

Abstract
Vimentin exhibits a complex pattern of developmental and tissue-specific expression regulated by such growth factors as TGFβ1, PDGF, FGF, EGF and cytokines. Vimentin is expressed in the more migratory, mesenchymal cell and its expression is often down-regulated to make way for tissue-specific intermediate filaments proteins such as desmin in muscle. Here, we suggest a mechanism to explain how TGFβ1 contributes to the up-regulation of vimentin expression while blocking myogenesis. TGFβ1 binds to serine/threonine kinase receptors resulting in the phosphorylation of Smad2 and Smad 3, followed by formation of a heteromeric complex with Smad4. The translocation of this complex to the nucleus modulates transcription of selected genes such as vimentin. However, the vimentin gene lacks a consensus TGFβ1 response element. By transient transfection analysis of vimentin’s various promoter elements fused to the CAT reporter gene, we have determined that tandem AP-1 sites surrounded by GC-boxes are required for TGFβ1 induction. Mutations within this region eliminated the ability of Smad3 to induce reporter gene expression. DNA precipitation and ChIP assays suggest that c-Jun, c-Fos, Smad3 and Sp1/Sp3 interact over this region, but this interaction changes during myogenesis with TGFβ1 induction.
Keywords: Smad3, c-Jun, c-Fos, AP-1 family, Sp1, TGFβ1, vimentin, myogenesis
Introduction
Transforming growth factor-beta1 (TGFβ1) is an important regulator of cell growth and differentiation in development and cancer where it is proposed to act as a tumor suppressor at least for early stages of cancer [1]. TGFβ1 functions by binding to a complex of transmembrane receptor serine/threonine kinases, which induces phosphorylation of the Type I receptor (GS segment) by the Type II receptor [2]. Receptor complex activation results in the C-terminal phosphorylation of Smad2 and Smad3 at selected serine residues. Two receptor-activated Smads (R-Smads) form heteromeric complexes with Smad4, a common Smad family member partner for the R-Smads. The Smad complex then translocates to the nucleus, where it activates transcription through physical interaction and functional cooperation with DNA-binding factors, such as Sp1 or AP-1 family members, or co-activators such as the CREB-binding protein (CBP) and p300 [2].
The TGFβ family has been shown to be potent inhibitors of terminal differentiation in cultured myoblasts [3,4]. This suppression blocks the expression of MyoD and myogenin, two members of the bHLH family, thereby inhibiting myogenesis in skeletal muscle cell lines [5]. Smad3 has been shown to bind to the bHLH region of MyoD interfering with MyoD/E protein dimerization and subsequent cooperative binding to E-box DNA [7]. Smad3 also has the ability to interact with MEF2. This interaction prevents the association of the MyoD/E-47 dimer with MEF2 resulting in the repression of muscle-specific gene expression following TGFβ1 treatment [8]. Not only does TGFβ1 treatment block the expression of MyoD and myogenin, but it also suppresses differentiation in systems where MyoD or myogenin is overexpressed, indicating an additional post-transcriptional means of repressing muscle-specific genes [5,6].
Of the three cytoskeletal networks in eukaryotic cells the intermediate filaments (IFs) are the most complex and least well understood. The IF protein family (IFP) includes a variety of proteins found in the cyptoplasm such as the cytokeratins, glial fibrillary acidic protein (GFAP), desmin, vimentin, neurofilaments, internexin, nestin and peripherin as well as the lamins found in the nucleus [9,10]. Within the IFP family, vimentin exhibits a complex pattern of tissue- and developmental-specific expression. During development vimentin is first expressed in mesodermal cells located between the primitive streak and the proximal endoderm [11]. Initially, it is widely expressed in the embryo, but is progressively restricted to fewer cell types during terminal differentiation. For example, it is expressed in early stages of muscle or astrocyte development, but is “turned off” during differentiation to make way for the tissue-specific IFPs, desmin and GFAP, respectively [12,13]. This down-regulation is thought to be due at least in part to the transcriptional repressor, ZBP-89 [14,15]. Vimentin expression is also regulated by cell-cycle [16,17], growth factors (TGFβ1, PDGF, FGF) [16,18] and cytokines such as INF-γ [19]. In addition, the vimentin gene is up-regulated in cells that have transgressed through the epithelial-mesenchymal transition and many metastatic tumors despite their embryological origin [20,21]. Thus, it is important to determine how the vimentin gene is selectively down-regulated during the terminal differentiation of some tissues like muscle, remains expressed in others, or is aberrantly expressed in many metastatic tumor cells. Moreover, how is the vimentin gene regulated by growth factors such as TGFβ1, which impact on cell differentiation?
At present several cis-elements and associated factors have been identified within the human vimentin promoter (see Fig. 2A). These include a TATA-box, eight putative GC-boxes [22], a NF-κB site (−239 to −224) [23], a PEA3-binding site (−173 to −159) [24], a DNA binding site referred to as Δ19 (−349 to −329) [25] and at least two copies of a negative element (PS: at −329 to −289 and −645 to 631) [14]. Further upstream (at −765 and −751) are tandem AP-1 binding sites [26] and the antisilencer element, ASE [27]. The AP-1 family is composed of basic, leucine zipper (bZIP) proteins that can form either homo- or heterodimers with other family members and bind to AP-1 sites in the vimentin promoter to directly activate gene expression [26]. In addition, c-Jun and its dominant negative mutant, TAM67, can further activate vimentin gene expression by interacting with Sp1 and enhance binding to GC-box1 [28]. Given this large number of interactive partners, the AP-1 family is able to mediate the expression of a vast number of genes that affect development as well as tumor progression. Ectopic expression of c-Jun, v-Jun and JunB in myogenic cells has been shown to inhibit muscle differentiation [29,30]. Therefore, the down-regulation of c-Jun during myogenesis and/or a change in its binding partners may be important to the myogenic program [31]. In the present study, we demonstrate that TGFβ1 treatment of skeletal C2C12 myogenic cells not only prevents the down-regulation of vimentin, but actually increases vimentin expression. Yet the vimentin gene does not contain any known TGFβ1 response element. An investigation into this regulatory mechanism suggests that tandem AP-1 elements located upstream of the transcription start site are required. However, this region binds a number of factors in addition to c-Jun and c-Fos notably Smad3 and Sp1/Sp3. We show that the unique association of these multiple factors is required to mediate the transcriptional changes in vimentin expression in response to TGFβ1 treatment.
