

Abstract
PACSIN family members regulate intracellular vesicle trafficking via their ability to regulate cytoskeletal rearrangement. These processes are known to be involved in trafficking of GLUT1 and GLUT4 in adipocytes. In this study PACSIN3 was observed to be the only PACSIN isoform that increases in expression during 3T3-L1 adipocyte differentiation. Overexpression of PACSIN3 in 3T3-L1 adipocytes caused an elevation of glucose uptake. Subcellular fractionation revealed that PACSIN3 overexpression elevated GLUT1 plasma membrane localization without effecting GLUT4 distribution. In agreement with this result, examination of GLUT exofacial presentation at the cell surface by photoaffinity labeling revealed significantly increased GLUT1, but not GLUT4, after overexpression of PACSIN3. These results establish a role for PACSIN3 in regulating glucose uptake in adipocytes via its preferential participation in GLUT1 trafficking. They are consistent with the proposal, which is supported by a recent study, that GLUT1, but not GLUT4, is predominantly endocytosed via the coated pit pathway in unstimulated 3T3-L1 adipocytes.
Keywords: adipocyte, endocytosis, GLUT1, GLUT4, PACSIN3, membrane trafficking, syndapin
The need to transport glucose from outside to inside the cell is a fundamental feature of cellular survival and growth. In mammals glucose transport of the facilitated diffusion type is mediated by a group of transporters known as the GLUT family (SLC2A gene symbol). The GLUT’s comprise a 13-member family of 12-trans-membrane spanning 50–60 kD proteins [1]. Among these, GLUT1 and GLUT4 are of central relevance to peripheral glucose disposal and have been extensively studied. Nevertheless, the mechanisms involved in controlling the trafficking of GLUT1 and GLUT4 to and from the plasma membrane, and the differences between the trafficking of the two GLUT’s, are not completely understood [2, 3]. It is possible that proteins involved in integrating cytoskeletal rearrangements with membrane trafficking participate in these mechanisms. Recent studies have identified adaptor proteins that do coordinate membrane trafficking events with alterations in the cytoskeleton. Major examples are the amphiphysin, cortactin, endophilin, intersectin and syndapin/PACSIN families [4–7]. These proteins contain multiple protein-protein interaction domains, which allow them to simultaneously interact with proteins involved in vesicle formation and cytoskeletal regulation [8–10].
In this study we have examined the role of the PACSIN family in the trafficking of GLUT1 and GLUT4 in 3T3-L1 adipocytes. There are three PACSIN isoforms. Expression of PACSIN1 is restricted to neural tissues, and PACSIN2 is ubiquitously expressed. PACSIN3 is primarily expressed in skeletal muscle and heart [4, 11]. Each member is known to participate in endocytosis [11, 12]. Overexpression of PACSIN isoforms in NIH 3T3 fibroblasts inhibits transferrin uptake [11]. Introduction of the SH3 domain of PACSIN results in filamentous actin accumulation and impairment in endocytosis, as assayed by apical plasma membrane accumulation of α-adaptin and clathrin [5]. Here we show that PACSIN3 participates in the trafficking of GLUT1, but not GLUT4, in unstimulated 3T3-L1 adipocytes.
Materials and Methods
Materials
Antibodies against the following proteins were purchased from: PACSIN1 (sc10412) and PACSIN2 (sc10417) (Santa-Cruz Biotechnology, Santa-Cruz, CA); PACSIN3 (ab2226) (Abcam, Cambridge, MA); myc-4A6 and Cbl-7G10 (Upstate Biotechnology, Lake Placid, NY); actin-AC-40 (Sigma, St. Louis, MO); adeno-V (NB 600-403) and Na+K+ATPase (NB 300-146) (Novus Biologicals, Littleton, CO); c-Cbl-pY700 (612304) (BD-Transduction Labs, Palo Alto, CA); Akt (9272), Akt-Thr308 (9275), Akt-Ser473 (9271), c-Cbl-pY774 (3555) (Cell Signaling, Beverly, MA). The anti-GLUT4 (829) and anti-GLUT1 (674e) antibodies have been described previously [13]. Biotinylated bis-glucose analogue, bio-ATB-BGPA (bio-ATB-BGPA, 4,4-O-[2-[2-[2-[2-[2-[6(biotinylamino)-hexanoyl]-amino]ethoxy]exoxy]ethoxy]-4-(1-azi-2,2,2-trifluoroethyl)benzoyl]amino-1,3propanediyl]bis-D-glucose), was generously provided by Geoffrey D. Holman (University of Bath, UK).
