

Abstract
Depriving sympathetic neurons in cell culture of nerve growth factor (NGF) causes their apoptotic death. Bax-induced release of cytochrome c from mitochondria and the subsequent activation of cytosolic caspases are central to this death. A Bax-dependent increase of mitochondrial-derived reactive oxygen species (ROS) that is an important component of the apoptotic cascade in these cells begins soon after NGF withdrawal. Here we report that Bax can also influence mitochondrial production of ROS in non-apoptotic sympathetic neurons. We determined ROS levels by using confocal microscopy to monitor changes in the fluorescence intensity of a redox-sensitive dye loaded into single cells. ROS levels were similar in NGF-replete bax wild-type neurons and neurons from which bax had been deleted. To enhance any effects that Bax might have on ROS levels in NGF-replete cells we exposed cultures to the ATP synthase inhibitor, oligomycin. This treatment hyperpolarizes mitochondrial membrane potential (ΔΨm), an event that can favor increased ROS production. NGF-replete neurons from mice in which bax had been deleted had much higher levels of mitochondrial-derived ROS when treated with oligomycin than did bax wild-type cells. Oligomycin treatment also caused greater hyperpolarization of ΔΨm in bax-deleted cells than in wild-type cells. These findings indicate that Bax can affect mitochondrial ROS production in non-apoptotic neurons and may do so by altering ΔΨm.
Sympathetic neurons undergo apoptotic death in vivo and in vitro when deprived of their required neurotrophic factor, NGF (Martin et al., 1988; Deckwerth and Johnson, 1993; Deshmukh et al., 1996; Neame et al., 1998; Martinou et al., 1999; Putcha et al., 2002). Apoptosis in these and many other types of neurons depends on the proapoptotic Bcl-2 family member, Bax (Deckwerth et al., 1996; Miller et al., 1997; White et al., 1998). Withdrawing NGF from sympathetic neurons causes Bax to become tightly associated with the outer membrane of their mitochondria where it induces release of cytochrome c and other apoptogenic substances from the mitochondrial intermembrane space into the cytoplasm (Liu et al., 1996; Wolter et al., 1997; Putcha et al., 1999; Susin et al., 1999; Du et al., 2000; Li et al., 2001). ROS levels are elevated in NGF-deprived sympathetic neurons in cell culture before they become committed to apoptotic death (Greenlund et al., 1995; Dugan et al., 1997). These ROS derive from the mitochondrial electron transport chain, lie downstream of Bax, and appear to be an important component of the apoptotic cascade (Kirkland et al., 2002). In the process of carrying out experiments on the effects of Bax on ROS in apoptotic neurons we discovered that Bax can also have a potent effect on production of ROS in non-apoptotic cells maintained in NGF-containing medium.
Sympathetic neurons were obtained from the superior cervical ganglia of newborn mice. bax wild-type (bax+/+), hemizygous (bax+/−knockout (bax−/−) mice were generated by mating mice hemizygous for the bax allele (Jackson Labs, Bar Harbor, ME; Knudson et al., 1995). Separate cultures were established for the ganglia from each pup of bax+/− X bax+/− matings. Genomic DNA was prepared from the tail of these pups and genotypes determined by PCR as described previously (Kirkland et al., 2002). Dissociation of neurons from the ganglia and plating of neurons in cell culture dishes were also done as described (Franklin et al., 1995; Deckwerth et al., 1996; Kirkland et al., 2002). Neurons were plated on #1 glass coverslips. These were placed in an Attofluor cell chamber (Molecular Probes, Eugene, OR) for microscopy. Cultures were incubated at the standard temperature for culturing sympathetic neurons (35° C; Johnson and Argiro, 1983) in an incubator having a 5% CO2 and 95% air atmosphere. They were maintained in Eagle’s minimum essential medium with Earle’s salts w/o L-glutamine (Life Technologies, Inc., Rockville, MD) and supplemented with 10 % fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μM fluorodeoxyuridine, 20 μM uridine, 1.4 mM L-glutamine, and 50 ng/ml 2.5S NGF. Cultures were 6–9 days old when used for experiments.
