

SUMMARY
Archaeal RNA polymerases (RNAPs) are most similar to eukaryotic RNAP II (Pol II) but require the support of only two archaeal general transcription factors, TBP (TATA-box binding protein) and TFB (archaeal homologue of the eukaryotic general transcription factors TFIIB) to initiate basal transcription. However, many archaeal genomes encode more than one TFB and/or TBP leading to the hypothesis that different TFB/TBP combinations may be employed to direct initiation from different promoters in Archaea. As a first test of this hypothesis, we have determined the ability of RNAP purified from Thermococcus kodakaraensis (T.k.) to initiate transcription from a variety of T.k. promoters in vitro when provided with T.k. TBP and either TFB1 or TFB2, the two TFBs encoded in the T.k. genome. With every promoter active in vitro, transcription initiation occurred with either TFB1 or TFB2 although the optimum salt concentration for initiation was generally higher for TFB2 (~250 mM K+) than for TFB1 (~200 mM K+). Consistent with this functional redundancy in vitro, T.k. strains have been constructed with the TFB1- (tfb1; TK1280) or TFB2- (tfb2; TK2287) encoding gene deleted. These mutants exhibit no detectable growth defects under laboratory conditions. Domain swapping between TFB1 and TFB2 has identified a central region that contributes to the salt sensitivity of TFB activity, and deleting residues predicted to form the tip of the B-finger region of TFB2 had no detectable effects on promoter recognition or transcription initiation but did eliminate the production of very short (≤ 5 nt) abortive transcripts.
Keywords: Archaea, transcription factor B, promoter recognition, RNA polymerase
Introduction
Archaea are prokaryotes, but the components of their transcription machinery are much more similar to their eukaryotic rather than bacterial counterparts 1; 2; 3. In Bacteria, a single RNA polymerase (RNAP) core enzyme (β′ βα2ω) forms 1:1 complexes with different sigma (σ) factors forming different RNAP holoenzymes that recognize and bind to different promoters. Bacterial promoter recognition and RNAP binding is dependent primarily on direct interactions between the σ-factor and promoter sequence, and σ-factor competitions dictate gene expression patterns in Bacteria 4. Most regulators of bacterial transcription either block (repressors) or stimulate (activators) RNAP binding to promoter sequences, and many function through interactions with the σ-factor 5. Eukaryotes, in contrast, have three distinct RNAPs, designated Pol I, II and III, dedicated to transcribing genes that encode rRNAs, proteins, and tRNAs and other small RNAs, respectively. These RNAPs do not directly recognize and bind to promoter sequences but are recruited to the appropriate promoters through associations with RNAP-specific general transcription factors. Pol II basal transcription initiation, for example, usually requires the participation of TBP/TFIID/SAGA, TFIIA, TFIIB, TFIIE, TFIIF, TFIIH, and involves direct interactions between Pol II and TFIIB 6.
Archaea have only one RNAP that, in terms of subunit sequences and composition, are most similar to Pol II 3 and studies with purified archaeal components have established that accurate transcription initiation in vitro requires only archaeal RNAP, TBP, and TFB (the archaeal homolog of TFIIB) 7; 8; 9; 10. A third transcription factor designated TFE (homologue of the α-subunit of eukaryotic TFIIE) has been shown to stimulate transcription from some promoters under non-optimal in vitro conditions 11;12; 13. Archaeal genomes do not encode homologues of the β-subunit of TFIIE, or homologues of the other Pol II general transcription factors but do often encode more than one TBP and/or TFB with some halophilic Archaea have the capacity to synthesize as many as 12 different TFBs and 8 TBPs 14; 15; 16; 17. This has led to the hypothesis that different TBP and TFB combinations may direct an archaeal RNAP to initiate transcription at different promoters. In essence, this is conceptually the same as different σ factors directing bacterial RNAPs to transcribe different groups of genes 15. Apparently consistent with this concept, most archaeal transcription regulators investigated to date function more like bacterial than eukaryotic regulators and directly block or enhance TFB/TBP binding to BRE-TATA region of an archaeal promoter 9. This very simple and attractive hypothesis, that different TBP/TFB combinations direct differential gene expression in Archaea as do different σ factors in Bacteria, has not been previously tested experimentally.
Progress in understanding archaeal transcription has been severely limited by the lack of genetics, and so an inability to validate results in vivo obtained in vitro. Several in vitro transcription systems have been established that use defined, purified components 18; 19; 20; 21; 22, but these have not been from Archaea with facile genetics. Recently, a simple gene knock-out technology was developed for Thermococcus kodakaraensis (T.k.)23, and as the genome of this marine hyperthermophilic archaeon encodes two TFBs (designated TFB1 and TFB2), this offered an opportunity to combine genetic and in vitro biochemical approaches to investigate if the two TFBs in an Archaeon directed transcription initiation from different promoters. To do so, we established in vitro transcription using purified T.k. RNAP and TBP and either TFB1 or TFB2, and assayed the ability of TFB1 and TFB2 to support transcription initiation from a variety of different T.k. promoters. Every promoter that was active in vitro, was active with both TFB1 and TFB2 and, consistent with this overlap of TFB1 and TFB2 functions in vitro, we were able to isolate T.k. mutants with the TFB1-encoding gene (TK1280) or TFB2-encoding gene (TK2287) deleted. These deletion mutants grow well, with no detectable negative phenotypes on both minimal and rich media. The optimum salt conditions for TFB1- versus TFB2-supported transcription in vitro do differ with the optimum salt condition for TFB1 being generally lower than those for TFB2. Domain swapping has identified a region involved in this salt dependence. To further probe the hypothesis that archaeal TFBs and bacterial σ factors are functional homologues, a TFB2 variant was generated with residues deleted from the tip of the B-finger region predicted to be structurally equivalent to the tip of region 3.2 of σ70. The tip of σ70 region 3.2 helps position the initiating NTPs for catalysis, but we demonstrate that the deletion variant of TFB2 supports transcription initiation with only a minimal defect in promoter clearance. Overall, the results reported are not consistent with TFB1 and TFB2 functioning in T.k. promoter selection as do alternative σ factors in Bacteria. This could, however, still be the case for the more divergent TFB and TBP isoforms present in other Archaea.
Results
Establishing, optimization and properties of the T.k. in vitro transcription system
Aliquots of purified T.k. RNAP and RNAP-HA were subjected to SDS-PAGE, and each of the sub-unit polypeptides was isolated and identified by MS-MS (Figure 1). The polypeptides present in RNAP-HA were identical to those in RNAP except for the RpoL-HA subunit that migrated more slowly during electrophoresis consistent with the addition of a 9 amino acid C-terminal hemagglutinin (HA) tag. The composition of the HA tag was confirmed by MS-MS. Purified RNAP and RNAP-HA had essentially the same activities in vitro in terms of promoter usage, abundance of transcripts synthesized, elongation rates and activities with TFB1 or TFB2. The subunit composition of RNAP-HA immunoprecipitated from lysates of T.k. (rpoL-HA) was the same as that of RNAP-HA purified by column chromatography (data not shown). Two polypeptides co-purified with T.k. RNAP, and these were identified by MS-MS as TFE, encoded by TK2024, and a CRISPR-associated protein (CSA2) 24, encoded by TK0462. TFE is also present in archaeal RNAP preparations that we have purified from Methanothermobacter thermautotrophicus (M. t.)20 hinting at a common, likely functional association of TFE with RNAP in Archaea. To date, we have not however detected any effect of adding additional purified TFE or CSA2 to in vitro transcription reaction mixtures containing T.k. components (data not shown).
Figure 1. Polypeptide subunit compositions of T.k. RNAP and RNAP-HA.