Construction of adenoviruses, cell culture and viral infection
PACSIN3 constructs were generated by polymerase chain reaction (PCR) amplification from wild-type PACSIN3 in pcDNA3.1 for subcloning in pShuttleCMV (Stratagene, La Jolla, CA) using HindIII and XbaI at the 5’ and 3’ restriction sites, respectively. A myc-tag (coding sequence GAGCAGAAGCTCATCTCGGAGGAGGACCTC) corresponding to amino acids 411–420 of human Myc (EQKLISEEDL) was added at the amino-terminus of each construct immediately preceded by the start codon (ATG). These constructs were digested with PmeI and recombined into the pAdEasy vector using BJ5183 E. coli. Adenoviral DNA constructs were subsequently transfected into 293HEK cells. Infected 293HEK cells lysed 12–14 days following the transfection. Crude viral lysates were isolated and used to infect additional 293HEK cells. Infected cells were isolated 24 h post-infection and lysates were analyzed with SDS-PAGE to test for PACSIN3 construct protein expression. Subsequent large-scale virus production was performed using forty 10 cm dishes of 293HEK cells.
3T3-L1 cells were maintained as fibroblasts in 20% calf-serum. Upon reaching confluence, cells were maintained in 20% calf-serum for two additional days. Cells were then differentiated as using 10% fetal bovine-serum supplemented with insulin (2.4 nM), 3-isobutylmethylxanthine-1-methyl (0.5 mM) and dexamethasone (250 nM). Two days following the initiation of differentiation, cells were fed with 10% fetal bovine-serum supplemented with insulin (2.4 nM). Adipocytes were subsequently maintained in 10% fetal bovine-serum.
Overexpression of PACSIN3 and its variants in 3T3-L1 adipocytes was achieved by adenovirus infection. Adenoviral constructs were first titered by infecting 3T3-L1 adipocytes at day 4–5 of differentiation with increasing amounts of virus, with beta-galactosidase (βgal) virus as a control. SDS samples of cells were prepared at 48 h post-infection, and immunoblotted for the viral hexon protein. The latter was quantitated using ECLPlus (Amersham Biosciences), and plots comparing the volume of viral stock to adenovirus protein expression were generated for all virus stocks (data not shown). Subsequently 3T3-L1 adipocytes at day 4 or 5 were infected with equal titers of the adenoviruses for the various PACSIN3 constructs.
Isolation of purified plasma membranes
3T3-L1 adipocyte plasma membranes were isolated as previously described [14]. In brief, 3T3-L1 adipocytes were serum starved in DMEM for several hours. Cells were then treated as indicated, washed with ice-cold fractionation buffer (50 mM Hepes, pH 7.4, 1 mM EDTA, 255 mM sucrose) supplemented with protease inhibitor cocktail (1 μg/ml leupeptin, 1 μg/ml antipain, 1 μg/ml benzamidine, 5 μg/ml trypsin inhibitor, 1 μg/ml chymostatin, 1 μg/ml pepstatinA, and 0.5 mM phenylmethylsulfonyl fluoride), and homogenized. Nuclei were pelleted by centrifugation at 2,000 rpm for 5 min. The post-nuclear supernatant was centrifuged at 11,500 rpm for 20 min in a SS34 rotor. The pellet was re-suspended in fractionation buffer and re-spun at 11,500 rpm in a SS34 rotor. The pellet was then re-suspended and loaded onto a sucrose cushion (50 mM Hepes, pH 7.4, 1 mM EDTA, 1.12 M sucrose, protease inhibitor cocktail) and spun at 25,000 rpm in a SW41 rotor for 60 min. The “fluffy” layer on top of the cushion was collected and re-suspended in a larger volume of fractionation buffer, followed by pelleting at 18,500 rpm for 20 min in a SS34 fixed angle rotor. This pellet is the plasma membrane (PM) fraction.