Relative ROS levels were determined by confocal microscopic imaging of neurons loaded with the redox-sensitive dye 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2 DCFDA; Molecular Probes) as described (Royall and Ischiropoulos, 1993; Kirkland et al., 2002). The intensity of this dye increases primarily upon oxidation by H2O2 and free radical products downstream of H2O2. Nerve growth factor 2.5S was from Harlan Bioproducts (Indianapolis, IN). All other reagents were from Sigma (St. Louis, MO). Confocal microscopy was done with a Nikon C1 laser-scanning confocal system (Southern Micro Instruments, Marietta, GA) attached to a Nikon Eclipse TE300 inverted microscope. Image capture and data analysis was accomplished by EZC1 software. Neurons were scanned at 512 X 512 pixel resolution. The dye was excited with the 488 nm line of the confocal lasers and the FITC photomultiplier of the confocal microscope was used for image acquisition. Confocal pinhole size was always the same within an experiment.
While most Bax resides in the cytoplasm of non-apoptotic sympathetic neurons and other non-apoptotic cell types, a substantial fraction of the cellular Bax pool is associated with the outer mitochondrial membrane (Putcha et al., 1999, 2002; Kaufmann et al., 2004). To determine whether this pool of Bax affects basal ROS levels in NGF-replete cells, we measured CM-H2DCFDA intensity in bax+/+, bax+/−, and bax−/− neurons maintained in NGF-containing media (Fig. 1). For each experiment the average CM-H2DCFDA intensity of NGF-maintained, bax+/+ neurons plated and loaded with dye at the same time as neurons of the other genotypes was used as control. bax−/− neurons do not have any detectable Bax protein while bax+/− cells have about half of the Bax concentration of bax+/+ neurons (Kirkland et al., 2002). The average CM-H2DCFDA intensity in the bax−/− neurons was 0.2 ± 0.07-fold less than the average in the bax+/+ cells (p = 0.013). To attempt to augment any differences in ROS levels in NGF-replete neurons with each of the three bax genotypes, we treated cultures with the macrolide antibiotic, oligomycin (5 μg/ml). This compound blocks passage of protons through the mitochondrial F1.FO ATP synthase. In normally functioning mitochondria, block of the synthase by oligomycin hyperpolarizes ΔΨm by increasing the proton gradient across the inner membrane. ATP synthesis is also blocked. Hyperpolarization of ΔΨm can increase leakage of electrons from the mitochondrial electron transport chain (Nicholls and Budd, 2000; Nicholls and Ward, 2002; Nicholls and Ferguson, 2002). These electrons then reduce molecular oxygen to superoxide which in turn is converted into ROS that can be detected by CM-H2DCFDA. Oligomycin, used at a concentration known to block most ATP production in these cells (Chang et al., 2003) greatly increased CM-H2DCFDA intensity in NGF-maintained bax+/+ neurons (Fig. 1A, B). The ROS levels in bax+/+ and bax+/− cells were increased by an equivalent amount (p = 0.44) by this treatment (Fig. 1A, B). Surprisingly, the oligomycin treatment caused a much larger increase of ROS in the bax−/− neurons than in the other two genotypes. Average CM-H2DCFDA intensity was about 2-fold higher in these cells than in the oligomycin-treated bax+/+ and bax+/− neurons (about 5-fold higher than control; Fig 1A, B). Therefore, Bax concentration had a profound effect on ROS levels in NGF-replete cells treated with oligomycin. These differences cannot be explained by differential dye loading as the same amount of CM-H2DCFDA loads into cells of each of the three bax genotypes (Kirkland et al., 2002).
Fig. 1. Oligomycin increased both ROS and ΔΨm more in NGF-replete bax−/− neurons than in neurons with bax+/+ and bax+/− genotypes.
