

Abstract
The ATM and ATR kinases function at the apex of checkpoint signaling pathways. These kinases share significant sequence similarity, phosphorylate many of the same substrates, and have overlapping roles in initiating cell cycle checkpoints. However, they sense DNA damage through distinct mechanisms. ATR primarily senses single stranded DNA (ssDNA) through its interaction with ATRIP, and ATM senses double strand breaks through its interaction with Nbs1. We determined that the N-terminus of ATR contains a domain that binds ATRIP. Attaching this domain to ATM allowed the fusion protein (ATM*) to bind ATRIP and associate with RPA-coated ssDNA. ATM* also gained the ability to localize efficiently to stalled replication forks as well as double strand breaks. Despite having normal kinase activity when tested in vitro and being phosphorylated on S1981 in vivo, ATM* is defective in checkpoint signaling and does not complement cellular deficiencies in either ATM or ATR. These data indicate that the N-terminus of ATR is sufficient to bind ATRIP and to promote localization to sites of replication stress.
Keywords: ATM, ATR, ATRIP, DNA damage, checkpoint
INTRODUCTION
Cellular responses to genotoxic stress in eukaryotic cells are coordinated by members of the phosphoinositide kinase related protein kinase (PIKK) family. Two members of this family, Ataxia-Telangiectasia mutated (ATM) and ATM and Rad 3-related (ATR), are particularly important in regulating cell cycle checkpoints in response to DNA damage and replication stress (15, 22). ATM is mutated in patients with the clinical disorder ataxia telangiectasia (A-T), a disease characterized by a number of debilitating symptoms, including chromosomal instability and cancer predisposition. ATR mutations are rare because ATR function is essential for cellular viability (6, 11). Hypomorphic mutations in ATR have been found in patients with Seckel syndrome (21). The ATM and ATR checkpoint signaling pathways may be significant tumorigenesis barriers (4, 5, 14).
ATM and ATR share significant sequence similarity, phosphorylate many of the same substrates, and have overlapping functional activities. However, they respond to different types of genotoxic stress. ATM primarily is activated in response to double strand breaks, while ATR responds to DNA damage or replication stress that exposes regions of single-stranded (ss) DNA (9). Consequently, ATM-deficient cells are sensitive to genotoxic agents that generate DNA double strand breaks (DSBs), such as ionizing radiation (IR) and radiomimetic chemicals, but they exhibit normal responses to most other types of DNA damaging agents including ultraviolet (UV) radiation and agents that stall replication forks such as hydroxyurea (HU). In contrast, ATR responds to DNA lesions that can initiate the uncoupling of polymerase and helicase activities at replication forks including UV radiation and HU (7).
A common step that regulates ATM and ATR signaling is localization of these kinases to sites of DNA damage or stalled replication fork. ATM localization is mediated by binding to the Nbs1 protein (13). Nbs1 is one component of the Mre11-Rad50-Nbs1 (MRN) complex that is critical for double strand break repair. ATR localization is dependent on the ATRIP protein, which serves as a subunit of the kinase (3, 11). ATRIP can recognize single stranded DNA via an interaction with the ssDNA binding protein RPA (26).
We reasoned that the ability of ATRIP to bind to RPA-coated ssDNA and ATR but not ATM provides ATR the ability to respond many types of genotoxic stress that ATM does not sense. To test this hypothesis we constructed an ATM protein (ATM*) that has gained the ability to bind to ATRIP. ATRIP binding allows ATM* to associate with RPA-ssDNA and to gain the ability to be efficiently recruited to sites of replication stress.
MATERIALS AND METHODS
DNA constructs and siRNA
HA-ATM was made from Flag-ATM. 3×HA tag was amplified by PCR and inserted into Not I/Sca I sites in Flag-ATM vector (24). Myc-Nbs1 is a gift from Dr. Stephen P. Jackson (Cambridge University, UK). An ATR N-terminal fragment containing amino acids 1–388 was subcloned upstream of the ATM open reading frame by PCR to make ATM*. HA-NLS-ATRIP and HA-NLS-ATRIP108-791 retroviral constructs and ATRIP siRNA were described previously (2, 3).
Cell Culture
HCT116 and ATRflox/− cells were maintained in McCoy’s medium supplemented with 10% fetal bovine serum. Deletion of the ATR gene from the ATRflox/− cells was performed as described previously (11). Stable cell lines in ATRflox/− cells were made by transfecting with HA-tagged ATR, ATM or ATM* Tetracycline-inducible expression vector and selected under 300 μg/ml Hygromycin B (Invitrogen) for single colonies. AT5BIVA (GM05849B) cells were obtained from the Coriell Cell Repositories and maintained in DMEM + 10% fetal bovine serum. To make stable cell lines for ATM and ATM* in AT5BIVA, ATM or ATM* expression vector was transfected into AT5BIVA cells by Fugene 6 (Roche), and selected either with 400 μg/ml G418 (for ATM) for single colonies or with 100 μg/ml Zeocine (for ATM*) for single colonies.