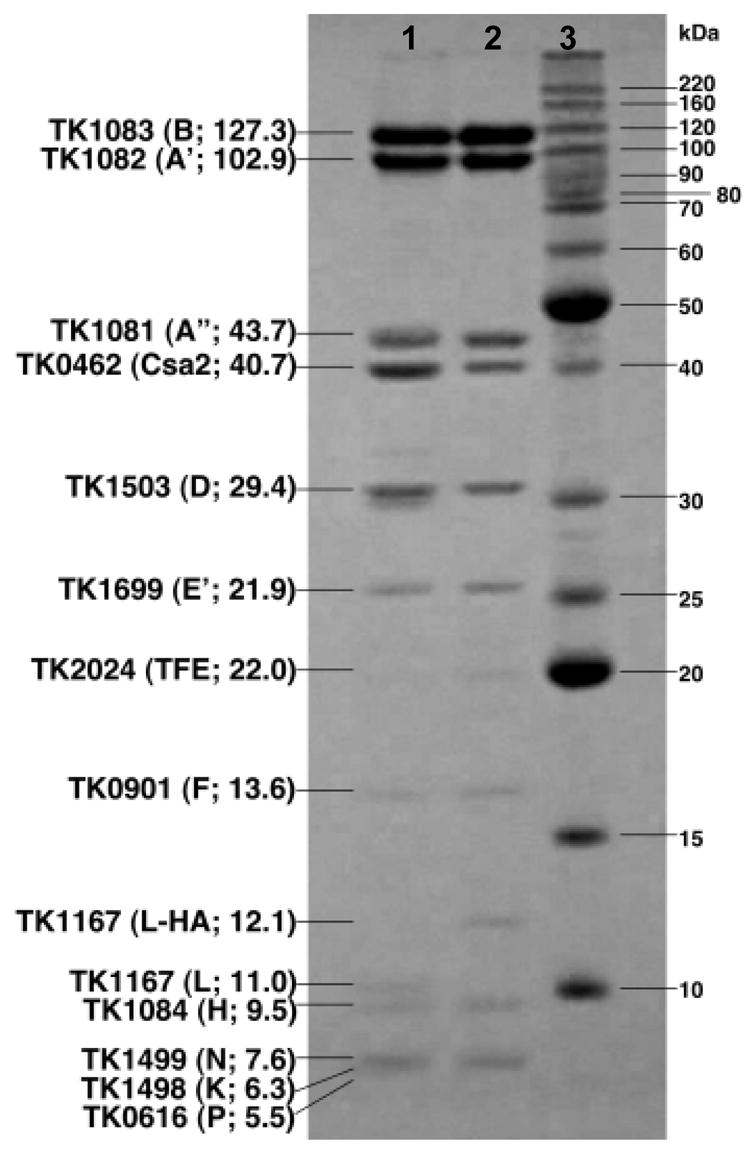
The polypeptides present in aliquots of purified RNAP (lane 1) and RNAP-HA (lane 2), separated by SDS-PAGE and Coomassie stained adjacent to molecular mass standards (lane 3). The encoding gene, single letter subunit designation, and molecular mass (in Kda) of each subunit are listed. The two proteins that co-purify with T.k. RNAP (TFE and Csa2) are identified.
Recombinant T.k. TBP was purified with the native sequence whereas the recombinant TFB1 and TFB2 preparations had N-terminal 6-histidine (6-his) extensions that facilitated their purification. A linear DNA template with the T.k. gdh (TK1431) promoter was used first to establish and then optimize promoter-dependent in vitro transcription using T.k. RNAP, TBP and TFB1 or TFB2. Transcription from this promoter (and all other T.k. promoters tested) by T.k. RNAP was found to be absolutely dependent on the presence of T.k. TBP and either T.k. TFB1 or TFB2 (Figure 2a). With both TFB1 and TFB2, transcription initiation occurred 22 bp downstream from the 3′-nucleotide of the TATA-box sequence (data not shown). Promoter-specific transcription did not occur when the TBP and/or TFB were from M.t. rather than from T.k.. Adding poly dI/dC or heparin at concentrations up to 80 μg/ml or 100 μg/ml, respectively, did not prevent re-initiation (data not shown) and multi-round transcription resulted in the linear accumulation of run-off gdh transcripts for at least 15 min at all temperatures from 50°C to 85°C. DNA template melting most likely imposed the upper temperature limit for transcription in vitro (Figure 2b). A T.k. protein designated TIP26 (TK1172) has been reported to influence T.k. TBP-DNA interactions 25; 26 and to form a complex with TBP+TFB2 bound to DNA 27. Adding recombinant TIP26 had no detectable effects on transcription in vitro by T.k. RNAP from several different promoters whether supported by TFB1 or TFB2 even when added at an ~16-fold molar excess over the DNA template (data not shown).
Figure 2. Optimization of the T.k. in vitro transcription system.
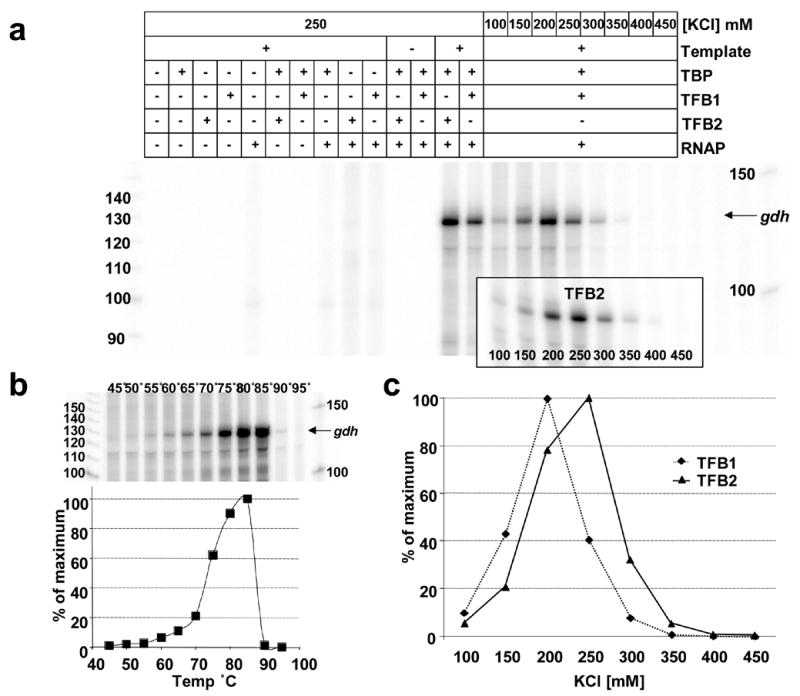
a. Electrophoretic separation of the run-off transcripts synthesized from a linear template initiated from the T.k. gdh promoter in reaction mixtures incubated at 85°C for 20 min. The +/− symbols above each lane identify the salt concentration and transcription factors present in that reaction mixture. The boxed insert shows the transcript synthesized in reaction mixtures that contained T.k. RNAP, TBP and TFB2 with the KCl concentration listed below the corresponding lane. b. Electrophoresis of the run-off transcripts synthesized from the gdh promoter during incubations for 20 min at the temperature listed above each lane in reaction mixtures containing TFB2. The graph shows the amount of transcript synthesized, measured by phosphorimaging, expressed as a percentage of the maximum synthesized. Transcript synthesis directed by TFB1 exhibited the same temperature dependent profile. c. The amounts of run-off transcript that accumulated during incubation for 20 min at 85°C in reaction mixtures that contained TFB1 (♦) or TFB2 (▲) at increasing KCl concentrations graphed as percentages of the maximum amount of transcript synthesized.
Pyrococcus woesei, a close relative of T.k., contains ~800 mM potassium di-myo-inositol-1,1′ phosphate and DNA binding by TBP from P. woesei requires 200 mM K+ 28. Salt concentration was therefore a key parameter for optimization and, as expected, transcription in vitro required a relatively high salt concentration. But, surprisingly, the optimum salt concentration for transcription from the gdh promoter was higher in reaction mixtures that contained TFB2 (250 mM KCl) versus TFB1 (200 mM KCl) (Figure 2c). The optimum salt concentration was subsequently found to differ for most promoters for TFB1- versus TFB2-dependent transcription (see below; Figure 4) and some T.k. promoters were active only with TFB2 at salt concentrations >250 mM KCl.
Figure 4. Salt dependence of promoter-directed transcription in reactions supported by TFB1 or TFB2.
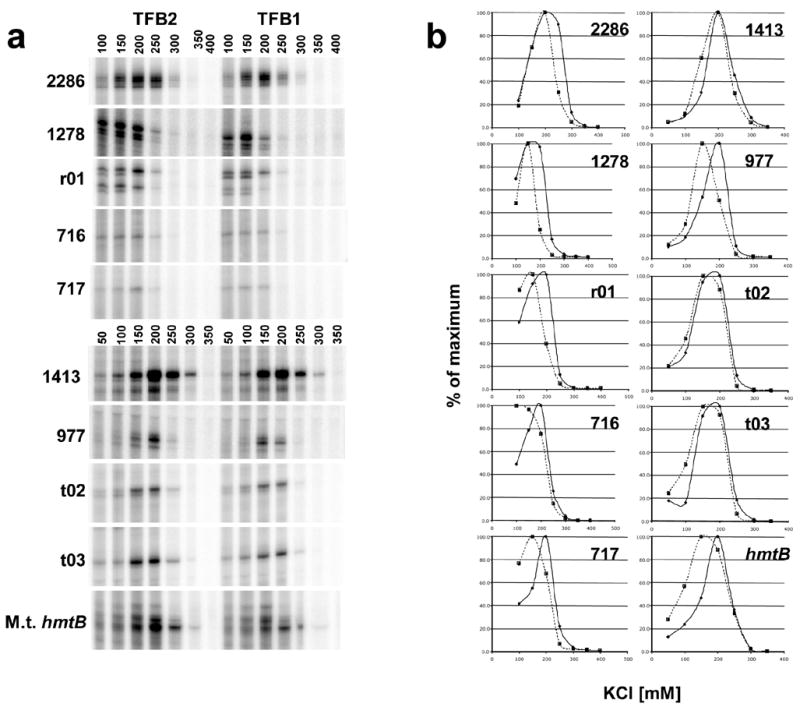
a. Electrophoretic separations of the run-off transcripts accumulated during 20 min incubation at 85°C in reaction mixtures that contained a template with the promoter for a T.k. protein encoding gene (numerical designations listed to the left of the image), or rRNA (rt02) or tRNA (t02, t03) gene. Transcripts were also synthesized from the heterologous promoter of the M.t. histone-encoding hmtB gene The KCl concentration (mM) in the reaction mixture is listed above the corresponding gel lane. b. The amounts of transcript accumulated at each KCl concentration are graphed as a percentages of the maximum amount synthesized in reaction mixtures supported by TFB1 (broken lines) or TFB2 (solid lines).