Glucose transport assay
Control and infected 3T3-L1 adipocytes were allowed to recover for three days following the infection protocol. Cells were then serum starved for several hours in DMEM prior to initiation of the uptake assay. The assay was performed as previously described [14]. In brief, the cells were incubated for 30 min in DMEM with or without 1 μM insulin for 30 min at 37° C. Cells were then washed in 37° C Krebs Ringers Phosphate (KRP) buffer followed by incubation in 37° C KRP buffer with or without 40 μM cytochalasinB (CB). Cells were immediately placed on a 37° C water bath shaker. [3H]-2-deoxyglucose was added sequentially to each dish, and uptakes were performed for 6 min. Reactions were quenched by drawing off media and washing 3x with ice cold KRP. Cells were lysed in 1% triton, and the incorporated 2-deoxyglucose measured by scintillation counting. The values for uptake in the presence of CB were subtracted from those in its absence to correct for nonspecific uptake.
bio-ATB-BGPA affinity photolabeling assay
3T3-L1 adipocytes were plated into either 35 mm or 20 mm wells. Adipocytes at day 4–5 of differentiation were infected as described above, or left uninfected, and allowed to recover for three days. Adipocytes were then photoaffinity labeled as previously described [15]. Cells were serum starved in DMEM at 37° C for several hours. Cells were then either stimulated with 1 μM insulin or not for 30 min at 37° C, followed by washing in KRBH (136 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 10 mM Hepes pH 7.4) at 18° C. Cells were then incubated in KRBH with or without 40 μM biotin-conjugated ATB-BGPA, and immediately UV irradiated for 1 min in Rayonet RPR100 (The Southern New England UV Company, Branford, CT). The cells were then washed with KRBH, 1% bovine-serum albumin in PBS and 0.1% bovine-serum albumin in PBS. The cells were next solubilized with 1 ml 1% Thesit (nonaoctaethylene-dodecyl-ether) in PBS supplemented with protease inhibitor cocktail (see fractionation buffer), and the lysate cleared by centrifugation at 16,000 rpm. The biotinylated proteins were collected by incubation of the supernatant with 50 μl streptavidin agarose (Sigma, St. Louis, MO) overnight. The beads were washed, and the bound biotinylated proteins were released with SDS sample buffer.
SDS-PAGE and western blotting
SDS samples were separated on Novex Precast Bis-Tris gels (10% or 4–12% acrylamide) (Invitrogen, Carlsbad, CA). Protein was transferred onto nitrocellulose or polyvinyldene difluoride membranes and probed with the suitable primary antibody and horse radish peroxidase-conjugated secondary antibody. Immunoblots were exposed to Hyperfilm (Amersham, Piscataway, NJ). Quantification was performed using ECL+ (Amersham, Piscataway, NJ) and STORM 840 Phosphorimager or by scanning densitometry of films exposed in the linear range of the chemiluminescence signal and Adobe Photoshop 7.0 software.