Drug Treatment, DNA Damage, and G2/M checkpoint assay
Hydroxyurea was purchased from Sigma, dissolved in water at 1 M, and stored frozen. Cells were treated with ionizing radiation from a 137Cs source at a dose rate of 1.8 Gy/min. UV radiation was administered with a Stratalinker (Stratagene) after cells were washed once with PBS. Unless otherwise indicated HU treatment was with 1mM HU for 3.5 hours, UV was with 50J/m2 followed by 3.5 hours incubation and IR was with 10 Gy followed by 1 hour incubation.
The G2/M checkpoint assay was performed essentially as described previously (8). In short, untreated or irradiated cells were harvest 1 hour after treatment. Cells were fixed, permeabilized and stained with anti-phosphoserine 10 histone H3 antibody and propidium iodide. Flow cytometry was performed on a BD Biosciences FACSCalibur and the percentage of mitotic cells was determined from the phopshoserine positive cells with 4n DNA content.
Antibodies
The ATRIP antibody was described previously (11). Antibodies to Chk1 and ATR were purchased from Santa Cruz Biotechnology. Chk1 P-S317 and Chk1 P-S345 antibodies were purchased from Cell Signaling Technology (Beverly, MA). ATM, Nbs1, and Mre11 antibodies were purchased from Novus Biologicals (Littleton, CO). ATM P-S1981 antibody was purchased from Rockland Immunochemicals (Gilbertsville, PA). Flag antibody was from Sigma (Sigma, MI), Myc and HA antibodies were purchased from Covance (Covance, CA). Fluorescein isothiocyanate- (FITC) and rhodamine-red–conjugated secondary antibodies were from Jackson Immuno-Research Laboratories (West Grove, PA).
Immunoblotting
Cells were lysed in 25 mM Tris (pH 7.5), 150 mM NaCl, 0.5% Igepal CA-630, 1 mM EDTA supplemented with 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM NaF, 20 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride. Lysates were cleared by centrifugation prior to determination of protein concentration (Bio-Rad). Lysates were boiled with equal volume 2×SDS sample buffer and separated by SDS-PAGE prior to immunoblotting.
RPA-ssDNA Binding Assay
Purification of recombinant RPA and binding to ssDNA were performed as previously (3). 293T cells were transfected with HA-ATRIP construct in combination with Flag-ATR, Flag-ATM or Flag-ATM* vectors. Cells were lysed in 25 mM Tris (pH 7.5), 50 mM NaCl, 1 mM EDTA, 0.75% CHAPS supplemented with 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM NaF, 20 mM β-glycerophosphate, 1 mM sodium vanadate.
Immunofluorescence
ATRflox/− cells lines stably expressing ATR, ATM, ATM* were cultured on glass slides and induced for exogenous expression with 1 μg/ml tetracycline for 24 hrs. Fixation and antibody staining were done as previously described (20). Images were obtained with a Zeiss Axioplan microscope equipped with a Zeiss camera and software.
IR Survival Assay
Cells were serially diluted to appropriate concentrations and seeded onto 100-mm tissue culture dishes in triplicate for 12 h. Ionizing radiation was applied to cells by using 137Cs source at a dose rate of 1.8 Gy/min. Viable colonies containing at least 50 cells were counted after 3 weeks incubation time, and survival fractions were calculated as the plating efficiency of treated cells relative to the plating efficiency of untreated control cells.
Kinase Assays
Kinase complexes were purified with Flag- or HA-agarose beads. Purifications were performed as described in (16) with some modifications. Purified immune complexes were washed with kinase buffer (10 mM HEPES pH7.5, 50 mM β-glycerophosphate, 50 mM NaCl, 10 mM MgCl2, 10 mM MnCl2) and incubated with recombinant GST-MCM2 (amino acids 79–138) or Brca1 (amino acids 1351–1152) (12) in the presence of 10 μM cold ATP and 0.5 μl γ-32P-ATP (3000Ci/mmol) at 30°C for 20 minutes. Reactions were terminated with 2X SDS-sample buffer and boiling.
