEN localization in murine and human cells.

(A) Endogenous Pten shows an import defect in ts20 cells after shift to the restrictive temperature for times indicated. Scale bars, 10μm.
(B) Endogenous Pten shows cytoplasmic accumulation after knockdown of Nedd4-1 in MEF cells. Scale bars, 10μm.
(C) Nedd4-1 siRNA efficiently reduces Nedd4-1 levels resulting in a Pten increase in MEFs.
(D) NEDD4-1 siRNA and overexpression also dictate localization of overexpressed gfp-PTEN in HeLa cells. Arrows point at cell nuclei. Note perinuclear PTEN accumulation upon NEDD4-1 knockdown. Scale bars, 10 μm.
(E) FRAP measurement of gfp-PTEN-Ub fusion reveals import defect of poly-ubiquitinated PTEN and its dominant cytoplasmic localization by IF (insert). Error bars show S.D. Scale bar, 10 μm.
(F) Fractionation and western analysis of gfp-PTEN and its fusion to ubiquitin (gfp-PTEN-Ub-KØ). Note that the mono-ubiquitination band is the dominant form of PTEN that is specifically enriched in the nuclear fraction (+mono-Ub).
To test if the Ub-mediated PTEN transport was indeed due to direct ubiquitination of PTEN we studied the role of the PTEN-targeting E3-ligase, NEDD4-1, in PTEN localization. Transient transfection of MEF cells with control siRNA showed dominant nuclear Pten and strong cytoplasmic Nedd4-1 localization (Figure 6B). Nedd4-1 knockdown, in contrast, led to a redistribution of Pten into the cytoplasm. Thus, we were able to phenocopy the effect of E1-ligase deficiency on PTEN localization by disruption of Nedd4-1. Importantly, Nedd4-1 mediated Pten import in addition to Pten degradation (see Figure 6C and Discussion). Similarly, knockdown of NEDD4-1 led to a cytoplasmic and perinuclear accumulation of exogenous PTEN in HeLa cells, while co-overexpression with gfp-PTEN resulted in strong nuclear PTEN (Figure 6D). Since NEDD4-1 in addition to PTEN poly-ubiquitination also mediates its mono-ubiquitination in vitro and in vivo (Wang et al., submitted) we conclude that PTEN mono-ubiquitination at discrete lysines including K13 and K289 directly regulates its import and shuttling. Consequently, the small pool of discretely shifted mono-ubiquitinated PTEN shown in Figures 4A–C represents the steady state PTEN fraction that is shuttling-competent.
Poly-ubiquitination in contrast resulted in cytoplasmic PTEN retention: the gfp-PTEN-Ub-KØ mutant, which was characterized by strong poly-ubiquitination in SDS-PAGE analysis (Figures 4D–E), showed import defects when assayed by FRAP and cytoplasmic accumulation in IF (Figure 6E). To determine nucleo-cytoplasmic distribution of the clearly visible ubiquitinated forms of this fusion, nuclear and cytoplasmic fractions of these cells were analyzed by western blotting. As expected (Figure 6F), poly-ubiquitinated PTEN adducts were exclusively cytoplasmic while the gfp-PTEN-UbKØ fusion itself displayed a distribution similar to gfp-PTEN. In contrast, the mono-ubiquitinated form, which largely depends on K13 and K289 availability (see Figure 4E), was specifically enriched in the nuclear fraction (10-fold compared to non-modified PTEN, see quantification in Supplemental Figure S1G), confirming that mono-ubiquitinated PTEN is shuttling most efficiently. Notably, these results also suggested that the pseudo-ubiquitination, which fuses ubiquitin in frame to PTEN (gfp-PTEN-Ub-KØ), does not increase PTEN shuttling and thus cannot mimic the effect of ubiquitination on the true target sites.