TFB1 and TFB2 do not exhibit promoter discrimination
Having established that transcription of the gdh gene that encodes an abundant housekeeping enzyme was supported by either TFB1 or TFB2, we considered it possible that TFB1 and TFB2 might differentially support transcription from promoters of protein- versus stable RNA-encoding genes, or of genes that encode proteins present in T.k. but not in a close relative (Pyrococcus abyssi) that has only one TFB, or of the genes that encode themselves and so exhibit autoregulation. However, as illustrated in Figure 3, in every case that a T.k. promoter was active in vitro, transcription was supported by either TFB1 or TFB2 and initiation always occurred at the same site (data not shown). Using the amount of run-off transcript that accumulated during 20 min at 85°C as the assay, every T.k. promoter tested had a different activity in vitro. Most were more active under optimized conditions with TFB2 than with TFB1, and optimization always involved establishing the salt-dependence of transcription for TFB1 and TFB2 (Figures 4A and 4B).
Figure 3. TFB1- and TFB2-dependent transcription from different T.k. promoters.

The run-off transcripts synthesized from linear templates carrying the promoter(s) for the T.k. genes listed, in reaction mixtures that contained TFB2 (in 250 mM KCl) or TFB1 (in 200 mM KCl) during incubations for 20 min at 85°C separated by gel electophoresis. Transcripts with the lengths expected for a run-off transcript(s) from the indicated promoter are indicated with arrows. TK0977 encodes a pullanase not present in closely related Archaea species that have only one TFB factor. Transcription of this gene in vivo is predicted to be repressed by substrate-sensing regulator 41. TK0716 and TK0717 are divergently oriented genes encoding transport proteins predicted to have been recently acquired by T.k. 40. TK1278 is the promoter-proximal gene (gar1) in the operon that encodes TFB1 (TK1280; see Figure 7A), TK2286 is the promoter proximal gene in the operon encoding TFB2 (TK2287; see Figure 7A), TK1431 encodes glutamate dehydrogenase and r01 encodes the 5S-1 ribosomal RNA. Transcription from the 5S template generated a transcript with length predicted for a 5S promoter-directed run-off transcript and a transcript with length (~240 nt) of an end-to-end transcript. Single-stranded DNA molecules were separated, as indicated, as size standards.
Promoter activity with T1D2 and T2D1
As shown in the alignment of TFB1 and TFB2 (Figure 5a), these proteins have very few amino acid differences in the domains predicted to interact with TBP, or to bind to the upstream or downstream BRE-promoter elements 29; 30. However, there are notable residue differences in the central region predicted to interact directly with RNAP 31 and to form a surface exposed for potential interactions with regulators (Figure 5b). We therefore generated hybrid proteins, designated T1D2 and T2D1, with the central region from TFB2 substituted in TFB1, and vice versa (Figure 5A). These hybrid transcription factors supported transcription from all the T.k. promoters tested (data not shown) although their maximum activities were ~70% of the parental TFBs. The salt-dependence of initiation was affected by swapping this region of the archaeal TFBs and both hybrid proteins exhibited optimum activity at a lower salt concentration than either TFB1 or TFB2 (Figure 5c). The lower promoter activities of T1D2 and T2D1 most likely reflects lower rates of RNAP recruitment during multi-round transcription given that TFB1 and TFB2 support very similar rates of abortive transcription initiation (see below) and RNAP elongation rate is unlikely to be dependent on the initiating TFB.
Figure 5. Amino acid sequences and predicted functional domains of TFB1 and TFB2, and salt dependent transcription by TID2 and T2D1.
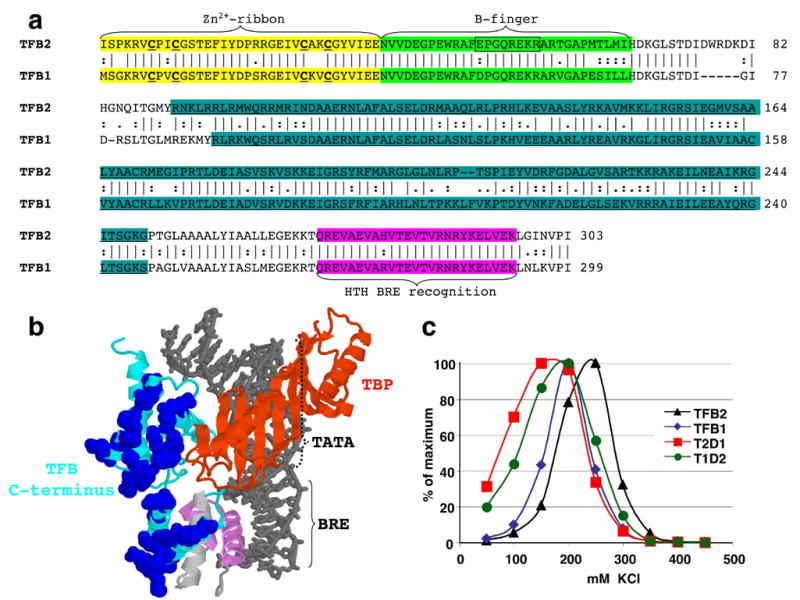
A. Alignment of the TFB1 and TFB2 sequences with identical (|), similar (:) and related (.) amino acid residues identified. The regions predicted to form the Zn2+-ribbon, B-finger and helix-turn-helix DNA-binding domains are shaded yellow, green and pink, respectively. The four cysteine residues predicted to participate in Zn2+-ribbon assembly are identified by bold font and underlined, and the residues deleted to generate TFB2Δ51-58 are boxed. The regions exchanged to create T1D2 and T2D1 are underlined and shaded in cyan. B. Structure determined for the Pyrococcus woesei TBP (brown), central and C-terminal DNA binding domains of P. woesei TFB (light blue and violet, respectively) with the BRE-TATA box region of an archaeal promoter 30. Residues on the surface of the TFB molecule that differ in charge in TFB1 and TFB2 from T.k. are shown space-filled and colored dark blue. C. Salt-dependence of run-off transcript synthesis from the gdh promoter, during incubation for 20 min at 85°C, in reaction mixtures containing T.k. RNAP, TBP and TFB1 (♦), TFB2 (▲), T1D2 (●) and T2D1 (■). The amounts of transcript synthesized in reaction mixtures containing different KCl concentrations are graphed as percentages of the maximum amount of transcript synthesized.
Transcription initiation supported by TFB2Δ51-58
The N-terminal region of TFB2 (and TFB1) almost certainly folds to form Zn2+-ribbon and B-finger domains (Figure 5), with the B-finger predicted to extend into the main channel of the archaeal RNAP and clash with a nascent transcript when the transcript reaches 4 or 5 nucleotides long. Single residue changes, and complete deletion of the B-finger region from the archaeal TFB from Methanocaldococcus jannaschii changed the NTP-concentration dependence of promoter-initiated transcription 11. These TFB variants also resulted in differences in abortive product synthesis from non-specific templates and, together, these results suggested that TFB also plays a role in transcription initiation after RNAP recruitment 11. Region 3.2 of bacterial σ70 forms a hairpin structure that similarly extends into the main channel of a bacterial RNAP and so clashes with a nascent transcript 32; 33;34. The tip of the σ70 hairpin does participate in binding the initiating nucleotides within the holoenzyme 35. Removal of the tip of region 3.2 of σ70 resulted in a variant that required a dinucleotide primer (ApU) for initiation, and this deletion also changed the ratio of long and short abortive transcripts generated by E. coli RNAP when initiating from the T7 A1 promoter. To probe this possible functional homology of TFB and σ70, we generated a TFB2 variant (designated TFB2Δ51-58; Figure 5) that lacks the B-finger residues predicted to be structurally equivalent to those deleted from the 3.2 region in the σ70 variant. TFB2Δ51-58 directed efficient transcription initiation in vitro from several different T.k. promoters (Figure 6; data not shown), with or without the addition of an initiating dinucleotide, and with no requirement for an increased concentration of initiating NTPs. The salt concentration optima for initiation with TFB2Δ51-58 were the same as with TFB2 (data not shown), but initiation supported by TFB2Δ51-58 resulted a minimal synthesis of very short abortive transcripts (≤ ~5 nt; Figure 6). The results with TFB2Δ51-58 were therefore consistent with a shorter B-finger not interfering with nascent transcript synthesis before the transcript was extended beyond 5 nt in length, but inconsistent with TFB2 participating in initial NTP binding and positioning for catalysis as documented for σ70.