RESULTS
Characterization of an ATR and ATM chimera
Unlike ATM, the ATR kinase can sense replication stress due its ability to associate with RPA-coated single stranded DNA. This ATR ability is dependent on the associated protein ATRIP (9). Since ATRIP does not bind to ATM we reasoned that it might act as a specificity determinant for ATR allowing it to respond to different genotoxic insults than ATM. If this hypothesis is correct, then it might be possible to confer some ATR properties onto ATM by allowing ATM to bind ATRIP. We previously determined by two-hybrid analysis that the N-terminus of ATR contain an ATRIP binding domain. Therefore, we attempted to test this hypothesis by creating chimeric ATM/ATR proteins in which portions of the ATR N-terminus were substituted for ATM N-terminal regions. These proteins were invariably mislocalized into the cytoplasm (data not shown). However, we were successful in creating a nuclear localized fusion protein containing amino acids 1–388 of ATR attached to full-length ATM (Fig. 1A). This fusion protein (ATM*) had a similar ability to bind ATRIP as ATR confirming that the first 388 amino acids of ATR are sufficient to bind to ATRIP (Fig. 1B). In contrast, wild-type ATM does not bind ATRIP.
Fig. 1.
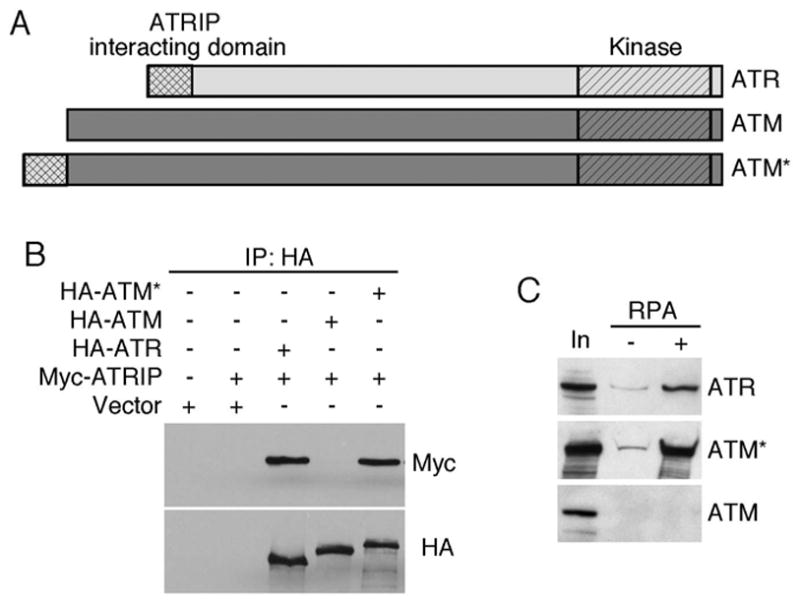
The N-terminus of ATR is sufficient to promote ATRIP and RPA-ssDNA binding properties onto ATM. (A) Schematic diagram of ATR, ATM, and ATM*. (B) 293T cells were transfected with HA-ATM, HA-ATR, HA-ATM* and Myc-ATRIP as indicated. Following lysis, HA-immunoprecipitates were separated by SDS-PAGE and blotted with Myc and HA antibodies. (C) Cell lysates containing ATR, ATM or ATM* were incubated with ssDNA bound to sepharose beads in the presence (+) or absence (−) of recombinant RPA. After washing, the bound proteins were eluted, separated by SDS-PAGE and immunoblotted. 10% of the lysate input (In) is included for comparison.
ATRIP binding promotes RPA-ssDNA association and localization of ATM* to stalled replication forks
If ATRIP is sufficient to allow ATR to sense RPA-coated ssDNA, ATM* should have gained this function. We tested whether ATM* can bind to RPA-ssDNA in vitro. Extracts from cells containing over-expressed HA-ATRIP in combination with Flag-ATR, Flag-ATM or Flag-ATM* were incubated with ssDNA or RPA-ssDNA bound to streptavidin beads. As expected, ATR-ATRIP complex bound preferentially to the beads when RPA was present (Fig. 1C). ATM did not bind to the RPA-ssDNA. However, ATM*-ATRIP had an equivalent ability to bind to the RPA-coated ssDNA as ATR-ATRIP (Fig 1C). Thus, the addition of the ATRIP binding domain of ATR onto ATM provided ATM the ability to bind to RPA-ssDNA.
To test whether ATM* also gained the ability to sense ssDNA in vivo we examined its ability to localize to sites of DNA damage and replication stress by indirect immunofluorescence. HA-ATR, HA-ATM, and HA-ATM* are localized diffusely throughout the nucleus in undamaged cells. After exposure to IR, ATR, ATM, and ATM* redistributed to intra-nuclear foci (Fig. 2A–C). These IR-induced HA-ATR foci, HA-ATM foci, or HA-ATM* foci are co-localized with ATRIP foci (Fig. 2A–C). Thus, localization of ATM, ATR, and ATM* to double strand breaks is nearly equivalent.