Nuclear PTEN is stable, active and rarely found in late stage colon cancer
We next sought to determine why loss of nuclear PTEN localization could be oncogenic. To this end we first determined PTEN half-life after forced localization to the nuclear or cytoplasmic compartment using PTEN-NLSSV-40 or PTEN-NESPKI (see Supplemental Figure S1A, lowest panels). While wild-type PTEN, which as demonstrated above is both nuclear and cytoplasmic, showed a half-life of roughly 7.5 hours (Figure 7A), forced cytoplasmic accumulation led to a clear decrease in stability (t1/2= 4.5 hrs), while nuclear localization resulted in increased stability (t1/2= 15 hrs) as predicted since NEDD4-1 is mostly cytoplasmic (see Figures 5B, 6B & D). To test nuclear PTEN activity, we next measured its ability to antagonize AKT. As shown (Figure 7B), transfection of increasing amounts of PTEN into PTEN-deficient PC3 cells resulted in 75 percent reduction (compared to the control) in the phospho-AKT/ AKT ratio. But surprisingly, forced nuclear PTEN was still able to antagonize AKT activation, similar to forced cytoplasmic (unstable) PTEN.
Figure 7. Nuclear PTEN is stable and active in vivo and reduced in late stage colon carcinoma.
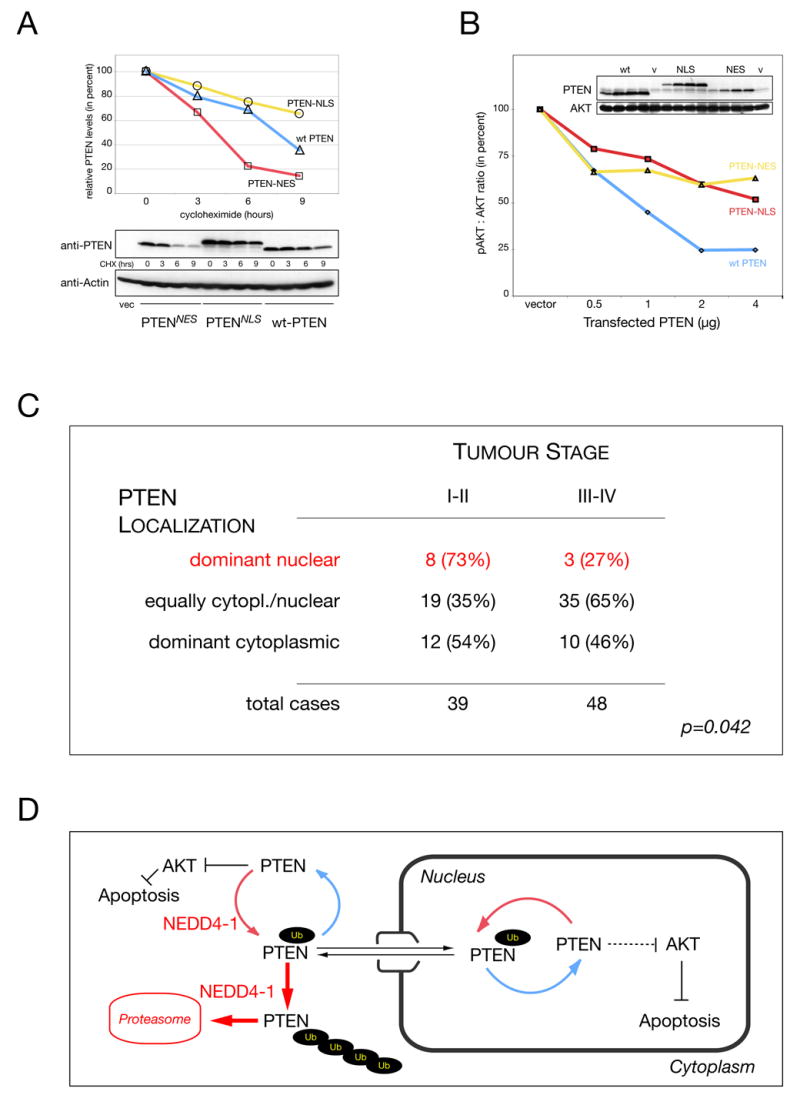
(A) Half-life of PTEN after forced nuclear (-NLS) or cytoplasmic overexpression (-NES) compared to wt PTEN in presence of cycloheximide (CHX). Densitometry quantification (PTEN : actin, upper panels) and raw data (lower panels). Control transfection is indicated (vec.).