Figure 6. Elongation of transcripts initiated in reaction mixtures containing TFB1, TFB2 or TFB2Δ51-58.
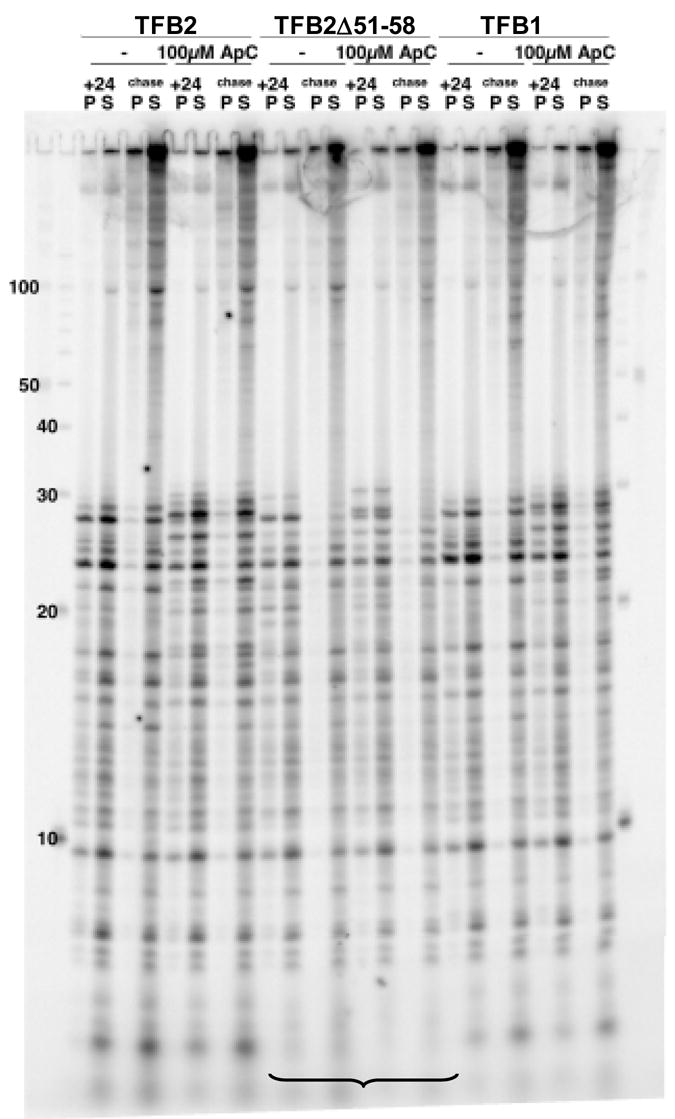
The transcripts synthesized from the M.t. hmtB promoter, initiated at the start of a 24 bp U-less cassette, in reaction mixtures that contained only ATP, GTP and [32P]-CTP lacking UTP, without (−) or with ApC added, and supported by TFB1, TFB2 or TFB2Δ51-58 are shown in the lanes labeled +24. The transcripts synthesized when all four NTPs were then added are shown in the lanes labeled chase. In both cases, the transcripts synthesized were separated into those fully terminated and released from the template (S; supernatant) and those that were arrested but remained attached to the template DNA (P, pellet). Under the electrophoresis conditions used, full-length run-off transcripts remained essentially within the gel loading wells. As identified by the bracket, very short (≤ 5 nt) abortive transcript products were not generated when initiation was dependent on TFB2Δ51-58. Single stranded DNA molecules were separated as size markers (nt) in the flanking lanes.
Deletion of the genes encoding TFB1 or TFB2
The gene encoding TBP (TK0132; tbp) is likely transcribed into a monocistronic transcript, whereas the genes encoding TFB1 (TK1280; tfb1) and TFB2 (TK2287; tfb2) appear to be located within multi-gene operons (Figure 7a). The two upstream genes (TK1278, TK1279) predicted to be co-transcribed with TK1280 are not widely conserved and do not encode proteins with identifiable functions. In contrast, the gene (TK2286) located immediately upstream from tfb2 encodes the Gar1 subunit of the H/ACA-box ribonucleoprotein particle 36 and, intriguingly, Gar1-encoding genes are similarly located immediately upstream and, as in T.k., also often partially overlap TFB-encoding genes in many Archaea. The gene (TK2288) downstream of tfb2 (TK2288) is predicted to encode a metallophosphoesterase but this remains to be determined experimentally. The DNA molecules constructed to replace tfb1 and tfb2 with trpE in the T.k. genome were designed to retain the integrity of the flanking genes and operon organizations (Figure 7a). In the donor DNA used to replace tfb1 (TK1280), trpE was positioned between TK1279 and TK1281 whereas the donor DNA used to replace tfb2 (TK2287) also deleted TK2288. We attempted to delete only tfb2 but were unsuccessful even with DNA constructs in which the trpE gene was positioned <1 kbp distant from TK2287, a procedure that did facilitate deletion of the T.k. nusA gene (TK1079; see Materials and Methods). The deletions of tfb1 and tfb2 were confirmed by Southern blots of restriction enzyme digested genomic DNAs (Figure 7b) and by PCR-amplification and sequencing of genomic DNAs using primers specific for trpE, tfb1, tfb2 and the flanking genes (data not shown). The T.k. mutants that lack tfb1 or tfb2 show no defects in laboratory growth on minimal or nutrient supplemented media (Figure 7c), or in survival for extended periods in stationary phase cultures.
Figure 7. Construction of T.k. mutants with TK1280 (tfb1) or TK2287 (tfb2) deleted and replaced by trpE.
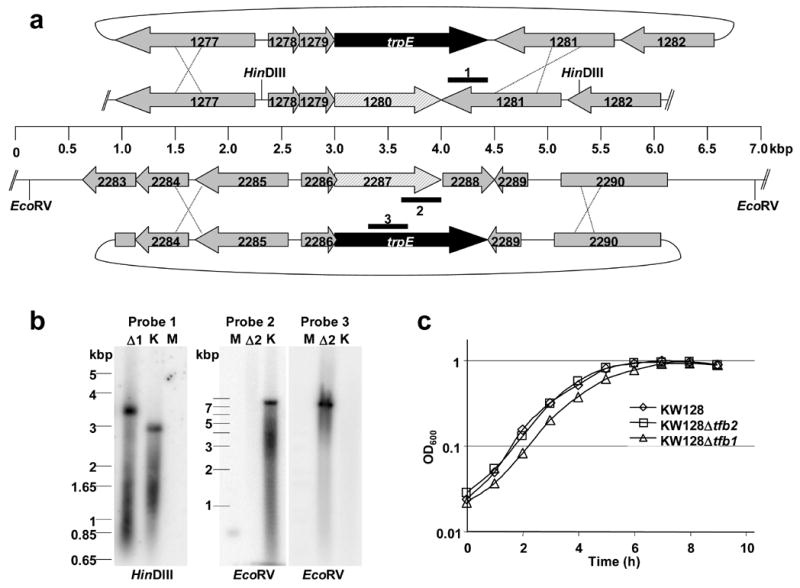
a. The plasmids (pLC57) and (pLC63) that were linearized and used as donor DNAs to replace TK1280 and TK2287, respectively, are shown positioned above and below the arrangement of the homologous genes in the T.k. genome. Recombination in the flanking regions (illustrated by the Xs) substituted trpE for TK1280 and for TK2287-TK2288. The heavy lines labeled 1–3 identify the regions used as hybridization probes in Southern blots of restriction enzyme digested genomic DNA. b. Southern blots of HindIII or EcoRV digested T.k. genomic DNA from T.k. KW128 (K), T.k. Δtfb1 (Δ1) and T.k. Δtfb2 (Δ2) hybridized with probes 1, 2 or 3. Size standards were separated in lanes M. c. Growth of T.k.KW128 (⋄), T.k.KW128Δtfb1 (△) and T.k. KW128Δtfb2 (□) cultures in MA-YT-S° medium incubated at 85°C.