Fig. 2. TGFβ1 and Smad3 promote vimentin reporter gene activity in C2C12 cells via the tandem AP-1 elements.
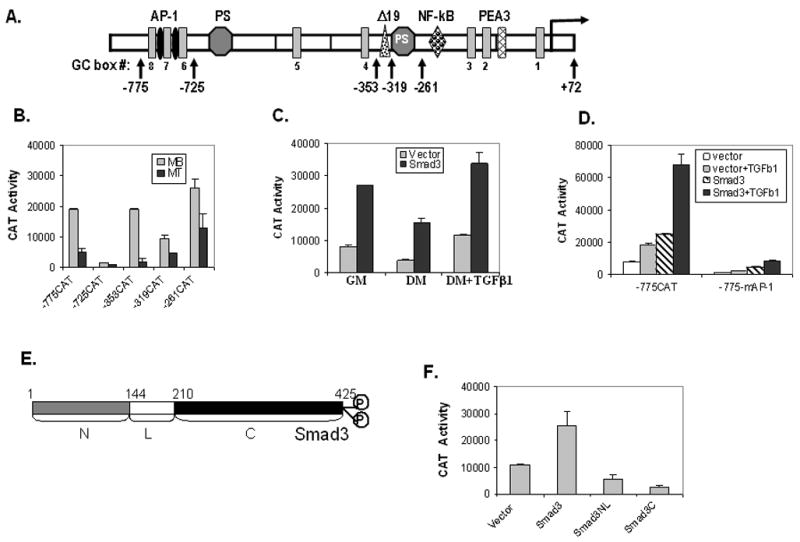
(A) Schematic diagram of the human vimentin promoter depicting its various cis-elements. The negative numbers (marked by a vertical arrow) indicate the 5′-end of different vimentin promoter constructs, which extend to +72 at the 3′-end, fused to the reporter gene, CAT. The transcriptional start site is noted by a horizontal arrow at +1. (B) Various vimentin promoter CAT constructs (-775CAT, -725CAT, -353CAT, -319CAT and -261CAT) were transiently transfected into C2C12 cells. Myoblast cells (MB) maintained in growth medium were harvested 48 h after transfection whereas myotubes (MT) were harvested 72 h after transfection and transfer to differentiation medium. Reporter gene activity was normalized to co-transfected beta-galactosidase as an internal control. Results are the average of three separate experiments performed in triplicate and bars represent the standard error. (C) Vimentin promoter construct -775CAT was transiently transfected into C2C12 cells with or without Smad3 expression plasmids as indicated. Cells were grown in either GM or DM without or with TGFβ1 as indicated. Reporter gene activity was measured as above. (D) The -775CAT or -775mAP-1CAT construct containing mutant tandem AP-1 sites was transfected into C2C12 myoblasts with either the empty vector or vector containing Smad3 without or with TGFβ1 as indicated. See Fig. 4B for sequence of the AP-1 and mAP-1 region. Reporter gene activity was measured as above. (E) Schematic structure of Smad3 indicating its deletion mutants. (F) The -775CAT construct was transiently transfected into C2C12 myoblasts with either the empty vector or vector expressing Smad3 or mutants thereof (Smad3NL or Smad3C) as indicated. Reporter gene activity was measured as above.
Materials and methods
Cell Cultures
C2C12 myoblast cells (ATCC) were maintained in high-glucose D-MEM (Invitrogen Corporation) growth medium (GM) with glutamine, supplemented with 15% FBS, penicillin (100 units/ml) and streptomycin (50 μg/ml). Sub-confluent cells were switched to differentiation medium (DM) containing 2% horse serum to initiate differentiation to myotubes. Cells were treated with 1 ng/ml TGFβ1 (Sigma) in either GM or upon switching to DM for 4 days prior to harvesting. COS-1 cells (ATCC) were maintained in high-glucose DMEM as above, but with 10% FBS.
RNA Extraction, Northern Analysis and Real Time PCR
Total RNA was extracted from C2C12 myoblasts, myotubes and TGFβ1 treated cells cultured as described above. Northern blots were carried out as previously described [32]. For PCR amplification, data acquisition and analysis were carried out by a LightCycler instrument using LightCycler 5.3.2 software (Roche, Mannheim, Germany). The SYBR green real-time PCR assay was carried out in 20 μl PCR mixture volume consisting of 0.3 μl Taq platinum DNA polymerase (Invitrogen), 0.5 μl SYBR, 0.4 μl of a 10 μM dNTP mix and 2 μl of cDNA. The primer sequences for actin were 5′-AAACTGGAACGGTGAAGGTG-3′ and 5v-AGAGAAGTGGGGTGGCTTTT-3′ and for vimentin 5′-AGGAAATGGCTCGTCACCTTCGTGAATA-3′ and 5′-GGAGTGTCGGTTGTTAAGAACTAGAGCT-3′. Actin gene amplification was carried out as follows: 95°C for 15 min, and then 50 cycles in 3 steps: 94°C for 15 sec, 55°C for 20 sec, 72°C for 15 sec. At the end of the amplification cycles, melting temperature analysis was carried out by a slow increase in temperature (0.1°C/sec) up to 95 °C. Amplification of the vimentin gene was the same as for the actin gene except for the annealing temperature (58°C) and elongation time (20 sec). Experiments were carried out in triplicate and the standard error is shown. PCR products were analyzed on a 2% agarose gel by ethidium bromide staining.
Plasmids
Various 5′-deletions (−775/+72, −725/+72, −353/+72, −319/+72, −261/+72) and site-mutated constructs (−775/+72 mAP-1) of the human vimentin promoter were fused to the CAT reporter gene as previously described (Fig. 2A) [14,28,33,34]. The numbered base defining the 5′-end of some of these constructs has been changed from previous publications to correct for approximately 50 bps incorrectly deleted from the first published sequence of the human vimentin promoter [16]. N-terminal Flag-tagged Smad3 and its truncated mutants (Smad3NL, Smad3LC, Smad3C) plus the constitutively active (TβRI-T204D) and dominant negative (TβRI-L45) TGF-β receptors were described previously [35,36]. Other plasmids were kindly provided as follows: pCMV-c-Jun and its mutants (TAM67, DBM-3, LZM-1) by Dr. Michael Birrer (NCI/NIH); E1A expression plasmids [E1A12S.WT, E1A.12S.Δ2–36, E1A.12S.Y/H(47/928)] [37] from Dr. Anton Krumm (Fred Hutchinson Cancer Research Center), and pcDNA3Z+/cFos from Dr. Tim Bos (Eastern Virginia Medical School).