Fig. 2.
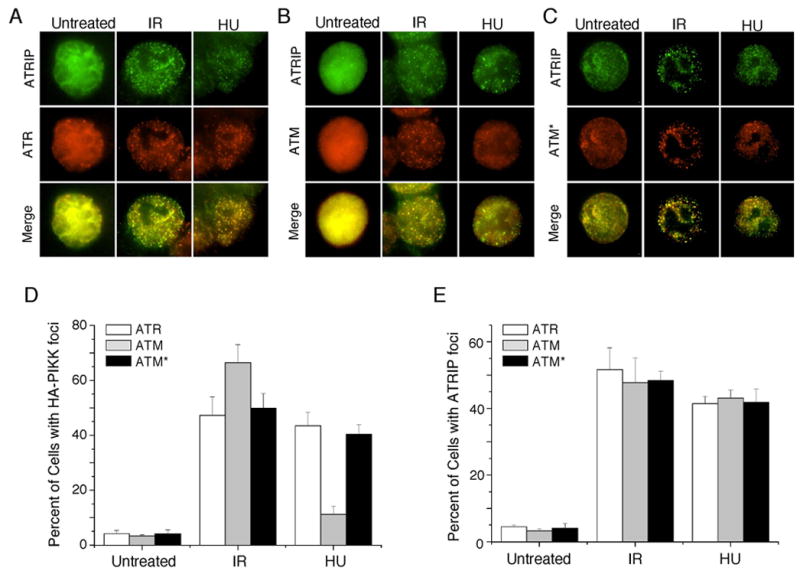
ATM* localizes to sites of double strand breaks and replication stress. ATRflox/−cells stably expressing (A) HA-ATR, (B) HA-ATM, or (C) HA-ATM* were treated with ionizing radiation (IR) or hydroxyurea (HU). 3.5 hours after treatment cells were stained with HA and ATRIP antibodies and appropriate fluorescent secondary antibodies. The percentage of cells with (D) HA or (E) ATRIP foci were scored. Error bars represent the standard deviation.
Unlike localization to double strand breaks, localization of ATM, ATR, and ATM* to stalled replication forks induced by HU treatment was significantly different. HA-ATM forms foci in only a small percentage (11%) of HU treated cells, which may represent those instances when the stalled fork collapses into a double strand break (Fig. 2D). In contrast, both HA-ATR and HA-ATM* are equally capable of localizing to sites of stalled replication forks. Approximately 40% of cells contained HA-ATR or HA-ATM* foci (Fig. 2D). These data suggest that conferring ATRIP binding capability onto ATM (ATM*) promotes a gain of function for ATM to allow it to efficiently localize to sites of stalled replication forks and sense regions of RPA-ssDNA in cells.
ATM* is an active protein kinase
To test whether ATM* retains kinase activity, wild-type ATM, a catalytically inactive ATM mutant and ATM* were tested in an in vitro kinase assay. The kinases were affinity purified from transfected 293T cells using the Flag epitope attached to the N-terminus of these proteins. Equivalent amounts of recombinant proteins were incubated with [γ-32P] adenosine triphosphate (ATP) and recombinant glutathione S-transferase (GST)-conjugated Brca1 protein containing amino acids 1351–1552 (GST-Brca11351–1552). GST-BRCA11351–1552 contains several phosphorylation sites for both ATM and ATR (12, 23). Both wild-type ATM and ATM* phosphorylated GST-Brca11351–1552 to nearly equivalent levels, whereas kinase-inactive ATM does not phosphorylate this substrate (Fig. 3A).
Fig. 3.