Discussion
We have established and optimized in vitro transcription using purified components from the genetically tractable archaeon Thermococcus kodakaraensis and, using this system, we have established that either of the two TFBs encoded in the T.k. genome, TFB1 and TFB2, is sufficient for basal transcription initiation. Efficient transcription initiation was obtained in vitro with either TFB1 or TFB2 from templates that carried promoters for rRNA, tRNA and mRNAs, including the gdh housekeeping gene, divergent promoters for genes (TK0716; TK0717) likely acquired recently by T.k.40, for TK0977 (encodes a pullanase) that is unique to T.k. and predicted to be regulated by substrate induction 41, and for the TFB1- and TFB2-encoding operons. In every case, transcription initiation occurred at the same site(s) in vitro with TFB1 and TFB2, and the products of promoter clearance assays demonstrated that TFB1- and TFB2-dependent initiation resulted in the same amounts and patterns of aborted transcripts. Optimum TFB2-dependent transcription initiation did occur at higher salt concentrations than were optimum for TFB1-dependent transcription initiation. Domain swapping established that a central segment of these transcription factors contributed to their salt requirements. Consistent with TFB1 and TFB2 having exchangeable and overlapping functions, viable mutants have been obtained with either of the two encoding genes (tfb1, TK1280; tfb2, TK2287) deleted, demonstrating that TFB1 or TFB2 alone is sufficient for T.k. viability under laboratory conditions. It is apparent therefore that the two TFBs in T.k. do not direct transcription exclusively from different promoters, and so do not function in promoter selection directly as do different σ-factors in Bacteria. T.k. TFB1 and TFB2 are however closely related proteins, with ~60% overall sequence identity and virtually identical residues constitute their C-terminal helix-turn-helix DNA-binding domains. The residues that constitute the BRE recognition and binding domains are, in fact, very highly conserved in almost all archaeal TFB. Nevertheless, it remains possible that some of the more divergent TFBs in Archaea with multiple TFBs do exhibit promoter specificity, for example PF0687 in Pyrococcus furiosus or MK0619 in Methanopyrus kandleri. It seems also possible that the halophilic Archaea with multiple TFBs and multiple TBPs might exploit this potential to assemble many different TFB+TBP combinations that, as partnerships, bind and direct transcription from different BRE-TATA-box sequences.
The TFB2Δ51-58 variant was constructed to investigate if the tip of the B-finger region of the archaeal TFB might play the same role as the tip of region 3.2 of σ70 in transcription initiation after RNAP recruitment. Shortening the B-finger region of TFB2 had no detectable effects on promoter recognition, the amounts of transcription, or the NTP concentration required for initiation, but did effectively eliminate the production of very short (≤ 5nt) abortive transcripts. The TFB2Δ51-58 results are therefore consistent with the tip of the B-finger region of TFB2 reaching into and clashing with a nascent transcript, but provide no evidence for this region also binding and positioning NTPs for catalysis as for σ70, or for participating in RNAP activity after recruitment.
Although the results obtained demonstrate that TFB1 and TFB2 have overlapping functions in basal transcription initiation in T.k., they could still facilitate differential gene expression. This now seems more likely to be accomplished through differences in surface residue interactions with RNAP and gene-specific regulators than simply by binding to different promoters. The quantitative differences in basal transcription initiation in vitro observed from the same promoter with TFB1 versus TFB2, and their different salt optima for initiation almost certainly result from differences in RNAP recruitment and pre-initiation complex assembly. The different TFBs (and possibly also different TBPs in some Archaea) could have different non-conserved surface residues/structures that interact with different regulators (Figure 5). Archaeal gene expression could also be regulated by differences in the abundance and genome sequestration of the different basal transcription factors. Given the availability of T.k. microarrays, practical genetics and mutants that lack TFB1 or TFB2, we can now determine the requirements for these alternative basal transcription factors in T.k. genome expression under different growth regimes, and test their roles in facilitating regulated gene expression.
Materials and Methods
Media and growth conditions for T.k
Cultures of wild type and mutant strains of T.k. were grown under anaerobic conditions at 85°C in MA-YT-S° or ASW-AA medium with or without added tryptophan (50 μg/ml), as described 23; 42. Cells competent for DNA uptake were prepared, as described 42, from cultures of T.k. KW128 (ΔtrpE::pyrF; ΔpyrF) grown for 12 h in MA-YT-S°. Transformants capable of growth in the absence of tryptophan were selected by growth on ASW-AA-trp plates solidified by incorporation of 1% Gel-rite (Sigma). Single colony isolates were re-streaked on plates lacking tryptophan, and passaged twice as liquid cultures grown in MA-YT-S° before genomic DNA was isolated (below) to confirm genetic constructions. Region(s) of interest were PCR amplified from the genomic DNA [the sequences of all primers used (purchased from Integrated DNA Technology and Operon) are available on request from TJS], and sequenced.
Plasmid constructions and deletion of TFB1 or TFB2 encoding genes
T.k. genes of interest were PCR amplified from T.k. KW128 genomic DNA and plasmids reconstructed as required by using standard molecular biology technologies. The plasmids constructed to over-express T.k. transcription factors in E. coli are described in detail below. To delete or alter genes on the T.k. chromosome, donor DNA plasmids were constructed using pUMT2 as the starting plasmid 42. Approximately 2 kbp of T.k. genomic DNA was cloned on either side of T.k. trpE gene to target recombination of the trpE gene into the T.k. KW128 genome. The plasmids constructed to delete TK1280 (tfb1; encodes TFB1) and TK2287 (tfb2; encodes TFB2) are shown in Figure 7, and the T.k. KW128 derivatives generated in which trpE has replaced TK1280 or TK2287-TK2288 are designated hereafter as T.k. Δtfb1 and T.k. Δtfb2, respectively.
Genomic DNA preparation
Cells were disrupted in 20 mM Tris-HCl (pH 8), 5 mM EDTA, 10% (v/w) sucrose by repetitive freeze-thawing, SDS and proteinase K (Sigma) added to final concentrations of 2% and 0.25mg/ml, respectively, and the mixture incubated for 30 min at 55°C. NaCl was added (1 M final concentration), the mixture was chilled and centrifuged to generate a clarified supernatant from which nucleic acids were precipitated by adding an equal volume of isopropanol. After centrifugation, the pellet was resuspended in 10 mM Tris-HCl (pH 8), 50 μg of RNaseA (GE Healthcare) was added and the mixture incubated at 37°C for 30 min. The DNA remaining was further purified by repetitive phenol: chloroform:isoamyl alcohol (25:24:1) extraction, and then alcohol-precipitated from the aqueous phase.
Southern blot analyses
Aliquots of genomic DNA (10 μg) were digested with either HindIII or EcoRV (New England Biolabs), the fragments generated were separated by electrophoresis through 0.75% (w/v) agarose gels, denatured and transferred to a Zeta-Probe membrane (BioRad). The membrane was incubated with a [32P]-labeled DNA probe, following the membrane manufacturer’s instructions, and the location of the probe visualized by phosphorimaging.
Construction of T.k. rpoL-HA
KW128 was transformed with pTS366 in which the T.k. trpE gene was cloned between TK1166 and TK1167 (rpoL). An in-frame DNA sequence was added to the 3′ terminus of rpoL so that the encoded protein (RpoL-HA) had a C-terminal 9 amino acid residue extension with the sequence of the HA epitope (tag). Trp+ transformants were screened for insertion of the trpE-rpoL-HA sequence by direct sequencing of genomic DNA and by sequencing PCR-amplified genomic DNA. RNAP was purified (see below) from one such T.k. transformant (T.k. KW128 trpE-rpoL-HA; ΔtrpE::pyrF), hereafter designated T.k. rpoL-HA. SDS-PAGE confirmed a size increase, and MS-MS analysis confirmed the specific addition of the HA epitope (tag) to the C-terminus of RpoL.
Construction of T.k. ΔnusA
A derivative of pUMT2 42 was constructed with the trpE gene positioned downstream from the T.k. nusA (TK1079) and adjacent gene (TK1080). Transformation with this DNA resulted in T.k. KW128 trp+ transformants, of which ~10% had the nusA gene deleted.