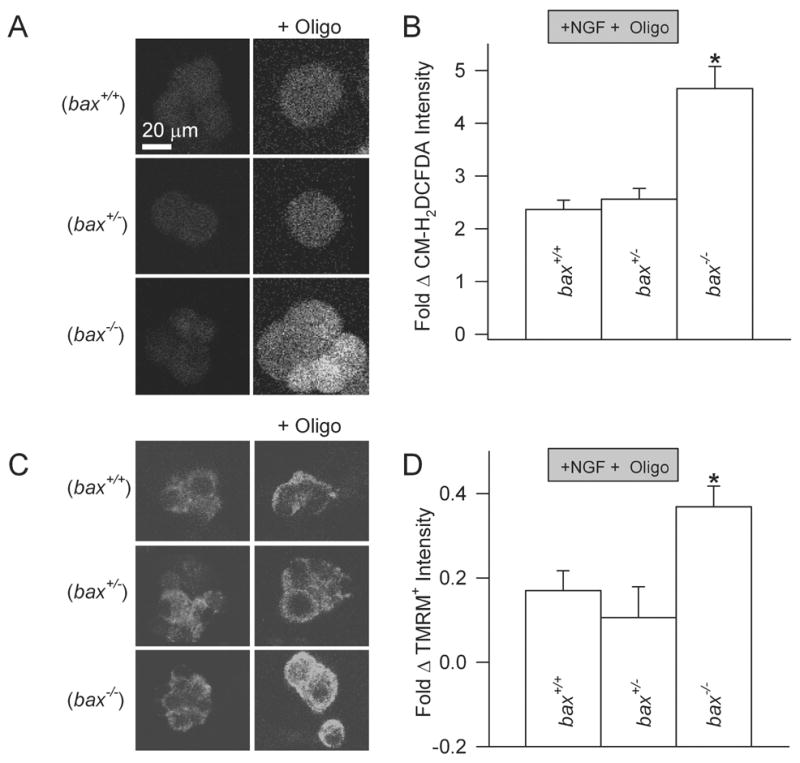
(A) Confocal micrographs showing NGF-replete neurons loaded with the redox-sensitive dye, CM-H2DCFDA. Left images show the very faint staining observed in bax+/+, bax+/−, and bax−/− neurons maintained in NGF-containing medium. Right images show neurons of the same three genotypes loaded with CM-H2DCFDA and exposed to Oligomycin (5 μg/ml). Note the increase in dye fluorescence compared to the neurons on the left indicating that the dye is oxidized.
(B) In NGF-replete cells, oligomycin (5 μg/ml) elevated ROS (CM-H2DCFDA intensity) to equivalent levels in bax+/+ and bax+/− neurons (p = 0.44). However, it increased ROS much more in NGF-replete bax−/− neurons than in neurons of the other two genotypes (p = 0.004 compared to the increase in bax+/+ cells). Treatment with oligomycin was done during the 20 min of incubation with CM-H2DCFDA. Oligomycin was also included in the L15 medium in which the cells were maintained during microscopy. n = 150–183 neurons. All experiments were done at least three times with neurons from at least three separate platings.
(C) Confocal micrographs of neurons loaded with the ΔΨm–dependent dye, TMRM+ (10 nM continuous exposure). Left images show NGF-replete bax+/+, bax+/−, and bax−/− neuronal somas loaded with TMRM+. Right images show NGF-replete bax+/+, bax+/−, and bax−/− neurons loaded with TMRM+ while also exposed to oligomycin (5 μg/ml). The increase in TMRM+ fluorescence intensity indicates hyperpolarization of ΔΨm.
(D) Oligomycin (5 μg/ml) increased ΔΨm to similar levels in NGF-replete neurons having either bax+/+ or bax+/− genotypes (p = 0.47) but had a greater effect on bax−/− cells (p < 0.0001 compared to the increase in bax+/+ cells). Neurons were incubated for 20 minutes with oligomycin and TMRM+ (10 nM). Both compounds were also included in the L15 medium used for microscopy. n = 133–180 neurons. All experiments were done at least three times with neurons from at least three separate platings.
To explore a possible relationship between ΔΨm and ROS in the oligomycin-treated neurons, we used the ΔΨm-sensitive dye, tetramethylrhodamine methyl ester (TMRM+; Molecular Probes) in non-quench mode (Nicholls and Budd, 2000; Nicholls and Ward, 2002; Nicholls and Ferguson, 2002). Cultures were incubated for 20–25 min at 35° C in the appropriate experimental medium containing TMRM+ (10 nM). Cultures were then washed 2X with Leibovitz’s L15 media containing the appropriate experimental treatments and TMRM+. They were left in the last wash for confocal microscopy. The dye was excited with the 543 nm line of the confocal lasers. The TRITC photomultiplier of the confocal microscope was used for image acquisition. Fig. 1C, D shows that oligomycin increased ΔΨm in neurons of each bax genotype. There were no differences in the ΔΨm in cells with each bax genotype without oligomycin treatment. However, Bax concentration was inversely related to ΔΨm in NGF-replete, oligomycin-treated neurons. The most hyperpolarized (highest) ΔΨm was found in bax−/− cells while the lowest ΔΨm was in bax+/− and bax+/+ cells. Therefore, higher Bax concentrations were associated both with lower ROS levels and lower ΔΨm in oligomycin-treated cells. These findings suggest that Bax had a depolarizing effect on ΔΨm in NGF-replete sympathetic neurons and that the augmentation of mitochondrial ROS production by bax deletion in oligomycin-treated cells may have been secondary to mitochondrial hyperpolarization resulting from the absence of this depolarizing effect.