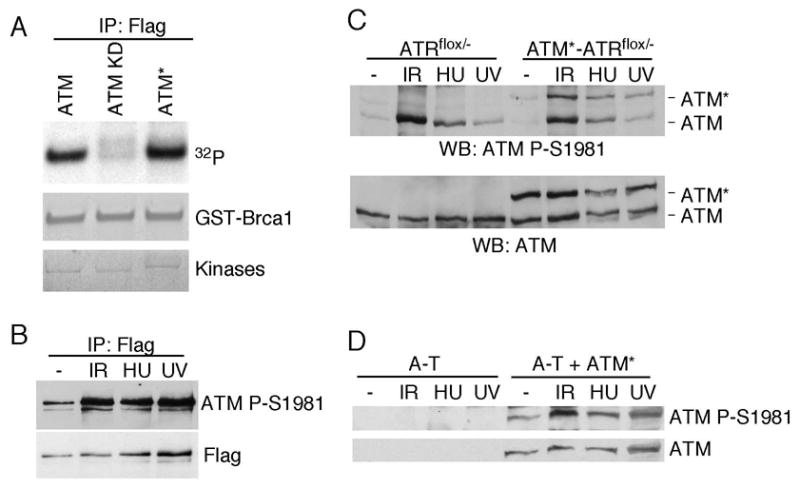
ATM* is an active kinase in vitro and autoophosphorylates in cells. (A) ATM, ATM kinase-dead (KD) or ATM* were immunoprecipitated from transiently transfected cells and immune-complex kinase assays were performed with recombinant GST-BRCA11351-1552 substrate. (B) Flag-ATM* was immunoprecipitated from transfected cells following treatment with IR, HU, or UV. Immunoprecipitates were blotted with antibodies to Flag and ATM phospho-S1981. (C) ATRflox/− cells or ATRflox/− cells stably expressing ATM* were treated with IR, HU, or UV. Cell lysates were immunoblotted with ATM or ATM phopsho-S1981 antibodies. (D) A-T cells or A-T cells expressing ATM* were treated with IR, HU, or UV. Cell lysates were harvested, separated by SDS-PAGE and blotted with ATM or ATM P-S1981 antibodies.
Following irradiation, wild-type ATM auto-phosphorylates on several serines including S1981 which correlates with its activation (1). To test if ATM* is also activated in cells, we measured phosphorylation using the S1981 phospho-peptide specific antibody. Flag-ATM* was expressed in 293T cells which were then exposed to IR, HU, or UV radiation. Flag-ATM* immunoprecipitates were recognized by the P-S1981 antibody and the amount of phosphorylation increased when Flag-ATM* was isolated from cells exposed to genotoxic stress (Fig. 3B). We also created stable ATRflox/− cell lines expressing ATM*. ATRflox/− cells are derived from HCT116 cells and have a conditional ATR allele (11). We observed similar phosphorylation of ATM* and endogenous wild-type ATM in these cells after ionizing radiation (IR), hydroxyurea (HU), and ultraviolet (UV) radiation treatment (Fig. 3C). To ensure S1981 phosphorylation on ATM* was not due to trans-phosphorylation by endogenous wild-type ATM, we confirmed that ATM* expressed in A-T cells is phosphorylated on S1981 phosphorylation (Fig. 3D). These data suggest that ATM* is an active kinase in vitro and can autophosphorylate in vivo.
ATM* cannot substitute for ATM
Since ATM* contains full-length ATM, forms radiation-induced foci, is an active kinase, and is phosphorylated on the activating S1981 site in response to DNA damage, we expected that it might function similar to wild-type ATM in cells. We tested this possibility by expressing either ATM or ATM* in an A-T cell line (AT5BIVA=GM05849B). Stable cell lines expressing comparable amounts of ATM and ATM* were created (Fig. 4A). After IR treatment, Chk2 is phosphorylated in ATM-complemented cells. However, Chk2 phosphorylation is severely attenuated in ATM*-complemented cells (Fig. 4B). A second ATM substrate, Kap1 (25), is phosphorylated to a much lesser extent in the ATM*-complemented cells compared to wild-type ATM-complemented cells. We also examined the ability of the ATM* protein to rescue the IR-sensitivity of A-T cells. While wild-type ATM significantly reduced the IR-hypersensitivity of A-T cells, the ATM* protein did not (Fig. 4C). In addition, A-T cells complemented with ATM* continue to have a defective G2/M checkpoint response to ionizing radiation and enter mitosis in the presence of DNA double strand breaks (Fig. 4D). Thus, despite localization to sites of DNA damage and phosphorylation on S1981, ATM* cannot substitute for wild-type ATM.
Fig. 4.