Purification of T.k. RNAP
The same protocol was used to purify RNAP from T.k. KW128 and RNAP-HA from T.k. rpoL-HA. Cultures (5 l) were grown at 85°C in MA-YT-S° medium with N2 bubbled slowly through the medium and mixing at 60 rpm. Near the end of the exponential growth phase, the ~15 g of accumulated wet-cell mass was harvested under anaerobic conditions and frozen. All subsequent steps were also undertaken under anaerobic conditions (5% H2 + 95% N2 atmosphere) at 23°C. The cells were resuspended in TMED [50mM Tris-HCl (pH 8), 10mM MgCl2, 0.1mM EDTA, 10mM DTT] containing 2 M KCl and ruptured by passage three times through a French pressure cell (20,000 psi). The resulting lysate was centrifuged at 16,000 rpm for 30 min at 4°C in a Beckman JLA16-250 rotor, the supernatant collected and 2 M KCl containing 30% PEG 8000 added to result in a final concentration of 6% PEG 8000. This mixture was stirred for 45 min, and the nucleic acids that precipitated were removed by centrifugation at 16,000 rpm for 75 min at 4°C in the JLA16-250 rotor. The supernatant was diluted by TMED addition to ~100 mM KCl, mixed gently with 100 g of DEAE-cellulose (Sigma) (swelled and de-fined in TMED containing 100 mM KCl) for 60 min. Proteins that bound to the DEAE-cellulose were washed extensively with TMED containing 100 mM KCl, and were then eluted by incubation in TMED containing 400 mM KCl. The resulting solution was diluted by TMED addition to ~120 mM KCl and then passed through a 17 ml Hi-Trap heparin column (GE Healthcare). Proteins that bound to the column matrix were eluted using a 150 to 800 mM linear gradient of KCl dissolved in TMED. Following this, and all subsequent column chromatographic separations, an aliquot of each eluted fraction was subjected to SDS-PAGE and Commassie blue staining to identify the fractions that contained T.k. RNAP. The fractions containing the majority of the RNAP were pooled, diluted with TMED to ~150 mM KCl, applied to a 1 ml MonoQ column (GE Healthcare), and bound proteins were eluted using a 200 to 800 mM linear gradient of KCl dissolved in TMED. The RNAP containing fractions were pooled and passed through a Superdex 200 16/60 column (GE Healthcare) equilibrated with TMED containing 300 mM KCl. The RNAP containing fractions were pooled, applied again to the MonoQ column and proteins that bound were eluted using a 200 to 750 mM linear gradient of KCl in TMED. The fractions that contained RNAP were pooled, and this solution was dialyzed extensively against 25 mM Tris-HCl (pH 8), 10 mM MgCl2, 0.1 mM EDTA, 200 mM KCl, 20 mM β-mercaptoethanol (ME), 50% (v/v) glycerol. Approximately 3.7 mg of purified RNAP was obtained per 15 g of wet cell mass. All of the RNAP subunits and co-purifying proteins were identified, after separation by SDS-PAGE and in-gel trypsin and chymotrysin digestion, by MS-MS at the Mass Spectrometry facility of the Ohio State University.
Cloning, expression and purification of recombinant T.k. transcription factors from E. coli
TBP
The encoding gene (TK0132) was amplified from KW128 genomic DNA with EcoRI and SalI restriction sites added that facilitated cloning into EcoRI+SalI digested pET30b (Novagen). The resulting plasmid (pTS364) was transformed into E. coli Rosetta2 (DE3) (Novagen), and cultures of the transformant were grown in LB (4 l) containing 34 μg chloramphenicol (cam)/ml and 40μg kanamycin (kan)/ml to an OD600 of 0.4 at 37°. Expression of the cloned gene was induced by addition of 0.25 mM IPTG and 1% sorbitol and incubation was continued at 22°C for 12 h. The cells were harvested (~4 g wet weight), resuspended in 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 0.2 mg lysozyme/ml and lysed by sonication. After centrifugation, the clarified supernatant was applied to a 5 ml Hi-Trap Q-FF (GE Healthcare) column, the bound proteins were washed with 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 0.1 M NaCl and eluted using a 0.1 to 2 M linear NaCl gradient. Fractions containing TBP were identified by SDS-PAGE, pooled, the mixture concentrated using a 10 kDa MWCO centrifugal concentrator (Ambion), and passed through a Superdex 200 16/60 column equilibrated with 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 100 mM NaCl. Fractions that contained the majority of the TBP were identified by SDS-PAGE, pooled, reapplied to the Hi-Trap Q-FF column, and bound proteins were eluted using a 200 to 900 mM linear NaCl gradient. The TBP containing fractions were identified, pooled and dialyzed into 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 100 mM NaCl, 50% glycerol, 5 mM ME. The yield was ~15 mg of purified recombinant TBP per 4 l culture.
TFB2
The encoding gene (TK2287) was amplified from KW128 genomic DNA with terminal NdeI and BamH1 sites added that facilitated cloning into NdeI+BamHI digested pET28a (Novagen). The resulting plasmid (pTS334) was transformed into E. coli Rosetta2 (DE3) and cultures of the transformant were grown in LB containing 34 μg cam/ml and 40 μg kan/ml to an OD600 of 0.5 at 37°C. Expression of the cloned gene was induced by addition of 0.25 mM IPTG and 1% sorbitol and incubation was continued at 22°C for 24 h. The cells were harvested (~13 g wet weight), resuspended in 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 100 mM NaCl, 0.2 mg lysozyme/ml and lysed by sonication. After centrifugation, the clarified supernatant was applied to a 5 ml Hi-Trap SP-FF column (GE Healthcare), the bound proteins were washed with 20 mM Tris-HCl (pH 8), 0.1 mM EDTA, 0.1 M NaCl and eluted using a 0.1 to 2 M linear NaCl gradient. The eluted fractions that contained TFB2 were identified by SDS-PAGE, pooled and the resulting solution was applied to a 5 ml Ni2+-charged chelating column (GE Healthcare). Bound proteins were washed extensively, and then eluted with a linear gradient of 500 mM NaCl and 5 mM imidazole to 100 mM NaCl and 500 mM imidazole dissolved in 20 mM Tris-HCl (pH 8). The TFB2-containing fractions were pooled and dialyzed into 20 mM Tris-HCl (pH 8), 100 mM NaCl, 0.1 mM EDTA, 5 mM ME, 50% glycerol. The yield was ~10 mg of purified TFB2 per 4 l culture.
TFB1
The encoding gene (TK1280) was amplified from KW128 genomic DNA with terminal EcoRI and BamH1 sites added that facilitated cloning into EcoRI+BamHI digested pQE-80L (Qiagen). The resulting plasmid (pTS371) was transformed in E. coli Rossetta 2 and cultures the transformant were grown in LB that contained 150 μg ampicillin (amp)/ml plus 34 μg cam /ml to an OD600 of 0.65 at 37°. Induced expression and purification of the encoded TFB1, with six histidine residues attached to the N-terminus, followed the procedure describe above for TFB2. The yield was ~8 mg of TFB1 per 4 l culture.
TIP26
The encoding gene (TK1172) was amplified from KW128 genomic DNA with terminal BamH1 and Sal1 sites added that facilitated cloning into BamHI+SalI digested pQE-82L (Qiagen). The resulting plasmid (pTS311) was transformed into E. coli Rosetta2, cultures were grown in LB that contained 150 μg amp/ml and 34μg cam/ml to an OD600 of 0.7 at 37°C before addition of 0.5 mM IPTG (final concentration) and incubation continued for 3 h at 37°. The cells were resuspended in 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10% glycerol, 0.2 mg lysozyme/ml and lysed by sonication. The lysate was clarified by centrifugation, the resulting supernatant incubated at 65°C for 30 min, clarified again by centrifugation and applied to a 5 ml HiTrap heparin column (GE Healthcare). Bound proteins were eluted using an 0.2 to 1 M linear gradient of NaCl dissolved in 25 mM Tris-HCl (pH 7.5), 10% glycerol. Fractions that contained the majority of the recombinant TIP26, identified by SDS-PAGE, were pooled and the resulting solution concentrated using a centrifugal concentrator (Millipore), and passed through a 16/60 S300 Sephacryl (GE Healthcare) column equilibrated with 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 10% glycerol. Fractions were collected at a rate of 0.3 ml/min, and those that contained TIP26 were pooled and dialyzed into 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 50% glycerol. The yield was ~5 mg of TIP26 per 4 l culture.