Results
PACSIN3 increases in expression with adipogenesis
Three PACSIN isoforms have been characterized and shown to be expressed differentially across tissue types [11]. The expression of these proteins in 3T3-L1 cells has not previously been examined. We examined their expression throughout the differentiation of 3T3-L1 fibroblasts into adipocytes (Fig. 1). Cells were isolated as fibroblasts or at various days after initiation of differentiation. PACSIN1 was not expressed at detectable levels at any of the stages, but was detectable in a rat brain sample. PACSIN2 expression was detectable in both 3T3-L1 fibroblasts and adipocytes; however, its expression decreased during differentiation. In contrast, PACSIN3 increased in expression during the course of 3T3-L1 differentiation. GLUT1 and GLUT4 were used as controls. As expected GLUT1 was expressed in both fibroblasts and adipocytes, whereas GLUT4 was only expressed after differentiation. Since PACSIN3 increased on differentiation, we investigated its potential participation in GLUT trafficking.
Fig. 1. Expression of PACSIN isoforms during 3T3-L1 differentiation.

3T3-L1 cells were isolated as fibroblasts (F) or following incubation in differentiation media for 0, 2, 4, 6, 8 or 10 days. SDS samples (25 μg) were immunoblotted for the stated proteins. Brain homogenate was included as a positive control (+) for PACSIN1. Blots are representative of two independent experiments.
PACSIN3 overexpression elevates glucose uptake
In order to assess the role of PACSIN3 in glucose transport, recombinant adenoviral constructs were generated based on the postulated protein-protein interaction domains present in PACSIN3 (Fig. 2A). N-terminal myc tagged constructs were generated for wild-type PACSIN3 (WT), a C-terminal truncation which deleted the SH3 domain (ΔC354), a C-terminal truncation that deleted the proline rich domain (PXXP) as well as the SH3 domain (ΔC329), and an N-terminal deletion construct that removed the polybasic region (also called the F-BAR domain [16]) (ΔN244). Upon infection of adipocytes, each construct was expressed and migrated on SDS gels at its predicted molecular weight (data not shown). The level of overexpression of each construct was two to three times that of endogenous PACSIN3 protein, as assessed by immunoblotting (data not shown). We also generated an adenoviral construct of myc-tagged SH3 domain (ΔN347). Its expression was significantly lower than that of the other constructs (data not shown), and consequently, it was not further characterized.
Fig. 2. Overexpression of PACSIN3 in 3T3-L1 adipocytes promotes glucose uptake.

(A) Schematic diagram of the PACSIN3 constructs expressed through adenoviral expression. (B) 3T3-L1 adipocytes at day 4–5 after differentiation were infected with equal viral titers of beta-galactosidase (βgal), mycWTPACSIN3 (WT), mycΔC354PACSIN3 (ΔC354), mycΔC329PACSIN3 (ΔC329) or mycΔN244PACSIN3 (ΔN244) constructs; 3 days post-infection, cells were incubated with or without 1 μM insulin for 30 min, followed by the measurement of 2-deoxy-glucose uptake. *, p < 0.05 and **, p < 0.01 compared to the corresponding βgal control by Student T-test. Values are the mean ± standard error from three independent experiments.
Adipocytes were infected with adenovirus for each PACSIN3 construct or the beta-galactosidase (βgal)-expressing control virus. Subsequently, glucose transport assays were then performed in the absence and presence of insulin (Fig. 2B). In the unstimulated state overexpression of either the WT or the ΔC354 construct resulted in increases in glucose uptake of approximately 3-fold and 2-fold, respectively, compared to βgal control. Expression of ΔC329 also resulted in a 25% increase in basal glucose transport. In contrast to the N-terminal containing constructs, expression of the ΔN244 construct resulted in no significant change in glucose transport in comparison to βgal control. In the insulin-stimulated state, overexpression of WT PACSIN3 elevated glucose transport by 30% in comparison to βgal control. Overexpression of the other PACSIN3 constructs did not significantly affect insulin-stimulated glucose transport. The effect of overexpression of the WT PACSIN3 on 2-deoxyglucose transport in the presence of insulin may reflect the contribution of the higher basal transport, although the ΔC354 construct, which raised basal transport, did not enhance transport in the presence of insulin.