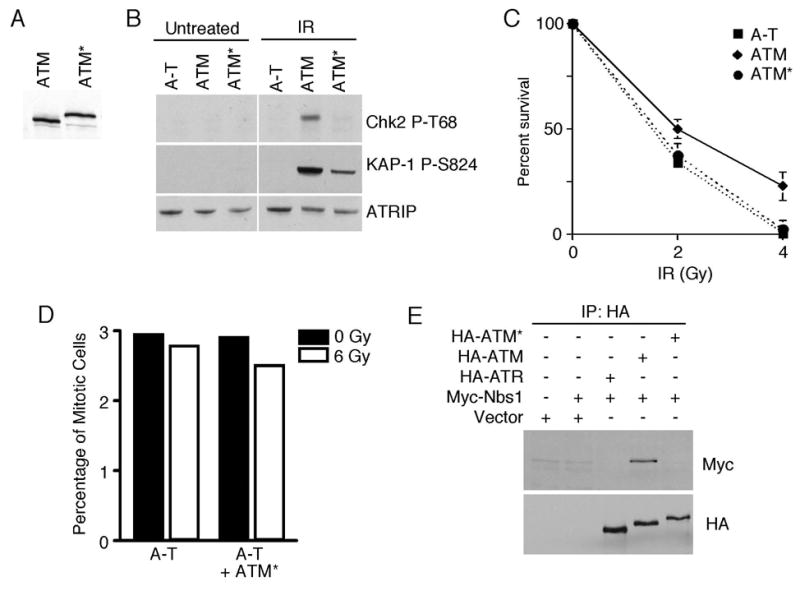
ATM* does not complement A-T cells. (A) Cell lysates from stable A-T cell lines expressing wild-type ATM, or ATM* were immuno-blotted with antibodies to ATM to demonstrate equal expression. (B) A-T, A-T/ATM or A-T/ATM* cell lines were treated with IR. Cell lysates were separated by SDS-PAGE and immunoblotted with the indicated antibodies. (C) A-T, A-T/ATM or A-T/ATM* cell lines were treated with 0, 2, or 4Gy of IR and colony formation was assessed after 3 weeks. Error bars represent standard deviations. (D) A-T or A-T/ATM* cell lines were treated with 6 Gy of IR and their ability to arrest at the G2/M checkpoint was assessed. The percentage of cells in mitosis with and without ionizing radiation is shown. (E) 293T cells were transfected with HA-ATM*, HA-ATM, HA-ATR, and Myc-Nbs1 expression vectors as indicated. HA-immunoprecipitates were separated by SDS-PAGE and blotted with Myc and HA antibodies.
To understand why ATM* cannot function equivalently to ATM we tested whether it binds to Nbs1 properly. Nbs1 is one of the components of the Mre11-Rad50-Nbs1 (MRN) complex, which is essential for DNA double-strand–break repair and genomic stability. It also promotes ATM localization to and perhaps activation at sites of double strand breaks. Wild-type ATM co-immunoprecipitates with Nbs1 as previously reported (13). However, ATM* did not co-immunoprecipitate with Nbs1 (Fig. 4E). The addition of the ATRIP binding domain onto ATM may block Nbs1 access, and may in part explain why ATM* cannot function equivalently to ATM in cells.
ATM* cannot substitute for ATR
ATM* binds ATRIP, is an active kinase, and localizes to sites of DNA damage and stalled replication forks. Therefore, we suspected that ATM* might be able to function like ATR and complement loss of endogenous ATR. We introduced ATM* or ATR into the ATRflox/− conditional knock-out cell line and selected for clonal cell lines that expressed these proteins under control of a tetracycline-inducible promoter. Infection of these cells with adenovirus encoding the Cre recombinase (Ad-Cre) deletes the endogenous ATR gene in approximately 90% of the cells in the population (11). Infection with adenovirus encoding GFP (Ad-GFP) serves as a control. Endogenous ATR can phosphorylate Chk1 after UV radiation treatment in cells infected with the control Ad-GFP (Fig. 5A). In ATRflox/− cells that are not complemented with a cDNA, Ad-Cre infection to delete ATR severely reduced UV radiation-induced Chk1 phosphorylation as expected. Ectopic expression of wild-type ATR but not ATM* restores phosphorylation of Chk1 on S345 or S317 after UV radiation in Ad-Cre infected cells. ATM* also does not phosphorylate Chk1 in cells treated with HU or IR (Fig. 5B). In addition, we found that ATM* is unable to support the viability of the ATRflox/− cells after deletion of endogenous ATR (data not shown). These data indicate that ATM*, despite being an active kinase, binding to ATRIP and localizing to sites of damage and stalled forks like ATR cannot substitute for ATR in cells.
Fig. 5.