(B) Titration of nuclear versus cytoplasmic PTEN activity. Quantification of pAKT : AKT ratio and PTEN expression levels (inserts). ‘v’ indicates vector control transfection. PTEN levels are shown and AKT is used as loading reference.
(C) PTEN localization in Tumor Tissue Microarray (TTM) with specimens of 87 colon cancer patients reveals positive correlation between nuclear PTEN localization and low tumor grade. Statistical significance is indicated (p, see also Experimental Procedures).
(D) Model for regulation of PTEN import and shuttling versus PTEN degradation. PTEN is ubiquitinated in the cytoplasm by NEDD4-1, which allows for PTEN import. Alternatively, PTEN is ubiquitinated further in the cytoplasm and degraded by the proteasome. Nuclear PTEN can shuttle back to the cytoplasm or after de-ubiquitination remains nuclear and protected from cytoplasmic degradation. Importantly, nuclear PTEN is not only protected but still able to antagonize AKT and cause apoptosis. Thus, PTEN specific Ubiquitin ligases and proteases regulate PTEN localization, function and stability.
Next, we tested if nuclear PTEN was also able to induce apoptosis. After transfection of PC3 cells, cleaved caspase-3 staining was performed and positive cells counted. PTEN-NES transfection into these p53-deficient cells resulted in an apoptotic index of 4.4 (+/− 1.7) positive cells per 1000 (at 48 hours), while nuclear PTEN was as efficient with 4.8 positive cells (+/− 1.3 per 1000), similar to PTEN transfection (3.1 +/− 1.0 per 1000) and in contrast to vector transfection (0.4 +/− 0.0 per 1000). In sum, our data showed that nuclear PTEN is more stable, can inhibit AKT and is able to induce p53-independent apoptosis.
Finally, to test the general relevance of nuclear PTEN in colon, we analyzed a tumor tissue micro-array with specimens of 87 colon cancer patients for tumor grade and PTEN localization (PTEN negative samples were significantly associated with advanced tumor stage, p < 0.022, and omitted for this analysis). As summarized in Figure 7C, we found that samples with high nuclear PTEN staining significantly associated with less advanced pathological stage. In addition, an increase in even nucleo-cytoplasmic PTEN distribution was associated with higher tumor stage, consistent with previous findings (Zhou et al., 2002). Taken together, these data were in agreement with the notion that tumors, which can retain a strong nuclear PTEN staining, remain of lower grade.
DISCUSSION
The molecular makeup of spontaneous cancers at the clinical stage usually presents a complex mixture of primary and consequential events, some of which are thought to contribute to essential aspects of tumor progression (Hanahan and Weinberg, 2000). Cancer syndrome associated mutations in contrast, often constitute well-defined primary tumorigenic events that are causal to tumor formation (Nagy et al., 2004). Specifically, germ-line mutation of PTEN has been shown to underlie the closely related autosomal dominant tumor susceptibility syndromes Cowden disease and Bannayan-Riley-Ruvalcaba syndrome (Eng, 2003). Therefore, we anticipated that PTEN germline mutations hold valuable information and novel insights into PTEN function in tumor initiation. While most of the described mutations result in truncation many PTEN missense mutations affect its catalytic activity. Through investigating instead how the enzymatically functional PTENK289E mutant could be at the root of inheritable cancer, we have come to several novel conclusions:
I) We have discovered that deficiency in PTEN import causes cancer susceptibility, independent of catalytic activity. While it has been previously shown that the PTENK289E mutant retains catalytic activity (Georgescu et al., 2000), its nuclear import defect has so far remained elusive. Correlation of the K289E import defect with the observed cytoplasmic accumulation in the relevant Cowden patient tissue thus represents the first example of a causal relation between PTEN localization and cancer initiation. Since no such certainty on the cause-and-effect relation exists in sporadic mutations like PTENK13E (which as published also suffers reduction in catalytic activity (Walker et al., 2004)), the K289E mutation allows us to unambiguously ascribe a tumor suppressive function to PTEN nuclear localization. In agreement with this finding we find significant correlation between low tumor stage in colon cancer and dominant nuclear PTEN localization.