PACSIN3 increases GLUT1 content in the plasma membrane
Glucose transport in 3T3-L1 adipocytes is due to the action of both GLUT1 and GLUT4 in the plasma membrane [17]. We utilized two approaches to examine the plasma membrane content of GLUT1 and GLUT4: subcellular fractionation and photoaffinity labeling of surface transporters. Because the effect on glucose transport activity was observed following overexpression of WT PACSIN3 but not overexpression of the ΔN244 construct, the ΔN244 construct was included as a control in addition to the βgal virus.
Subcellular fractionation of infected 3T3-L1 adipocytes revealed that there was a significant increase in GLUT1 in the plasma membrane fraction upon WT PACSIN3 overexpression in both the basal and insulin-stimulated states, whereas there was no effect of the ΔN244 construct (Fig. 3A). Quantitation showed that WT elevated the amount of plasma membrane GLUT1 by 2.8 ± 1.0 (n = 5) and 1.9 ± 0.3 (n = 2) relative to the βgal control in the basal and insulin-stimulated states, respectively. Overexpression of WT PACSIN3 had no significant effect on plasma membrane GLUT4 (Fig. 3B). Consistent with its known function in inhibiting □transferrin uptake [11], WT PACSIN3 overexpression also elevated transferrin receptor in the plasma membrane by 2.3 ± 0.03 (n = 5) and 1.3 ± 0.2 (n = 3) in the basal and insulin-stimulated states, respectively, relative to the βgal control (Fig. 3C). As expected from earlier studies [2, 18], in the βgal control, insulin caused a substantial increase in plasma membrane GLUT1 (1.7 ± 0.2, n = 2) and GLUT4 (2.1 ± 0.6, n = 3), and a smaller increase in plasma membrane transferrin receptor (1.4 ± 0.1, n = 3) (Fig. 3A–C). The Na+K+ATPase, which was immunoblotted as a loading control, showed no change under any condition (Fig. 3D). Throughout, the cells with the ΔN244 construct showed the same distribution of GLUT1, GLUT4, transferrin receptor and ATPase as the βgal control (Fig. 3).
Fig. 3. PACSIN3 overexpression increases GLUT1 and transferrin receptor in the plasma membrane.

3T3-L1 adipocytes were infected with equal titers of βGal, WT or ΔN244 viruses. Three days post-infection, cells were either left untreated (−) or were stimulated with 1 μM insulin (+) for 30 min and fractionated as described in Materials and Methods; purified plasma membrane fractions were isolated. Protein was then separated by SDS-PAGE. For (A) GLUT1 and (B) GLUT4, 0.5 μg of protein was loaded per lane. For (C) transferrin receptor (Tfr), and the (D) Na+K+ATPase, 25 μg of protein was loaded per lane. Representative immunoblots are depicted. The values of the relative intensities on the immunoblots have been normalized to the value for the basal βgal control. *, p < 0.05 compared to the corresponding βgal control as determined by Student T-test . Values represent mean ± standard error from two to five experiments.