The effect of Bax concentration on ROS in the NGF-replete neurons was the opposite of that observed in NGF-deprived cells where higher Bax protein concentration is associated with elevated ROS levels (Kirkland et al., 2002). One possible explanation for this apparent discrepancy is that, during apoptosis, Bax-induced release of cytochrome c from the mitochondria causes activation of caspases that then attack mitochondrial respiratory complexes causing increased ROS production (Ricci et al., 2004). However, Bax also has pro-oxidant effects in apoptotic neurons that are separable from effects related to cytochrome c release (Kirkland and Franklin, 2001; Kirkland et al., 2002). Another possible explanation for the apparent discrepancy is that Bax also has a pro-oxidant effect in NGF-replete neurons causing them to upregulate antioxidant defense mechanisms. This possibility is suggested by the slightly elevated ROS in bax+/+ cells as compared to bax−/− neurons. While the suppression of basal ROS by bax deletion did not achieve high statistical significance, it is possible that it reveals a trend that would be made more apparent with additional data. The elevated ROS observed in oligomycin-treated bax−/− as compared to bax+/+ neurons might then be explained by a diminished ability of the former neurons to detoxify increased ROS. However, neurons of all three genotypes exhibit an identical, linear relationship between H2O2 concentrations and CM-H2DCFDA intensities over the range of CM-H2DCFDA intensities reported here (Kirkland et al., 2002). This finding suggests it is unlikely that the observed differences in ROS levels in neurons of the three bax genotypes can be explained by differences in their abilities to detoxify ROS. Moreover, the data presented here shows that oligomycin increased ΔΨm more in the bax−/− than in the bax+/+ neurons which cannot be explained by differences in cellular antioxidant capacity. Elevated production of ROS by mitochondria is favored by increased ΔΨm because of thermodynamic considerations (Nicholls and Budd, 2000; Nicholls and Ward, 2002; Nicholls and Ferguson, 2002). Therefore, the most likely explanation for the effects of Bax deletion on elevated ROS in the oligomycin-treated neurons is that Bax influences ΔΨm. How Bax could exert this type of effect is unknown. Bax causes mitochondrial depolarization in digitonin-permeabilized astrocytes (Carvalho et al., 2004). This effect may be secondary to Bax-induced depletion of cytochrome c from the mitochondrial electron transport chain. That cannot be the explanation for the data reported here as the cells were not apoptotic and Bax levels should be unrelated to amount of cytochrome c retained in mitochondria. Confirming this statement, we have found that there are no differences in the amounts of cytochrome c in neurons with each of the three bax genotypes (data to be reported elsewhere). Therefore, Bax can affect ΔΨm and ROS levels even in non-apoptotic sympathetic neurons. How Bax does this is unknown but could involve effects on mitochondrial respiration or on the proton gradient of the inner mitochondrial membrane.
Acknowledgments
This work was supported by National Institutes of Health grant NS37110.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carvalho ACP, Sharpe J, Rosenstock TR, Teles AFV, Kowaltowski AJ, Youle RJ, Smaili SS. Bax affects intracellular Ca2+ stores and Ca2+ wave propagation. Cell Death Differ. 2004;11:1265–1276. doi: 10.1038/sj.cdd.4401508. [DOI] [PubMed] [Google Scholar]
- Chang KL, Schmidt RE, Johnson EM., Jr Alternating metabolic pathways in NGF–deprived sympathetic neurons affect caspase-independent death. J Cell Biol. 2003;162:245–256. doi: 10.1083/jcb.200302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Elliot JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome-c dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Creedon DJ, Johnson EM, Jr, Holtzman DM. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: Requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Johnson MI, Argiro V. Techniques in tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Schinzel A, Borner C. Bcl-w(edding) with mitochondria. Trends Cell Biol. 2004;14:8–12. doi: 10.1016/j.tcb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Franklin JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci. 2001;21:1949–1963. doi: 10.1523/JNEUROSCI.21-06-01949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced prooxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002;22:6480–6490. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtelotte WG, Brown GAJ, Korsmeyer SJ. Bax- deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I, Desagher S, Eskes R, Antonsson B, André E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2001;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics. Vol. 3. Academic Press; London, UK: 2002. [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and cell death: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, Bcl-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM., Jr Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–453. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H. Evaluation of 2′, 7′-dichlorofluorescin and dihydrorhodamine123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]