II) We show that nuclear PTEN import is regulated by mono-ubiquitination, which to our knowledge represents the first example of target protein ubiquitination enhancing nuclear import (and shuttling). PTEN poly-ubiquitination, in contrast leads to its cytoplasmic retention and degradation (Ciechanover, 2005) and is therefore oncogenic in nature (see also(Wang et al., submitted). Thus PTEN mono-ubiquitination and subsequent nuclear import has a protective function, a highly relevant finding for the discussion of NEDD4-1 in tumorigenesis (see below). As mentioned, ubiquitin charging of some E2-ligases has been shown to regulate their nuclear import (Plafker et al., 2004), and intriguingly de-ubiquitination of the Bcl-3 transcription factor has been recently shown to cause its cytoplasmic accumulation (Massoumi et al., 2006). Yet, Bcl-3 cytoplasmic accumulation due to loss of nuclear (Ub-mediated) retention cannot be excluded, in contrast to PTEN where ubiquitination dictates the actual transport rates.
Mono-ubiquitination has previously been implied in p53 nuclear export (Li et al., 2003; Lohrum et al., 2001), but p53 also contains several well-characterized NLS and NES sequences that enhance transport (Michael and Oren, 2003). PTEN in contrast, may rely entirely on the ubiquitination system for shuttling since no transferable NLS-like sequences have been demonstrated for it and we have shown that the K13 cluster does not rely on charge for import. It will be interesting to see if PTEN shares ubiquitination-specific import receptors with the above-mentioned cargoes.
III) We identify K13 and K289 as two major and conserved sites for PTEN ubiquitination and we show that their mutation leads to a constitutive shuttling defect that can be overcome by forced mono-ubiquitination. Importantly, PIP2 phospholipid binding and allosteric PTEN activation functions have also been ascribed to the basic patch containing K13 (for a review see (Leslie and Downes, 2004)) and it will be interesting to see how these functions affect ubiquitination and vice versa.
Since further sites on PTEN can also be ubiquitinated both in vitro and in vivo, we hypothesize that mono-ubiquitination efficiency is a limiting step of PTEN shuttling, and that ubiquitin proteases reverse this process and antagonize shuttling because the nuclear pool of PTEN is not quantitatively mono-ubiquitinated. Taken together, our data suggest a model (Figure 7D), in which cytoplasmic mono-ubiquitination of PTEN leads to either PTEN import or alternatively poly-ubiquitination and PTEN degradation (by NEDD4-1). Nuclear PTEN-Ub can shuttle back to the cytoplasm or after de-ubiquitination remains nuclear and protected from cytoplasmic degradation until it is again mono-ubiquitinated and exported.
IV) We demonstrate that PTEN nuclear localization is coupled to activity of the only known PTEN E3-ligase, NEDD4-1, which can both mono- and poly-ubiquitinated PTEN in vitro and in vivo. Since we here show that mono-ubiquitination is essential for PTEN function, Nedd4-1 effectively has both oncogenic (PTEN degradation) and tumor suppressive (PTEN shuttling) potential. Several scenarios how PTEN levels and localization are regulated can therefore be envisioned: (i) NEDD4-1 modification/ protein association could switch it from mono- to poly-ubiquitination mode, (ii) NEDD4-1 localization could dictate PTEN levels in a particular compartment (in poly-ubiquitination mode) and/ or transport out of the compartment (in mono-ubiquitination mode). In this respect it is interesting to note that although many cells show dominant cytoplasmic NEDD4-1, some cell lines and certain murine tissues show strong nuclear NEDD4-1 (Trotman and Pandolfi, unpublished). Significantly, the above implications for NEDD4-1 localization and activity apply equally to PTEN de-ubiquitinases, which we propose to be essential for control of PTEN residence times and stability within the nuclear and cytoplasmic compartments as depicted (Figure 7D).