A limitation of subcellular fractionation is that it only assesses the total amount of protein associated with the plasma membrane rather than protein inserted in the plasma membrane. Likewise, it does not offer insight into the functional conformation of the glucose transporter. The only technique that assesses both exofacial plasma membrane content and the functional conformation of the glucose transporter is photoaffinity labeling [19]. We directly examined the effect of PACSIN3 on GLUT1 and GLUT4 using the photoaffinity label bio-ATB-BGPA. This reagent is a membrane-impermeant, glucose derivative with a photoactivateable diazirine molecule and a biotin group; hence, the amount of transporter isolated with immobilized streptavidin after photoactivation provides a measure of the relative amount in the plasma membrane [15]. Consistent with the fractionation findings, PACSIN3 overexpression resulted in a 2.1-fold elevation in the plasma membrane content of GLUT1 in the basal state as compared to βgal control (Fig. 4A). Overexpression also elevated insulin-stimulated GLUT1 exofacial plasma membrane accessibility by approximately 50% in comparison to βgal control. These differences were similar to the increases observed for both basal and insulin-stimulated glucose transport following PACSIN3 overexpression (Fig. 2B) and to the results of the subcellular fractionation (Fig. 3A). Overexpression ofΔN244 did not affect GLUT1 photolabelling in comparison to the βgal control (Fig. 4A). Basal and insulin-stimulated GLUT4 photolabelling were not significantly affected by PACSIN3 or ΔN244 overexpression, in comparison to the βgal control (Fig. 4B). As expected [2], insulin treatment elevated both GLUT1 and GLUT4 at the cell surface in the β-gal control (Fig. 4). In sum, these results show that overexpression of PACSIN3 elevates glucose transport by increasing the localization of GLUT1 at the plasma membrane.
Fig. 4. PACSIN3 overexpression promotes GLUT1 but not GLUT4 exofacial presentation at the plasma membrane.
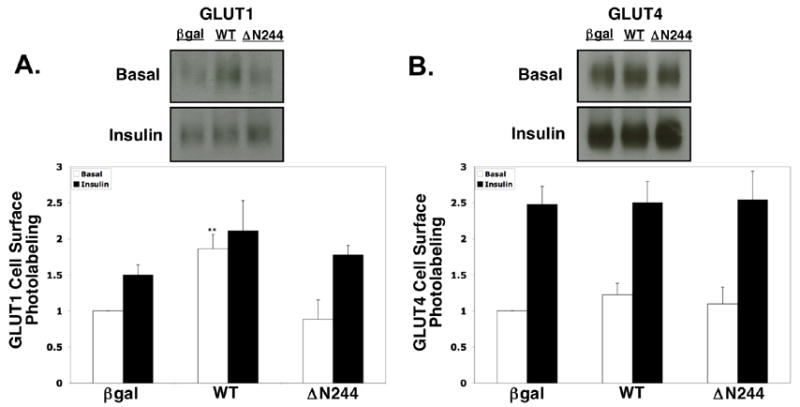
3T3-L1 adipocytes were infected with equal titers of βgal, WT or ΔN244 viruses. Three days post-infection, cells were treated with or without 1 μM insulin for 30 min and then UV irradiated for 1 min in the presence of 40 μM bio-ATB-BGPA as described in Materials and Methods. Equal volumes of each sample were separated by SDS-PAGE and immunoblotted with (A) anti-GLUT1 and (B) anti-GLUT4 antibodies. Immunoblots were quantified and relative labeling was calculated by setting basal βgal values as 1.0, Values within respective GLUT species were then calculated relative to the βgal basal signal. **, p < 0.01 compared to the corresponding βgal control. Values represent mean ± standard error from three independent experiments.
The increase in plasma membrane GLUT1 caused by PACSIN3 overexpression could potentially be due to an increase in the total cellular amount of GLUT1 rather than an effect on GLUT1 trafficking. However, immunoblotting SDS lysates for GLUT1 showed that PACSIN3 overexpression did not alter total cellular GLUT1 content (data not shown). We also examined the effect of PACSIN3 overexpression on total cellular GLUT4 and on insulin activation of the protein kinase Akt, which is required for GLUT4 translocation [3]. Immunoblotting for GLUT4 and for activated Akt with antibodies against the phosphorylated Thr308 and Ser473 showed that PACSIN3 had no effect on GLUT4 amount or Akt activation (data not shown).