Cell Transfection and CAT assay
COS-1 and C2C12 cells were transiently transfected using the calcium phosphate/DNA co-precipitation methods. Cells (2 × 105) were plated in a six-well plate, incubated at 37°C overnight, and transfected with DNA (1 μg), keeping the total amount of transfected DNA constant by adding empty vector. Forty-eight h after transfection, cell lysates were prepared using the freeze/thaw method. pCMV-β-gal was co-transfected to serve as an internal control for transfection efficiency. Β-galactosidase activities and CAT assays were performed according to the established protocol of Promega.
Whole Cell Extract Preparation
Whole Cell Extracts (WCE) were prepared from COS-1 cells transiently transfected with Flag-Smad3, c-Jun, or c-Fos by the freeze/thaw method. WCEs for immunoprecipitation and western blots were prepared by resuspending the cells in 1X lysis buffer [20 mM Hepes, pH 7.9, 150 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 10% glycerol (vol/vol), 0.5 mM phenylmethylsulfonylfluoride, 1 mM orthovanadate and protease inhibitor cocktail (Sigma) at 10 μl/1ml extract]. The protein concentration was measured using the BCA Protein Assay Kit (Pierce).
Immunoprecipitation and Western Blots
WCE (1 mg) was mixed with antibody (5 μg) and 50 μl of protein A/G PLUS-agarose in 1X lysis buffer as described above and incubated with rocking overnight at 4°C. The beads were washed three times with RIPA buffer (50 mM Tris-HCl, pH 7.5, 1% NP-40 (vol/vol), 150 mM NaCl, and 0.5% sodium deoxycholate). Antigen-antibody complexes were separated from the beads by boiling in 1X SDS sample buffer. The proteins were separated on a 10% SDS-PAGE gel and electrophoretically transferred to nitrocellulose. The membrane was blocked in 1X TBST buffer (25 mM Tris, pH 7.5, 137 μM NaCl, and 2.7 mM KCl) with 5% non-fat milk for 1 h at room temperature, and then incubated with primary antibody for 1 h at 37°C. The membrane was washed three times in 1X TBST buffer and incubated with horseradish peroxidase conjugated secondary antibody. The specific antigen-antibody interactions were detected using an ECL kit (Amersham). Antibodies used for immunoprecipitation and western blot (1:1000) are as follows: monoclonal anti-β-actin antibody (A1978), anti-Flag antibody (F3165), and anti-vimentin (V6630) from Sigma-Aldrich; and polyclonal anti-c-Jun (D) (sc-44), anti-MHC (sc-20641), anti-myogenin (sc-576), anti-tubulin (sc-9104) and anti-c-Fos (sc-1694) from Santa Cruz.
DNA Precipitation Assays
COS-1 cells transfected with plasmids containing Flag-tagged Smad3, pCMV-c-Jun or pCMV-c-Fos were harvested in 1X lysis buffer (10 mM Hepes, pH 7.9, 100 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT, 0.1% Nonidet P-40, and phosphatase and protease inhibitor cocktails from Sigma-Aldrich). Cell debris was removed by centrifugation twice for 10 min at 12,000×g at 4°C. Cell extracts were incubated with 1 μg of biotinylated double-strand oligonucleotides (dsDNA) corresponding to the wild-type or mutant AP-1 site in the vimentin promoter. Oligonucleotide sequences used are as follows: WT/AP-1; 5′-GGGCGCGGTGAGTCACCGCCGGTGACTAAGCGACCCC-3′, mAP-1; 5′-GGGCGCGGCACGTCACCGCCGGCACCTAAGCGACCCC-3′, and mSp1; 5′-GATCAGTATGAGTCAATGAAGATGACTAAGTACTAGA-3′. All oligonucleo-tides were labeled with biotin at the 5′-end. The DNA affinity precipitation assay (DAPA) was performed as described above, except that StreptAvidin MagneSphere Paramagnetic Particles (SA-PMPs) replaced streptavidin-agarose.
ChIP Assay
C2C12 myoblast cells were either harvested or allowed to differentiate for 4 days without or with TGFβ1 treatment (1 ng/ml). Proteins were cross-linked to DNA with 1% formaldehyde at 37°C for 15 min. Cross-linking was stopped by the addition of glycine to a final concentration of 125 mM at room temperature for 5 min. Cell pellets were resuspended in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, with protease inhibitor cocktail from Sigma-Aldrich). Pellets were sonicated 4 times at 30 sec each at a submaximal input followed by centrifugation for 10 min at 12,000 rpm. Supernatants were collected and diluted in dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM Tris-HCl, pH 8.1) followed by immunoclearing with salmon sperm DNA/Protein A agarose for 2 h at 4°C. Immunoprecipitation was performed overnight at 4°C with 1 μg of anti-Smad3 antibody (Zymed Lab Inc. Cat # 51–1500), c-Jun (sc-1694X), c-Fos (sc-52X), Sp3 (sc-644X), Sp1 (sc-59X) or normal rabbit IgG antibody from Santa Cruz. Following immunoprecipitation, 60 μl of salmon sperm DNA/Protein A agarose was added, and then incubated for another 1 h at 4°C. Sepharose beads were washed sequentially for 10 min each in low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 500 mM NaCl), and LiCl immune complex wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1). Beads were then washed 3 times with TE buffer and extracted twice with a total of 500 μl of elution buffer (1% SDS, 0.1 M NaHCO3). 5 M NaCl (20 μl) was added to the eluants, which were then heated at 65°C for 4 hr to reverse the formaldehyde cross-linking. After reversing the cross-linking, 10 μl of 0.5 M EDTA, 20 μl of 1 M Tris-HCl, pH 6.5 and 2 μl of 10 mg/ml Proteinase K was added to the eluants and incubated for 1 h at 45°C. DNA recovery was performed by phenol/chloroform extraction and ethanol precipitation with the addition of glycogen (20 μg) as inert carrier. The pellets were then washed with 70% ethanol and allowed to air dry. The pellets were resuspended in 20 μl TE buffer. For PCR, 1 μl out of the 20 μl of extracted DNA was used in 30 cycles of amplification. The primer sequences were as follows: 5′-GGGCTCCATGAGTCATATCC-3′ and 5′-ATCTGGCTCAAGACCTTTGC-3′. The resulting PCR product was 160 bp in length.