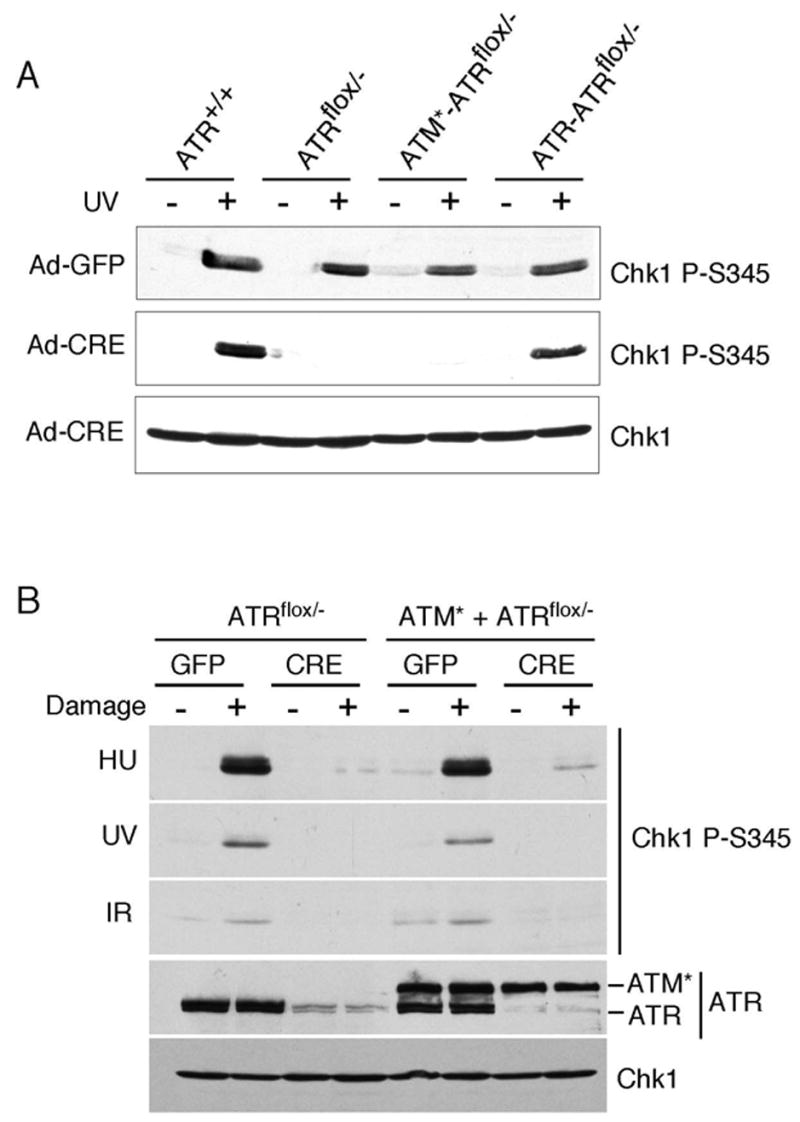
ATM* does not complement ATR-deficient cells. (A) ATR+/+, ATRflox/−, or ATRflox/−cells expressing ATM* or ATR were infected with adenovirus expressing either GFP as a control or CRE to delete the “floxed” ATR allele. Four days after infection cells were treated with 50J/m2 of UV, incubated for 1 hour, and harvested. Following SDS-PAGE separation, cell lysates were immunoblotted with the indicated antibodies. (B) ATRflox/−, or ATRflox/− cells expressing ATM* were infected with GFP or CRE adenovirus then treated with HU, UV, or IR. Cell lysates were separated by SDS-PAGE and blotted with the indicated antibodies.
To understand why the ATM*-ATRIP complex cannot function like ATR we asked whether it can be activated by TopBP1. TopBP1 is a direct activator of ATR-ATRIP complexes and is required for phosphorylation of all ATR substrates (16, 19) HA-ATM, HA-ATR, or HA-ATM* protein complexes were purified by HA affinity purification from stable ATRflox/− cell lines that inducibly express these proteins. In vitro kinases assays with GST-MCM2 as a substrate were performed (10). ATR activity towards MCM2 was increased by a TopBP1 C-terminal fragment containing the ATR activation domain but not by a control fragment that does not bind to ATR (Fig. 6 and data not shown). However, neither ATM activity (data not shown) nor ATM* activity towards substrate is changed by TopBP1. These data indicate that although ATM* is an active kinase in vitro and can bind to ATRIP, the kinase activity of ATM*-ATRIP complexes cannot be stimulated by TopBP1 like wild-type ATR-ATRIP complexes. This defect may in part explain the inability of ATM* to complement loss of ATR.
Fig. 6.