Discussion
In this report we show that increased GLUT1 plasma membrane content is the basis for the effect of PACSIN3 overexpression on glucose transport. GLUT1 is known to recycle continuously between intracellular membranes and the plasma membrane [2]. Most likely, overexpression of PACSIN3 causes an inhibition of GLUT1 endocytosis, and thereby causes the increase in the plasma membrane. Previously, PACSIN3 overexpression has been shown to inhibit transferrin uptake, which proceeds via the transferrin receptor by the clathrin-coated pit pathway of endocytosis [11, 20]. In agreement with this finding, we found that PACSIN3 overexpression caused an increase in transferrin receptor in the plasma membrane. PACSIN3 appears to function as a protein that couples vesicle budding to actin polymerization associated with endocytosis in the clathrin-coated pit pathway [8, 11]. Thus, it might seem unexpected that overexpression of it inhibits endocytic processes. Most likely the explanation is that the alteration in the stoichiometry between PACSIN3 and other protein components of the endocytic machinery upon overexpression of PACSIN3 is inhibitory, possibly because the excess PACSIN3 sequesters a required protein. Whatever the detailed mechanism of this effect, it very likely does not depend on the SH3 domain of PACSIN3, since the construct with this domain deleted increased basal glucose transport to almost the same extent as overexpression of WT PACSIN3.
GLUT4 is also known to recycle to and from the plasma membrane in unstimulated 3T3-L1 adipocytes, albeit at a lower rate than GLUT1 [2]. Thus, a priori it is somewhat unexpected that PACSIN3 overexpression did not cause a significant shift of GLUT4 to the cell surface as well. However, while this manuscript was in preparation, a study appeared showing that GLUT4 endocytosis from the plasma membrane in unstimulated 3T3-L1 adipocytes occurs primarily by a cholesterol-dependent pathway that is distinct from the clathrin-coated pit pathway [21]. Hence, the difference between the response of GLUT1 and GLUT4 to PACSIN3 overexpression can be explained by the proposal that GLUT1, like the transferrin receptor, is primarily endocytosed via the clathrin-coated pit pathway, whereas GLUT4 is endocytosed by another pathway that does not involve PACSIN3.
Acknowledgments
This work was largely performed in the lab of Mike Mueckler at Washington University in St. Louis, and we are deeply indebted to Dr. Mueckler for his guidance. We are grateful to Geoffrey Holman for his generous gift of bio-ATB-BGPA. We would like to acknowledge Christopher Wolin for his insightful suggestions, and Matt Storck and Richard Hresko for their technical assistance, and Gus Lienhard for helpful advice. This work was supported by a scholarship to W.G.R. from the Lucille P. Markey foundation, and by grants to Mike Mueckler (NIH RO1 DK38495, NIH RO1 DK067229), and a grant from the American Diabetes Association (to M.M.). M.P. is supported by the Deutsche Forschungsgemeinschaft (DFG PL233/3-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joost H, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Shurmann A, Seino S, Thorens B. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab. 2001;282:E974–E976. doi: 10.1152/ajpendo.00407.2001. [DOI] [PubMed] [Google Scholar]
- 2.Yang JH, Holman GD. Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J Biol Chem. 1993;268:4600–4603. [PubMed] [Google Scholar]
- 3.Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev. 2004;25:177–204. doi: 10.1210/er.2003-0011. [DOI] [PubMed] [Google Scholar]
- 4.Wasiak S, Quinn CC, Ritter B, de Heuvel E, Baranes D, Plomann M, McPherson PS. The Ras/Rac guanine nucleotide exchange factor mammalian Son-of-sevenless interacts with PACSIN 1/Syndapin I, a regulator of endocytosis and the actin cytoskeleton. J Biol Chem. 2001;276:26622–26628. doi: 10.1074/jbc.M100591200. [DOI] [PubMed] [Google Scholar]