Results
Tandem AP-1 elements are involved in blocking the decline in vimentin expression by TGFβ1 in C2C12 cells during myogenesis
Previously, TGFβ1 has been shown to inhibit the differentiation of C2C12 myoblasts into myotubes [3,4]. Normally, vimentin gene expression is “turned off” during myogenesis to make way for the muscle-specific IFP, desmin [13,38]. Thus, we wanted to determine if TGFβ1 blocks the normal decline in vimentin gene expression [39]. C2C12 cells were cultured in growth medium with high serum (GM) to mimic myoblast conditions or in low serum (DM) to promote myotube differentiation for 4 days minus or plus TGFβ1 (Fig. 1A). In the absence of TGFβ1, C2C12 myoblasts begin to fuse to myotubes by day 2 and continue fusing at day 4. The inclusion of TGFβ1 in DM greatly reduces myogenic differentiation both in 2 and 4 day cultures. Western blots of WCEs confirm the induction of muscle-specific proteins such as myosin heavy chain (MHC) and myogenin with myotube formation (Fig. 1B) whereas there is little change noted in tubulin expression. However, TGFβ1 treatment blocks the expression of the myogenic proteins. As expected, vimentin expression remains high in myoblasts grown in GM minus or plus TGFβ1, decreases drastically upon myogenic differentiation, but remains expressed in myotubes treated with TGFβ1. Both real-time PCR and Northern blot analysis confirm that the reduction in vimentin gene expression upon differentiation to myotubes is alleviated by TGFβ1 treatment whereas the control GAPDH showed little difference (Figs. 1C and 1D). In fact, exposure to TGF β1 slightly increased the level of vimentin mRNA expression in both myoblasts and myotubes.
Fig. 1. TGFβ1 reverses the decline in vimentin gene expression during C2C12 differentiation.
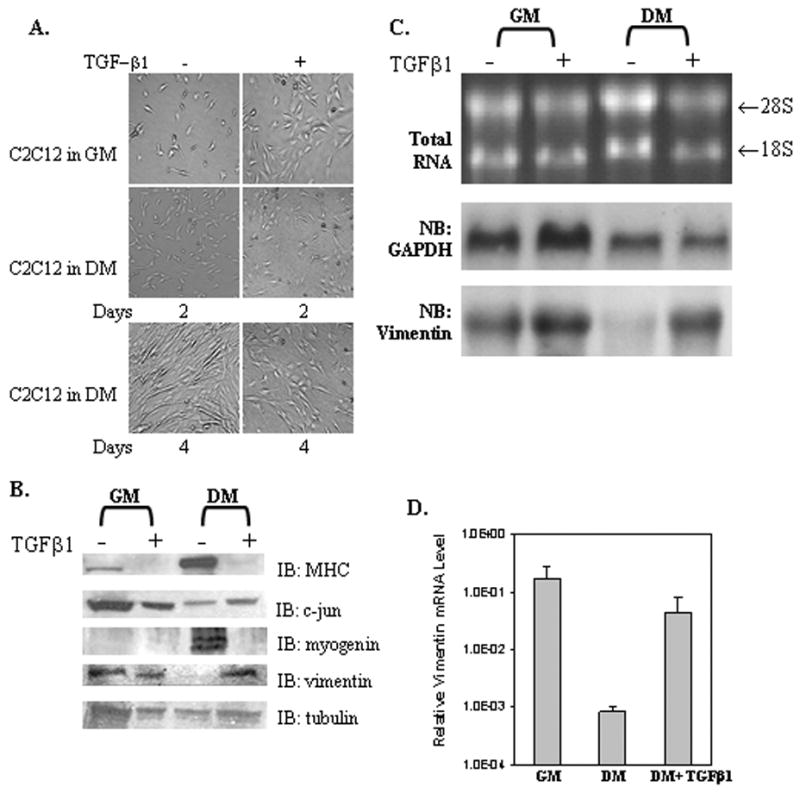
(A) C2C12 cells grown in DMEM medium plus 15% FBS (growth medium, GM) or 2% horse serum (differentiation medium, DM) without or with TGFβ1 (1 ng/ml) for 2 or 4 days. (B) Western blot analysis of WCEs (50 ug) isolated from C2C12 cells as grown in panel A for 4 days and described in Materials and Methods. (C) Northern blot analysis of total RNA (20 μg) isolated from C2C12 cells grown in GM or DM minus or plus TGFβ1 for 4 days as previously described [32]. The nitrocellulose membrane was first hybridized with a 32P-labeled human vimentin cDNA probe (bottom) and then the membrane was stripped and re-probed with a 32P-labeled GADPH cDNA (middle). Another sample of total RNA (5 μg) was stained with ethidium bromide to verify the integrity of the RNA (Top). (D) Total RNA (100 ng) was reverse-transcribed into cDNA using poly-dT as a primer followed by the use of 1/20 of this sample for real-time PCR analysis. The data was normalized to adolase gene expression. The Y-axis represents the relative mRNA levels of the vimentin gene. Results are the average of three separate experiments performed in triplicate with bars representing the standard error.
Although the vimentin gene is up-regulated by TGFβ1 treatment, a screen of the human vimentin promoter region failed to locate a canonical TGFβ1 response element (Fig. 2A). Therefore, transient transfections of various vimentin gene promoter fragments (defined as a negative number representing the unique 5′-end nucleotide to +72 and identified as vertical arrows) fused to the reporter gene CAT were performed in C2C12 myoblasts (MBs) versus myotubes (MTs) (Fig 2B). In myoblasts a typical expression pattern was noted as detected in nonmuscle cells like HeLa [14]. The proximal promoter region in construct -261CAT gave substantial reporter gene activity. Inclusion of the negative element, PS, in -319CAT showed reduced activity. Considerable activity was restored by the addition of the adjacent Δ 19 enhancer element [13,14], which was completely eliminated by inclusion of another copy of the PS element found in -725CAT [40]. The addition of DNA up to −775 restored CAT activity almost to those levels displayed by the proximal promoter region alone. Thus, it appears that the positive factors, Sp1/Sp3 and c-Jun, which have been shown to bind to their respective elements and promote vimentin gene expression as well as the repressor ZBP-89, which binds the PS elements are present in myoblasts [14,33,41]. However, considerably lower expression is noted in myotubes. Only the proximal promoter region yields substantial reporter gene activity. Thus, either these positive-acting factors are not present in myotubes or they are unable to induce gene expression [14,42,43].