V) We find that nuclear PTEN is not only protected but still able to antagonize AKT and cause apoptosis. These findings are in line with our observation of dominant nuclear PTEN localization in early stage but not advanced stage colon cancer. Together with our observations in the Cowden patient tissue, these data suggest that retention of the nuclear import capability of PTEN is a critical tumor-suppressive determinant and it will be interesting to see if other patient derived PTEN mutations also target PTEN import instead of catalytic activity.
Taken together, we have through an integrative approach, linked the genetics of a cancer syndrome with the biochemistry and cell biology of protein modification to demonstrate how ubiquitination regulates nuclear import and tumor progression through PTEN. We therefore propose that ubiquitin is a decisive regulator of this major tumor suppressor and that further defining this process is crucial to understanding and developing ubiquitination-related cancer therapies.
EXPERIMENTAL PROCEDURES
Plasmids and protein purification
PTEN cloned into pcDNA3.1 (Lee et al., 1999) was subcloned into pEGFP-C2 and its derivatives constructed as described in the Supplemental Data section. The wt Ubiquitin plasmid contains 8 tandems of HA-ubiquitin under the CMV promotor. For protein production PTEN, PTENK289E and PTENRKR were expressed via baculovirus as detailed in the Supplemental Data.
In vitro ubiquitination assays
Mono-ubiquitination reactions of wild-type PTEN and K289E mutant were carried out at 30 °C for 2 hours in 40mM Tris-HCl, pH7.5, 2mM DTT, 5mM MgCl2, using 10μg of lysine-free ubiquitin mutant (Boston Biochem, Cat# UM-NOK), 40nM human E1 (Boston Biochem, Cat# E302), 8μM UbCH5c, 5mM ATP (Sigma, Cat#A-7699), 300ng of recombinant PTEN or GST-PTEN-RKR, and 2μl purified NEDD4-1 (see (Wang et al., submitted)). See Supplemental Data for detailed information.
Cells, Western Blotting and Immunocytochemistry
For immunofluorescence, cells were prepared using standard preparation and confocal analysis procedures detailed in Supplemental Data. The ts20 cell line was a kind gift from Dr. H Ozer (New Jersey Medical School). Nedd4-1 knockdown was performed in p53−/−MEFs using the pSuper-Retra-LacZ (LacZi) or pSuper-Retra-Nedd4-1 (iN4A) plasmids (see (Wang et al., submitted) and Supplemental Data). For protein half-life, cells were treated with 30μg/ml cycloheximide as previously described (Chen et al., 2005). Half-life extrapolation, nucleo-cytoplasmic fractionation, apoptosis assays and immunoprecipitations were carried out using the standard methods described in the Supplemental Data section.
Photobleaching in vivo import/export assays
PC3 cells were plated, transfected with Lipofectamine 2000 and observed in LabTekII chambers (Nalgene) at 24 hours post transfection (identical results were obtained at 36 hrs, data not shown). Import/export assays were performed on a Zeiss LSM 510 confocal microscope and evaluated as described (see references and detailed methods in Supplemental Data).
Mass spectrometry
Gel-resolved proteins “PTEN RKR” (control) and “Ub-PTEN RKR” derived from the ubiquitination reactions were digested with trypsin, batch purified on a reversed-phase micro-tip, and resulting peptide pools individually analyzed by matrix-assisted laser desorption/ionization reflectron time-of-flight (MALDI-reTOF) mass spectrometry (UltraFlex TOF/TOF; BRUKER; Bremen, Germany) for peptide mass fingerprinting as described in Supplemental Data.