- 5.da Costa SR, Sou E, Xie J, Yarber FA, Okamoto CT, Pdigeon M, Kessels MM, Mircheff AK, Schechter JE, Qualmann B, Hamm-Alvarez SF. Impairing actin filament or syndapin functions promotes accumulation of clathrin-coated vesicles at the apical plasma membrane of acinar epithelial cells. Mol Biol Cell. 2003;14:4397–4413. doi: 10.1091/mbc.E03-05-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schafer D. Coupling actin dynamics and membrane dynamics during endocytosis. Curr Opin Cell Biol. 2002;14:76–81. doi: 10.1016/s0955-0674(01)00297-6. [DOI] [PubMed] [Google Scholar]
- 7.Simpson HN, Qualmann B, Kelly RB, Kay BK, McPherson PS, Schmid SL. SH3-domain-containing proteins function at distinct steps in clathrin-coated vesicle formation. Nat Cell Biol. 1999;2:119–124. doi: 10.1038/10091. [DOI] [PubMed] [Google Scholar]
- 8.Kessels M, Qualmann B. The syndapin protein family: linking membrane trafficking with the cytoskeleton. J Cell Sci. 2004;117:3077–3086. doi: 10.1242/jcs.01290. [DOI] [PubMed] [Google Scholar]
- 9.McPherson P. Regulatory role of SH3 domain-mediated protein-protein interactions in synaptic vesicle endocytosis. Cell Signal. 1999;11:229–238. doi: 10.1016/s0898-6568(98)00059-x. [DOI] [PubMed] [Google Scholar]
- 10.Kessels M, Qualmann B. Syndapin oligomers interconnect the machineries for endocytic vesicle formation and actin polymerization. J Biol Chem. 2006;281:13285–13299. doi: 10.1074/jbc.M510226200. [DOI] [PubMed] [Google Scholar]
- 11.Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci. 2000;113:4511–4521. doi: 10.1242/jcs.113.24.4511. [DOI] [PubMed] [Google Scholar]
- 12.Qualmann B, Kelly R. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J Cell Biol. 2000;148:1047–1062. doi: 10.1083/jcb.148.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haney P, Slot JW, Piper RC, James DE, Mueckler M. Intracellular targeting of the insulin-regulatable glucose transporter (GLUT4) is isoform specific and independent of cell type. J Cell Biol. 1991;114:689–699. doi: 10.1083/jcb.114.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hresko RC, Heimberg H, Chi MMY, Mueckler M. Glucosamine-induced insulin resistance in 3T3-L1 adipocytes is caused by depletion of intracellular ATP. J Biol Chem. 1998;273:20658–20668. doi: 10.1074/jbc.273.32.20658. [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto M, Hatanaka Y, Yang J, Dhesi J, Holman GD. Synthesis of biotinylated bis(D-glucose) derivatives for glucose transporter photoaffinity labelling. Carbohydr Res. 2001;331:119–127. doi: 10.1016/s0008-6215(01)00025-8. [DOI] [PubMed] [Google Scholar]
- 16.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Liao W, Nguyen MTA, Imamura T, Singer O, Verma IM, Olefsky JM. Lentiviral short hairpin ribonucleic acid-mediated knockdown of Glut4 in 3T3-L1 adipocytes. Endocrinol. 2006;147:2245–2252. doi: 10.1210/en.2005-1638. [DOI] [PubMed] [Google Scholar]
- 18.Tanner L, Lienhard G. Insulin elicits a redistribution of transferrin receptors in 3T3-L1 adipocytes through an increase in the rate constant for receptor externalization. J Biol Chem. 1987;262:8975–8980. [PubMed] [Google Scholar]
- 19.Yang J, Clark AE, Kozka IJ, Cushman SW, Holman GD. Development of an intracellular pool of glucose transporters in 3T3-L1 cells. J Biol Chem. 1992;267:10393–10399. [PubMed] [Google Scholar]
- 20.Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983;97:329–339. doi: 10.1083/jcb.97.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blot V, McGraw TE. GLUT4 is internalized by a cholesterol-dependent nystatin-sensitive mechanism inhibited by insulin. The EMBO Journal advance online publication. 2006:1–11. doi: 10.1038/sj.emboj.7601462. [DOI] [PMC free article] [PubMed] [Google Scholar]