Previously, Smad3 had been shown to inhibit myotube differentiation by blocking muscle bHLH transcription factors [7,8]. Could Smad3 be involved in the induction of vimentin gene expression? To address this question, Smad3 was co-transfected with the -775CAT construct, which contains most of the human vimentin promoter studied to date. In addition, this construct contains tandem AP-1 elements (located at −765 and −751), which have been previously shown to bind the transcription factor c-Jun, a known binding partner of Smad3 [44]. C2C12 cells were cultured in growth (GM) or differentiation (DM) medium for 4 days (Fig. 2C). In all cases co-expression with Smad3 produced a greater than 3-fold increase in reporter gene activity. Interestingly, myoblasts grown in high serum GM yield comparable CAT activity to cells treated with TGFβ1 for 4 days (Fig. 2C). Additional transient transfection analyses demonstrate that Smad3 induction is dependent upon the tandem AP-1 elements as mutation of these elements reduces gene expression considerably with or without TGFβ1 treatment or Smad3 co-transfection (Fig. 2D). Not only is Smad3 important to TGFβ1 induction, but full-length Smad3 is required for expression of the vimentin promoter (Figs. 2E and 2F). Deletion of either the Smad3 NL- or C-domains gave activity below that of the vector alone.
Smad3 is required for the induction of vimentin expression in COS-1 cells
To further determine what factors might be required for the TGFβ1 induction of vimentin expression, we chose to switch to COS-1 cells. Since COS-1 cells are known to express low levels of some Smad family members, we could modify Smad content and determine what is required for TGFβ1 induction (Fig. 3A, lane 1). In examining Smad2 and Smad3 levels, only endogenous Smad2 could be detected on a western blot. However, transfection with Smad2, 3, or 4 expression plasmids yields a substantial increase in Smad protein levels (Fig. 3A, lanes 2, 3 and 4). Since Smad4 was detected by a Flag-tag, its endogenous level could not be monitored in lane 1. We next determined the effect of Smad co-expression on reporter gene activity with the -775CAT construct (Fig. 3B). CAT activity was the highest with Smad3, less with Smad4, but returned to Smad3 levels with co-expression of both Smad3 and 4. Obviously, ectopic expression of Smad3 is required, but the additional expression of Smad2 or Smad4 did not seem to be required to attain the highest expression level under these culture conditions. Transient transfection analysis of vimentin’s other promoter constructs in COS-1 cells alone or with Smad3 co-expression reveals low levels of reporter gene activity, only the -775CAT construct yields high levels of activity dependent on Smad3 co-expression, which is considerably reduced with mutation of the AP-1 sites (Fig. 3C). Further transient transfection analyses were performed to determine what part of Smad3 might be important for activity of the -775CAT construct (Fig. 3D). As found for TGFβ1 induction of vimentin expression in C2C12 cells, full-length Smad3 is necessary, as co-transfection of either the C-, LC- or NL-terminal truncated Smad3 mutants (Fig. 3D) results in little activity compared to full-length Smad3. Moreover, CAT activity with Smad3 was synergized by the addition of the constitutively activated TGFβ1 receptor (TβRI-T204D), but reduced by co-transfection with a dominant negative TGFβ1 receptor (TβRI-L45). Since it has been shown previously that the tandem AP-1 sites in vimentin’s promoter bind and activate transcription via c-Jun, we wanted to determine what role c-Jun might play in Smad3 activation [28]. Co-Immunoprecipitation of c-Jun with Smad3 demonstrates that there is in fact an interaction between these two proteins (Fig. 3E, lane 4).
Fig. 3. TGFβ1 and Smad3 promote vimentin reporter gene activity via the tandem AP-1 elements in COS-1 cells.
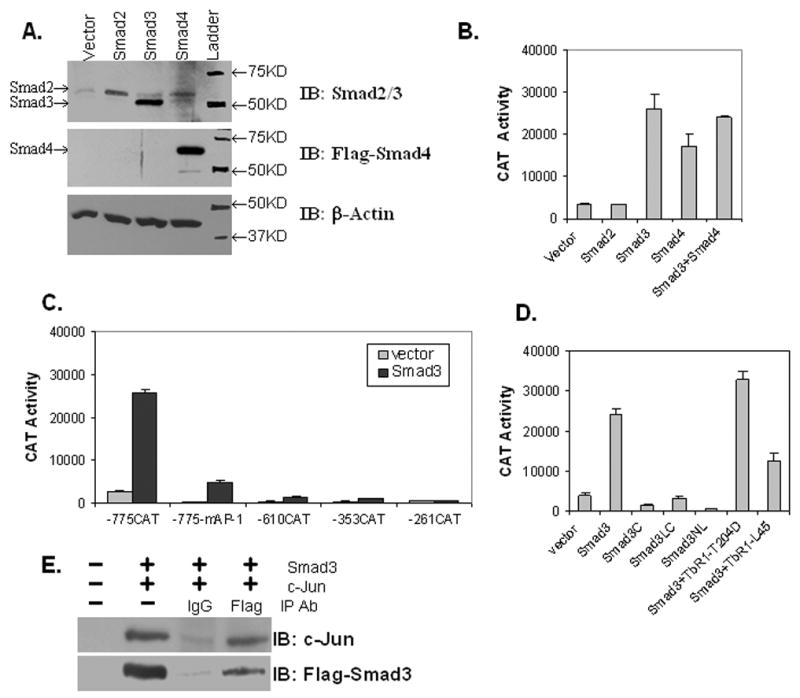
(A) Western analysis of WCEs (40 μg) isolated from COS-1 cells transfected with vector alone (lane 1) or vector containing Smad2, Smad3, or Smad4 cDNAs. Antibodies used for immunoblotting (IB) are indicated and actin is included as a loading control. (B) The -775CAT construct was transiently transfected into COS-1 cells with empty vector or vector expressing Smad2, Smad3 or Smad4 as indicated. Reporter gene activity was measured as above. (C) Constructs -775CAT, -775mAP-1CAT, -725CAT, -353CAT, -319CAT, or -262CAT were transiently co-transfected with either empty vector or the Smad3 expression plasmid into COS-1 cells. Cells were harvested 48 h after transfection. Reporter gene activity was measured as above. (D) The -775CAT construct was transiently co-transfected into COS-1 cells with empty vector, or vector expressing Smad3 or deletion mutants thereof (as described in Fig. 2E), and Smad3 plus a constitutively active (TβRI-T204D) or dominant negative (TβRI-L45) TGFβ1 receptor. Reporter gene activity was measured as above. (E) Co-immunoprecipitation of Smad3 and c-Jun from COS-1 WCEs as described in Materials and Methods. COS-1 cells were transiently transfected with empty vector (lane 1) or Flag-Smad3 and c-Jun expression plasmids. Cells were harvested 48 h after transfection and protein expression verified in WCEs (40 μg) in lane 2. WCEs (1 mg) were immunoprecipitated with IgG or Flag-antibodies and immunoblotted (IB) with either c-Jun or Flag-Smad3 antibodies as indicated.