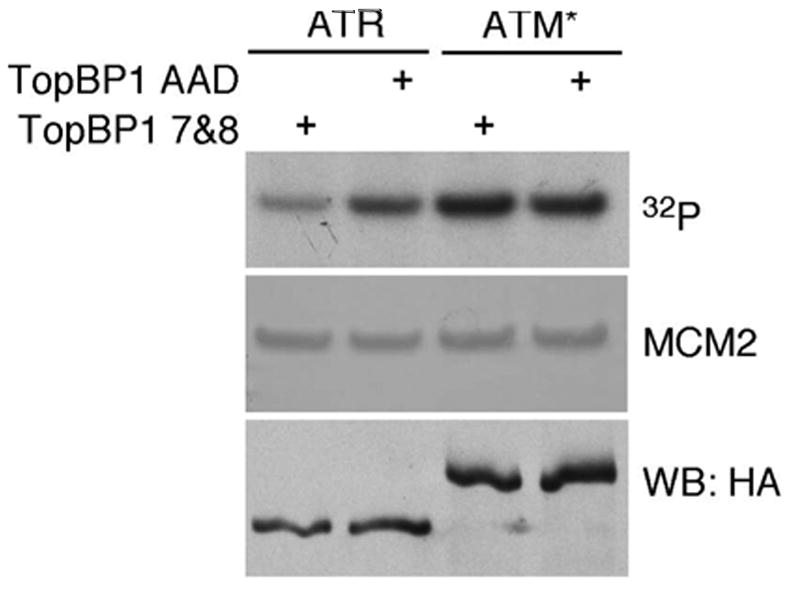
ATM* is not activated by TopBP1. HA-ATR-ATRIP or HA-ATM*-ATRIP complexes were purified from cells and placed in immune-complex kinase reactions with GST-MCM2 as a substrate. Recombinant TopBP1 containing the ATR-activation domain and BRCT repeats 7–8 (TopBP1 AAD) or just BRCT repeats 7–8 (TopBP1 7&8) was added to the reaction as indicated. Following separation by SDS-PAGE the incorporation of 32P into MCM2 was measured by autoradiography. Equal levels of MCM2 substrate was confirmed by coomassie staining and levels of HA-ATM or HA-ATM* were analyzed by immunoblotting with HA antibody.
DISCUSSION
In this study, we have tested if ATRIP could confer ATR properties to ATM by fusing the N-terminal ATRIP binding domain of ATR onto full-length ATM. The chimeric ATM* protein has the same ability to bind ATRIP as wild-type ATR. It also has gained the ability to bind RPA-ssDNA and localize to sites of replication stress similarly to wild-type ATR. In addition, ATM* is an active kinase in vitro and is phosphorylated on the activating site S1981 in cells. However, ATM* cannot substitute for either ATR or ATM in cells. Our data indicate that the N-terminus of ATR is sufficient to bind to ATRIP and promote localization of a PIKK kinase to sites of replication stress. However, localization and autophosphorylation of ATM is not sufficient to promote checkpoint signaling.
Although ATM* localizes to sites of DNA damage it is not functional. ATRIp binding to ATM* may interfere with Nbs1 binding. Nbs1 binding to ATM promotes both its localization to double strand breaks (13) as well as its activation (17, 18). Our immunofluorescence studies indicate that ATM* can localize to damage-induced foci likely through its interaction with ATRIP. Although it localizes to irradiation induced foci, the architecture of the checkpoint complexes at the double strand break in the presence of ATM* is likely to be different than with wild-type ATM. This may influence its ability to phosphorylate Chk2. Nbs1 binding may also be important for another step in ATM* activation beyond S1981 phoshorylation and localization. The inability of ATM* to function as ATR may be due to the lack of TopBP1-dependent regulation of the kinase.
Our data indicate that the N-terminal domain of ATR functions as a localization signal by mediating binding to ATRIP. Our data also provide further support for the idea that PIKK kinases are regulated by steps in addition to localization. An implication of our data is that neither the localization of PIKK kinases nor their autophosphorylation may be sufficient assays for their activation.
Acknowledgments
We thank William Dunphy (California Institute of Technology, USA) and Stephen Jackson (Cambridge University, UK) for reagents. This work was supported by National Cancer Institute grant R01CA102729. D.C. also is supported by the Pew Scholars Program in the Biological Sciences sponsored by the Pew Charitable Trusts.
References
- 1.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 2.Ball HL, Cortez D. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem. 2005;280:31390–31396. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball HL, Myers JS, Cortez D. ATRIP Binding to RPA-ssDNA Promotes ATR-ATRIP Localization but Is Dispensable for Chk1 Phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 5.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 6.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 7.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem. 2003;278:37139–37145. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- 9.Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005;19:1007–1012. doi: 10.1101/gad.1316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 12.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 13.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 14.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 15.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, Paull TT. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 18.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26:6056–6064. doi: 10.1128/MCB.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Driscoll M, V, Ruiz-Perez L, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 22.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 23.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziv Y, Banin S, Lim DS, Canman CE, Kastan MB, Shiloh Y. Expression and assay of recombinant ATM. Methods Mol Biol. 2000;99:99–108. doi: 10.1385/1-59259-054-3:99. [DOI] [PubMed] [Google Scholar]
- 25.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 26.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]