Immunohistochemistry on patient samples and tumor tissue microarrays
Cowden patient colon sample slides were stained for PTEN using the monoclonal 6H2.1 (Cascade, Figure 1D) and polyclonal Ab-2 (Nemoarkers) as previously described (Trotman et al., 2003). Tissue microarray construction, its staining and scoring methods are detailed in Supplemental Data.
Statistical analysis, sequence alignments and PTEN-modeling
Statistical significance calculations, PTEN sequence alignments and modeling are described in the Supplemental Data section
Supplementary Material
Acknowledgments
We would like to thank Q. Li, S. Mabon and M. S. Jiao for help with insect cell culture, FRAP and pathology analysis, respectively. P. Bonner for data management, L. Lopez and E. Iskidarova for technical assistance in tissue micro-array construction, I. Linkov and M. Asher and the Pathology Core Laboratory at MSKCC for help with their expertise in IHC and A. Nazarian for help with mass spectrometry. This research was supported by the NIH Grant R01-CA-82328 to P.P.P. and the NCI Cancer Center Support Grant P30-CA-08748 to P.T. and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
ABBREVIATIONS
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PIP3
Phosphatidylinositol 3,4,5,-trisphosphate
- Wt
wildtype
- S.D
standard deviation
Missense mutation nomenclature uses amino acid single letter code of wt residue followed by position, followed by missense mutant amino acid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat. 2000;16:109–122. doi: 10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SG, Kim HJ, Park BJ, Min HJ, Park JH, Kim YW, Dong SH, Kim BH, Lee JI, Chang YW, et al. Mutational abrogation of the PTEN/MMAC1 gene in gastrointestinal polyps in patients with Cowden disease. Gastroenterology. 1998;115:1084–1089. doi: 10.1016/s0016-5085(98)70078-2. [DOI] [PubMed] [Google Scholar]
- Chook YM, Blobel G. Karyopherins and nuclear import. Curr Opin Struct Biol. 2001;11:703–715. doi: 10.1016/s0959-440x(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A. PTEN mutations in gliomas and glioneuronal tumors. Oncogene. 1998;16:2259–2264. doi: 10.1038/sj.onc.1201756. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033–7038. [PubMed] [Google Scholar]
- Gorlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6:776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Lian Z, Di Cristofano A. Class reunion: PTEN joins the nuclear crew. Oncogene. 2005;24:7394–7400. doi: 10.1038/sj.onc.1209089. [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–8532. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Behrendt N, Chen Z, Noda T, Hino O, Cordon-Cardo C, Pandolfi PP. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005;19:1779–1786. doi: 10.1101/gad.1314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, Lindboe CF, Fryns JP, Sijmons RH, Woods DG, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–1387. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- Plafker SM, Plafker KS, Weissman AM, Macara IG. Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J Cell Biol. 2004;167:649–659. doi: 10.1083/jcb.200406001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Cristofano AD, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Dyke TV, et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Pandolfi PP. PTEN and p53: who will get the upper hand? Cancer Cell. 2003;3:97–99. doi: 10.1016/s1535-6108(03)00022-9. [DOI] [PubMed] [Google Scholar]
- Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J. 2004;379:301–307. doi: 10.1042/BJ20031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Chen Z, Koppie T, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. Identification of the Proto-Oncogenic Ubiquitin Ligase for PTEN. doi: 10.1016/j.cell.2006.11.039. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman DC, Zhou XP, Cummings MC, Pavey S, Hayward NK, Eng C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int J Cancer. 2002;99:63–67. doi: 10.1002/ijc.10294. [DOI] [PubMed] [Google Scholar]
- Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, Aaltonen LA, Eng C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol. 2002;161:439–447. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.