Smad3, c-Jun, c-Fos and Sp1 interact in a multi-protein complex on vimentin’s tandem AP-1 elements
Next, we wanted to verify that c-Jun and Smad3 were binding to the promoter at the tandem AP-1 elements by a second approach. Here, we choose DNA oligonucleotide precipitation assays (DAPA) to determine what other possible proteins in addition to c-Jun could be binding to this region. For example, other AP-1 family members such as c-Fos have been linked to the TGFβ1 response via an AP-1 element [45]. Indeed, c-Jun, c-Fos and Flag-tagged Smad3 were all found to bind to the tandem AP-1 sites (Fig. 4A). In fact overexpression of c-Fos was not required to detect c-Fos binding as a small amount of endogenous c-Fos was found bound with Smad3 (Fig. 4A, lane1). To further explore requirements for protein:DNA binding, DAPA assays were repeated with wild type (WT) and mutant tandem AP-1 sites (mAP-1) as defined earlier [28]. Surprisingly, when both of the tandem AP-1 sites were mutated, there was still a measurable amount of Smad3 binding to this region (Fig. 4B). In fact, binding was comparable with WT or mAP-1 sites. However, it is noteworthy that c-Jun overexpression did appear to lower the amount of Smad3 bound to the WT compared to the mAP-1 site, additional evidence that Smad3 and c-Jun interact off DNA [46]. A further examination of the region around the tandem AP-1 elements revealed three possible GC-boxes located immediately upstream, in between, and downstream of the tandem AP-1 sites (upstream: 5′-GGCGCGG-3′, in between 5′-CCGCCGG-3′ and downstream 5′-GCGACCCC-3′). However, there was still no apparent Smad binding site. Since Sp1 has also been linked to TGFβ1 induction of gene expression via interaction with Smad3, perhaps Sp1 and/or Sp3 is also involved in expression of the vimentin gene [47]. Additional DAPAs were performed with DNA mutated for all putative GC-boxes (Fig. 4C, mSp1) using WCEs co-transfected with Flag-tagged Smad3, Sp1, and c-Jun. Sp1 and Smad3 as well as c-Jun were found bound to the WT AP-1 elements, but Smad3 and Sp1 binding to the mSp1 sequence was considerably reduced whereas c-Jun binding remained constant. Additional co-IP assays confirm the interaction of Sp1, c-Jun and Smad3 (data not shown) as detected previously [36].
Fig. 4. Smad3, c-Jun, c-Fos and Sp1 assemble into a multiprotein complex on vimentin’s tandem AP-1 sites.
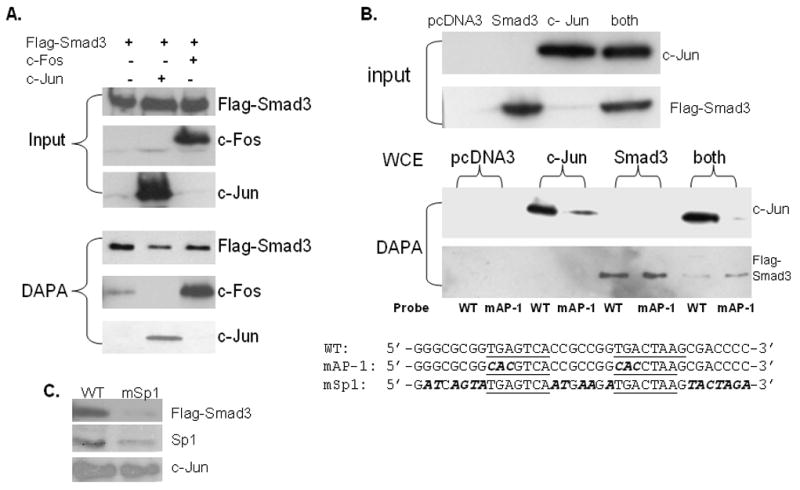
(A) WCEs were prepared from COS-1 cells transiently transfected with the Smad3 expression plasmid alone (lane 1), plus c-Jun (lane 2) or c-Fos (lane 3). Protein expression was verified by western blot of WCE (40 μg) as shown in the input panel. DNA precipitation assays (DAPAs) were performed with 5′-end biotinylated dsDNA containing wild-type (WT) tandem AP-1 sites (sequence shown at bottom of panel B) incubated with WCE (500 μg) from transiently transfected COS-1 cells as indicated. After washing, bound proteins were eluted and their content analyzed by western blot with Smad 3 (Flag-tag), c-Jun or c-Fos antibodies (lower panel. (B) COS-1 cells were transiently transfected as described in panel A with pcDNA3 vector alone or vectors containing Smad3 or c-Jun or co-transfected as labeled. Protein content of WCEs (40 μg) was analyzed in the input panel. DAPAs were carried out with a 5′-biotinylated dsDNA containing wild type (WT) or double mutant (mAP-1) sites as shown below. Tandem AP-1 elements are underlined. Bases changed in either mAP-1 or mSp1 are in bold and italicized. (C) DAPAs with 5′-biotinylated dsDNA containing either WT or triple mutant Sp1 (mSp1) AP-1 binding sites (sequences shown at the bottom of panel B) were conducted with WCEs isolated from COS-1 cells transiently transfected with Smad3, c-Jun, or Sp1 separately. Western blots with the appropriate antibody confirmed protein expression (data not shown). Bound proteins were eluted and their content analyzed by western blot with Flag-Smad3, Sp1 and c-Jun antibodies as indicated.