Gene construction and purification of the encoded T1D2, T2D1 and TFB2Δ51-58
The tfb1 and tfb2 sequences in pTS371 and pTS334 respectively (see above), were changed using a Quikchange kit (Stratagene) to introduce a SpeI site near the 3′-terminus of each gene. The resulting plasmids were designated pCJ206 and pCJ207. The tfb2 gene in pCJ207 was PCR amplified and cloned into EcoRI+BamH1 digested pQE80-L resulting in plasmid pCL207b. Plasmids pCJ206 and pCJ207b were digested with BsrGI plus SpeI. The BsrGI-SpeI fragment released from pCJ206 was cloned into the BsrGI+SpeI digested pCJ207b, and vice-versa. This resulted in pCJ208 that encodes T2D1, a protein with the N- and C-terminal regions of TFB2 flanking the central domain of TFB1, and pCJ210 encodes T1D2, a protein with the N- and C- terminal regions of TFB1 flanking the central domain TFB2. Plasmid pCJ208 was transformed in E. coli Rosetta2 and T2D1 generated and purified as described above for TFB1 and TFB2 with a yield of ~5 mg of T2D1 per 15 g of cells. Plasmid CJ210 was transformed into E. coli Rosetta2 and cultures grown in LB that contained 150μg amp/ml plus 34μg cam/ml to an OD600 of 0.4 at 37°. IPTG and sorbitol were added (final concentrations of 0.25 mM and 1%, respectively) and incubation continued for 3 h at 20°C. The cells were harvested, resuspended in 20 mM Tris-HCl (pH 6.8), 20 mM ME, 200 mM NaCl, 0.1 mM EDTA, 15% glycerol and lysed by sonication. The lysate was clarified by centrifugation and the supernatant applied to a 5 ml HiTrap heparin column. Bound proteins were eluted using a 0.2 to 2 M linear NaCl gradient. Fractions that contained T1D2, identified by SDS-PAGE, were pooled, the solution concentrated using a centrifugal concentrator (Millipore) and passed through a 16/60 S100 Sephacryl column, eluted at 0.1 ml/min with 20 mM Tris-HCl (pH 6.8), 20 mM ME, 550 mM NaCl, 0.1 mM EDTA, 15% glycerol. Fractions that contained T1D2 were pooled, the solution diluted to ~150 mM NaCl and applied to a 5 ml HiTrap SP-FF column. Bound proteins were eluted using a 0.15 to 2 M linear gradient of NaCl. Fractions that contained the majority of the T1D2 were pooled, applied to a 5ml HiTrap chelating column charged with Ni2+, and T1D2 was eluted using an imidazole gradient. The fractions containing T1D2 were pooled and dialyzed into 20 mM Tris-HCl (pH 8), 250 mM NaCl, 0.1 mM EDTA, 5 mM ME, 50% glycerol. The yield was ~2mg of T1D2 from 4l culture.
The DNA sequence in pTS334 encoding amino acids 51 to 58 of TFB2 (see Figure 5) was deleted using the Quikchange system. The resulting plasmid, pCJ208, was transformed into E. coli Rossetta2 and the encoded transcription factor, TFB2Δ51-58, was generated and purified as described above for TFB1. The yield was ~7 mg of TFB2Δ51-58 per 4 l culture.
Transcription templates
Intergenic regions, predicted to contain T.k. promoters, were amplified by PCR from KW128 genomic DNA, purified by band-excision after agarose gel electrophoresis, and quantified by fluorometry. Templates were generally <500 bp. The transcription templates used that carry the promoter for the hmtB gene from M.t. have been described previously 20.
In vitro transcription
Linear DNA molecules that carried the promoter for the T.k. glutamate dehydrogenase (TK1431; gdh) gene were used as templates to establish promoter-dependent in vitro transcription using T.k. components. After optimization, transcription reaction mixtures (usually 20 μl) in which the DNA template (10 nM) was the limiting reagent contained 20 mM Tris-HCl (pH 8; measured at 22°C), 5 mM MgCl2, 50 to 500 mM KCl, 5 to 10% glycerol, 10 mM DTT, 40 nM RNAP, 80 nM TBP, 80 nM TFB1 or TFB2, 200 μM ATP, GTP, CTP, 20 μM UTP and 10 to 50 nCi/μl [32P]-α-UTP. They were incubated at 85°C for 20 min before the reaction was stopped and the [32P]-labeled transcripts synthesized were purified, visualized and quantified after separation by gel electrophoresis as previously described 20. Stalled elongation complexes containing the T.k. RNAP positioned at +24 relative to site transcription initiation downstream from the hmtB were generated as described for complexes containing the M.t. RNAP 20. The buffer was optimized for the T.k. system (250 mM or 200 mM KCl for reaction mixtures containing TFB1 or TFB2, respectively) and transcript elongation to +24 was allowed for only 2 min at 85°C. The reaction mixtures also contained 500 μM ATP, GTP, 20 μM CTP, [32P]-α-CTP and in some cases also 100 μM ApC (Sigma).
Identification of sites of transcription initiation
Unlabeled transcripts synthesized in vitro were purified and reverse transcribed as previously described 43. The labeled products were subjected to electrophoresis in lanes adjacent to DNA sequence ladders generated by thermal-ddNTP sequencing using the same primer and Taq polymerase.
Antibody precipitation of RNAP-HA from lysates of T.k (rpoL-HA)
Cultures (50 ml) of T.k. KW128 and T.k. (rpoL-HA), grown at 85°C in MA-YT-S° medium to mid-exponential phase cells were chilled rapidly. The cells were harvested by centrifugation, resuspended in 1 ml of 25 mM Tris-HCl (pH 8), 10 mM MgCl2, 0.1 mM EDTA, 10 mM ME, 100 mM KCl, and lysed by three cycles of freezing in liquid N2 and slow thawing. The lysates were clarified by centrifugation at 4°C for 20 min at 14,400g, and the supernatant was retained as a “soluble RNAP” fraction. The sedimented material was resuspended in 1ml of the same buffer, sonicated to fragment the DNA and, after centrifugation, the resulting supernatant was retained as likely to contain RNAP bound in “elongation complexes”. Beads (35 μl) coated with anti-HA antibody (ProFound HA Tag IP/Co-IP system; Pierce) were mixed and shaken slowly with aliquots of the soluble RNAP and elongation complexes for 24 h at 4°C. The beads were recovered by centrifugation through a microcolumn, washed 3 times with 25 mM Tris-HCl (pH 7.2), 0.15 M NaCl and the proteins washed from the beads were precipitated by trichloroacetic acid (TCA) added to a final 15% concentration. Proteins bound to the beads were eluted (3 times) using 166 μl of 1 M glycine (pH 2.8), the eluants were neutralized by immediate mixing with 8.3 μl of 1 M Tris-HCl (pH 9.5), combined, and TCA added to precipitate the eluted proteins. The TCA-precipitated proteins were dissolved and denatured by incubation in gel-loading buffer, separated by DISC SDS-PAGE (15% polyacrylamide gels), and silver stained using a mass spectrometry compatible staining kit as described by the manufacturer (Invitrogen).
Acknowledgments
This work was supported by research grants from the Department of Energy (DE-FG02-87ER13731) and National Institutes of Health (GM53185) to JNR, and a National Institutes of Health fellowship (1F32-GM073336-01) to TJS. We thank Drs. H. Atomi for T.k. strains, K. Murakami and his laboratory for instruction in working with T.k., A. Hatoum for providing a genomic DNA sequencing protocol, and our colleagues at Ohio State University for their help and advice.