After determining that c-Jun, c-Fos, Smad3 and Sp1/Sp3 could all be involved in the TGFβ1 induction of vimentin expression, we wanted to determine if these factors could be found on the endogenous vimentin promoter via ChIP assays (Fig. 5). Western blots confirm that levels of Smad2, 3 and 4 do not appear to vary during differentiation of myoblasts into myotubes or with TGFβ1 treatment (data not shown). Previously, Sp1 and Sp3 levels have been shown to decline during myogenesis [42]. Western blots (Fig. 1B) confirm that c-Jun levels do decline during C2C12 myogenesis. Thus, it is possible that a switch in binding partners could occur, which in turn would contribute to a decrease in transcription as seen for vimentin in myotubes (Fig. 2B) [43,48]. Likewise, Western blots reveal an increase in c-Jun levels in myotubes grown in DM containing TGFβ1 (Fig. 1B). Chip assays reveal that binding of Smad3, c-Jun and c-Fos to vimentin’s endogenous promoter does decline when C2C12 myoblasts differentiate to myotubes, whereas upon TGFβ1 treatment binding increases substantially (Fig. 5). Similarly, Sp1 and Sp3 binding appears to increase on the endogenous promoter following TGFβ1 treatment; however, no decline is detected during myotube differentiation despite the reported decline in Sp1/Sp3 protein levels [42]. In fact, Sp3 binding appears to increase in myotubes compared to myoblasts.
Fig. 5. ChIP assays detect differential binding of Smad3, c-Jun, c-Fos and Sp1/Sp3 to vimentin’s tandem AP-1 sites.
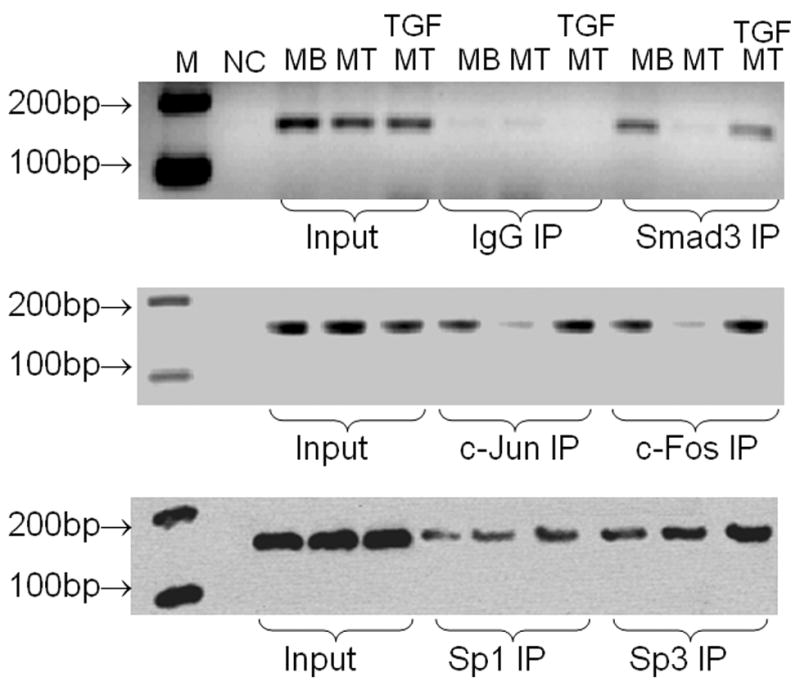
MB, MT and MT cells grown in the presence of TGFβ1 1 ng/ml) for 4 days were harvested and used for ChIP assays as described in Materials and Methods. Input lanes verify equal amounts of DNA were used for the initial immunoprecipitation. Antibodies used for immunoprecipitation (IP) are noted and IgG was included as a negative control. Immunoprecipitated DNA was extracted and used for PCR with primers encompassing the tandem AP-1 binding sites within the vimentin promoter. NC is a negative control where template DNA was left out of the PCR reaction. The resulting PCR products (160 bp) were separated on a 2% agarose gel and visualized with ethidium bromide staining. The motility of reference DNA markers is indicated.
Since c-Jun and c-Fos were both found on the tandem AP-1 elements via DAPA assays, we wanted to determine what would be the effect of expressing Smad3, c-Jun, c-Fos or combinations thereof on the -775CAT construct in COS-1 cells. Surprisingly, overexpression of c-Jun alone or along with Smad3 led to repression while overexpression of c-Fos alone or along with Smad3 activated vimentin reporter gene expression (Fig. 6A). Expression of these various proteins was verified by western blots in the bottom panel. Since c-Fos is expressed at high levels in myoblasts, perhaps the over-expression of c-Fos in COS-1 cells mimics the activity seen in the C2C12 myoblast reporter gene assays [31]. It has been shown that c-Fos does not directly interact with Smad3 or 4 whereas all Jun family members, c-Jun, JunB and JunD, do interact [46]. Moreover, we determined which domains of c-Jun might be important for repressing Smad3 activation. Co-transfection experiments with the -775CAT construct reveals that the transactivation (TAM67) or DBD-3 domains could be deleted and/or mutated with little effect on the ability of c-Jun to repress CAT activity. Only deletion of the leucine zipper domain (LZM-1) had much of an effect (Fig. 6B). However, the expression of JDP2, an inhibitor of c-Jun transcriptional activation, completely repressed Smad3 activation attesting to the overall involvement of c-Jun [49,50].
Fig. 6. Overexpression of c-Jun represses while c-Fos synergizes vimentin expression.