Abbreviations
- TFB
archaeal transcription factor B
- TFE
transcription factor E
- TBP
TATA-box binding protein
- RNAP
RNA polymerase
- Pol
sigma factor, σ, polymerase
- T.k
Thermococcus kodakaraensis
- M.t
Methanothermobacter thermautotrophicus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baumann P, Qureshi SA, Jackson SP. Transcription: new insights from studies on Archaea. Trends Genet. 1995;11:279–283. doi: 10.1016/s0168-9525(00)89075-7. [DOI] [PubMed] [Google Scholar]
- 2.Kyrpides NC, Ouzounis CA. Transcription in archaea. Proc Natl Acad Sci USA. 1999;96:8545–8550. doi: 10.1073/pnas.96.15.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langer D, Hain J, Thuriaux P, Zillig W. Transcription in archaea: similarity to that in eucarya. Proc Natl Acad Sci U S A. 1995;92:5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grigorova IL, Phleger NJ, Mutalik VK, Gross CA. Insights into transcriptional regulation and sigma competition from an equilibrium model of RNA polymerase binding to DNA. Proc Natl Acad Sci USA. 2006;103:5332–5337. doi: 10.1073/pnas.0600828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hochschild A, Dove SL. Protein-protein contacts that activate and repress prokaryotic transcription. Cell. 1998;92:597–600. doi: 10.1016/s0092-8674(00)81126-5. [DOI] [PubMed] [Google Scholar]
- 6.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 7.Cramer P, Bushnell DA, Fu J, Gnatt AL, Maier-Davis B, Thompson NE, Burgess RR, Edwards AM, David PR, Kornberg RD. Architecture of RNA polymerase II and implications for the transcription mechanism. Science. 2000;288:640–649. doi: 10.1126/science.288.5466.640. [DOI] [PubMed] [Google Scholar]
- 8.Bell SD, Magill CP, Jackson SP. Basal and regulated transcription in Archaea. Biochem Soc Trans. 2001;29:392–395. doi: 10.1042/bst0290392. [DOI] [PubMed] [Google Scholar]
- 9.Geiduschek EP, Ouhammouch M. Archaeal transcription and its regulators. Mol Microbiol. 2005;56:1397–1407. doi: 10.1111/j.1365-2958.2005.04627.x. [DOI] [PubMed] [Google Scholar]
- 10.Bell SD, Jackson SP. Mechanism and regulation of transcription in archaea. Curr Opin Microbiol. 2001;4:208–213. doi: 10.1016/s1369-5274(00)00190-9. [DOI] [PubMed] [Google Scholar]
- 11.Werner F, Weinzierl RO. Direct modulation of RNA polymerase core functions by basal transcription factors. Mol Cell Biol. 2005;25:8344–8355. doi: 10.1128/MCB.25.18.8344-8355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanzelka BL, Darcy TJ, Reeve JN. TFE, an archaeal transcription factor in Methanobacterium thermoautotrophicum related to eucaryal transcription factor TFIIEalpha. J Bacteriol. 2001;183:1813–1818. doi: 10.1128/JB.183.5.1813-1818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell SD, Brinkman AB, van der Oost J, Jackson SP. The archaeal TFIIEalpha homologue facilitates transcription initiation by enhancing TATA-box recognition. EMBO Rep. 2001;2:133–138. doi: 10.1093/embo-reports/kve021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baliga NS, Bonneau R, Facciotti MT, Pan M, Glusman G, Deutsch EW, Shannon P, Chiu Y, Weng RS, Gan RR, Hung P, Date SV, Marcotte E, Hood L, Ng WV. Genome sequence of Haloarcula marismortui: a halophilic archaeon from the Dead Sea. Genome Res. 2004;14:2221–2234. doi: 10.1101/gr.2700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baliga NS, Goo YA, Ng WV, Hood L, Daniels CJ, DasSarma S. Is gene expression in Halobacterium NRC-1 regulated by multiple TBP and TFB transcription factors? Mol Microbiol. 2000;36:1184–1185. doi: 10.1046/j.1365-2958.2000.01916.x. [DOI] [PubMed] [Google Scholar]
- 16.Ng WV, Kennedy SP, Mahairas GG, Berquist B, Pan M, Shukla HD, Lasky SR, Baliga NS, Thorsson V, Sbrogna J, Swartzell S, Weir D, Hall J, Dahl TA, Welti R, Goo YA, Leithauser B, Keller K, Cruz R, Danson MJ, Hough DW, Maddocks DG, Jablonski PE, Krebs MP, Angevine CM, Dale H, Isenbarger TA, Peck RF, Pohlschroder M, Spudich JL, Jung KW, Alam M, Freitas T, Hou S, Daniels CJ, Dennis PP, Omer AD, Ebhardt H, Lowe TM, Liang P, Riley M, Hood L, DasSarma S. Genome sequence of Halobacterium species NRC-1. Proc Natl Acad Sci USA. 2000;97:12176–12181. doi: 10.1073/pnas.190337797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goo YA, Yi EC, Baliga NS, Tao WA, Pan M, Aebersold R, Goodlett DR, Hood L, Ng WV. Proteomic analysis of an extreme halophilic archaeon, Halobacterium sp. NRC-1. Mol Cell Proteomics. 2003;2:506–524. doi: 10.1074/mcp.M300044-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Darcy TJ, Hausner W, Awery DE, Edwards AM, Thomm M, Reeve JN. Methanobacterium thermoautotrophicum RNA polymerase and transcription in vitro. J Bacteriol. 1999;181:4424–4429. doi: 10.1128/jb.181.14.4424-4429.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hausner W, Thomm M. Purification and characterization of a general transcription factor, aTFB, from the archaeon Methanococcus thermolithotrophicus. J Biol Chem. 1993;268:24047–24052. [PubMed] [Google Scholar]
- 20.Santangelo TJ, Reeve JN. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J Mol Biol. 2006;355:196–210. doi: 10.1016/j.jmb.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 21.Wettach J, Gohl HP, Tschochner H, Thomm M. Functional interaction of yeast and human TATA-binding proteins with an archaeal RNA polymerase and promoter. Proc Natl Acad Sci USA. 1995;92:472–476. doi: 10.1073/pnas.92.2.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qureshi SA, Bell SD, Jackson SP. Factor requirements for transcription in the Archaeon Sulfolobus shibatae. EMBO J. 1997;16:2927–2936. doi: 10.1093/emboj/16.10.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T. Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea. 2004;1:263–267. doi: 10.1155/2004/204953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto T, Matsuda T, Inoue T, Matsumura H, Morikawa M, Kanaya S, Kai Y. Crystal structure of TBP-interacting protein (Tk-TIP26) and implications for its inhibition mechanism of the interaction between TBP and TATA-DNA. Protein Sci. 2006;15:152–161. doi: 10.1110/ps.051788906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuda T, Morikawa M, Haruki M, Higashibata H, Imanaka T, Kanaya S. Isolation of TBP-interacting protein (TIP) from a hyperthermophilic archaeon that inhibits the binding of TBP to TATA-DNA. FEBS Lett. 1999;457:38–42. doi: 10.1016/s0014-5793(99)01005-4. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda T, Fujikawa M, Haruki M, Tang XF, Ezaki S, Imanaka T, Morikawa M, Kanaya S. Interaction of TIP26 from a hyperthermophilic archaeon with TFB/TBP/DNA ternary complex. Extremophiles. 2001;5:177–182. doi: 10.1007/s007920100193. [DOI] [PubMed] [Google Scholar]
- 28.O’Brien R, DeDecker B, Fleming KG, Sigler PB, Ladbury JE. The effects of salt on the TATA binding protein-DNA interaction from a hyperthermophilic archaeon. J Mol Biol. 1998;279:117–125. doi: 10.1006/jmbi.1998.1743. [DOI] [PubMed] [Google Scholar]
- 29.Deng W, Roberts SG. A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev. 2005;19:2418–2423. doi: 10.1101/gad.342405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kosa PF, Ghosh G, DeDecker BS, Sigler PB. The 2.1-A crystal structure of an archaeal preinitiation complex: TATA-box-binding protein/transcription factor (II)B core/TATA-box. Proc Natl Acad Sci USA. 1997;94:6042–6047. doi: 10.1073/pnas.94.12.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bushnell DA, Westover KD, Davis RE, Kornberg RD. Structural basis of transcription: an RNA polymerase II-TFIIB cocrystal at 4.5 Angstroms. Science. 2004;303:983–988. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- 32.Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- 33.Murakami KS, Masuda S, Darst SA. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 A resolution. Science. 2002;296:1280–1284. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
- 34.Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 35.Kulbachinskiy A, Mustaev A. Region 3.2 of the sigma subunit contributes to the binding of the 3′-initiating nucleotide in the RNA polymerase active center and facilitates promoter clearance during initiation. J Biol Chem. 2006;281:18273–18276. doi: 10.1074/jbc.C600060200. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Ye K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006;443:302–307. doi: 10.1038/nature05151. [DOI] [PubMed] [Google Scholar]
- 37.Soppa J. Transcription initiation in Archaea: facts, factors and future aspects. Mol Microbiol. 1999;31:1295–1305. doi: 10.1046/j.1365-2958.1999.01273.x. [DOI] [PubMed] [Google Scholar]
- 38.Xie Y, Reeve JN. In vitro transcription assays using components from Methanothermobacter thermautotrophicus. Meth Enzymol. 2003;370:66–72. doi: 10.1016/S0076-6879(03)70006-8. [DOI] [PubMed] [Google Scholar]
- 39.Xie Y, Reeve JN. Transcription by an archaeal RNA polymerase is slowed but not blocked by an archaeal nucleosome. J Bacteriol. 2004;186:3492–3498. doi: 10.1128/JB.186.11.3492-3498.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukui T, Atomi H, Kanai T, Matsumi R, Fujiwara S, Imanaka T. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 2005;15:352–363. doi: 10.1101/gr.3003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van de Werken HJ, Verhees CH, Akerboom J, de Vos WM, van der Oost J. Identification of a glycolytic regulon in the archaea Pyrococcus and Thermococcus. FEMS Microbiol Lett. 2006;260:69–76. doi: 10.1111/j.1574-6968.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- 42.Sato T, Fukui T, Atomi H, Imanaka T. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol. 2003;185:210–220. doi: 10.1128/JB.185.1.210-220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wickstrum JR, Santangelo TJ, Egan SM. Cyclic AMP receptor protein and RhaR synergistically activate transcription from the L-rhamnose-responsive rhaSR promoter in Escherichia coli. J Bacteriol. 2005;187:6708–6718. doi: 10.1128/JB.187.19.6708-6718.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]