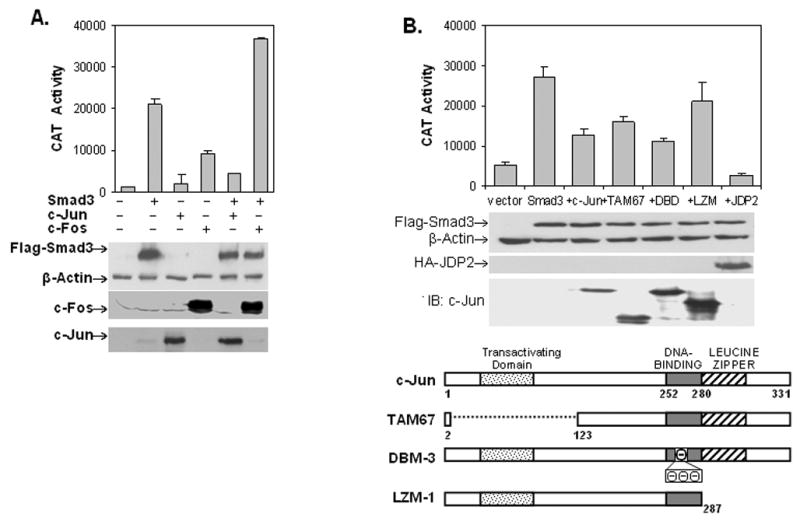
(A) The -775CAT construct was co-transfected with vector alone or expression plasmids containing Smad3, c-Jun or c-Fos alone or Smad3 plus c-Jun or c-Fos into COS-1 cells. Reporter gene assays were measured as in Fig. 2. Western blots (shown below) confirm protein expression from the various transfections with β-actin as a loading control (B) COS-1 cells were transiently transfected with the -775CAT construct with vector alone or vector plus Smad3 alone or co-transfected with c-Jun, c-Jun mutants or JDP2 expression plasmids. Reporter gene assays were measured as above. Western blots confirm protein expression with β-actin serving as a loading control. A schematic diagram of c-Jun and its various mutants is shown below. The dominant-negative TAM67 lacks the transactivating domain. DBM-3 contains an alanine to aspartic acid substitution at residue 265, followed by a three amino-acid insertion of negatively charged amino acids. LZM-1 lacks the C-terminal, leucine-zipper domain.
P300 is involved in Smad3’s induction of vimentin gene expression
Smad3 has been shown to interact with the co-activator p300 to activate gene expression following TGFβ1 treatment [35,51]. In this interaction the C-terminus of Smad3 is important for regulating the binding of CBP/p300 and this activation can be inhibited with E1A, which interferes with p300 function. To explore this possible relationship the -775CAT construct was co-transfected with empty vector or vector containing Smad3 alone or plus vector containing E1A/WT (wild-type) or its various mutants in COS-1 cells (Fig. 7A). Inclusion of E1A/WT results in repression of Smad3 activation, while E1AMut (Δ2–36), which lacks the N-terminal amino acids 2–36 and is defective in interacting with p300, fails to repress Smad3 induction of the vimentin promoter [37,52]. On the other hand, mutant E1AY/H(47,928), which lost its ability to bind Rb, but can still bind p300, represses Smad3 activation [37,52]. Similar results were found in C2C12 myoblasts (Fig. 7B). Furthermore, the constitutively activated receptor, TβRI-T204D, promotes activity equal to that of co-transfection with Smad3 alone as seen previously in COS-1 cells (Fig. 3D). Moreover, co-transfection of p300 alone yields a 3-fold activation over the vector only control further substantiating a role for p300 in enhancing vimentin expression. Western blot analysis using WCE from these same CAT assays confirms expression of E1A and its mutants (data not shown).
Fig. 7. p300 is involved in Smad3’s activation of vimentin gene expression.
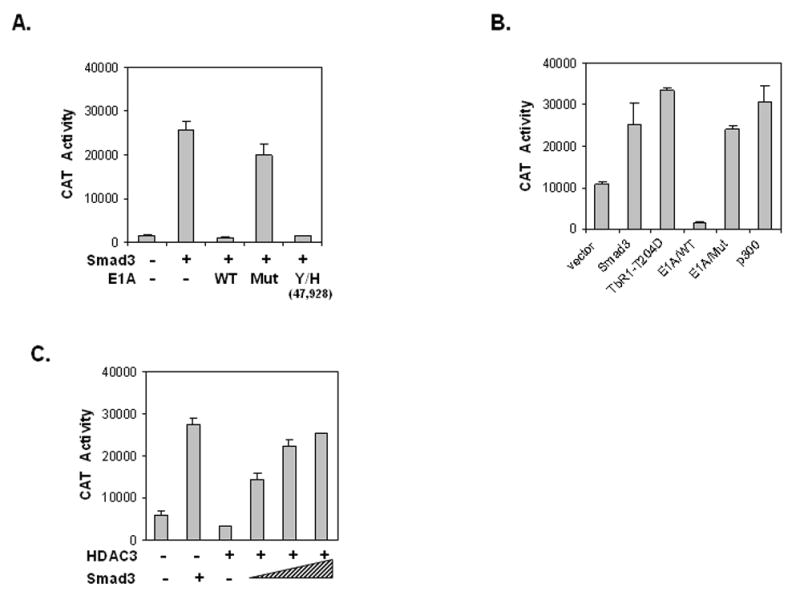
(A) The -775CAT construct was co-transfected into COS-1 cells with vector alone or vector containing Smad3 alone or plus plasmids expressing E1A/WT, E1A/Mut (Δ2–36) or E1AY/H(47,928). E1AMut (Δ2–36) is deleted for the N-terminal amino acids 2–36 whereas mutant E1AY/H(47,928) contains two point mutants at amino acid position 47 and 928, which obliterates p300, but retains Rb binding. Reporter gene activity was measured as above and results are from three separate experiments performed in triplicate. (B) The -775CAT construct was co-transfected into C2C12 myoblast cells as in panel A with the addition of the expression plasmid containing the constitutive active TGFβ1 receptor (TbRI-T204D) or p300. Reporter gene activity was measured as in panel A. (C) The -775CAT construct was co-transfected into COS-1 cells with Smad3 alone or expression plasmids encoding HDAC3. Reporter gene activity was measured as in panel A.
The c-Jun repressor, JDP2, has been shown to recruit HDAC3, which can in turn repress AP-1 transcriptional activation [53]. Here, we show that HDAC3 can interact with regulatory factors bound to the AP-1 element and repress gene activity of the -775CAT construct (Fig. 7C). However, this repression can be overcome by transfecting COS-1 cells with increasing amounts of a Smad3 expression plasmid. A western blot using WCE from these same CAT assays confirms expression of Smad3 and HDAC3 (data not shown).
Discussion
Vimentin exhibits a complex pattern of developmental- and tissue-specific expression regulated by growth factors such as TGFβ1. Normally, vimentin levels decline during myogenesis to make way for the muscle-specific IFP, desmin (Fig. 1). However, TGFβ1 treatment of C2C12 cells prevents the reduction of vimentin gene expression as well as blocking myogenesis. Until now, how vimentin expression is regulated during myogenesis and the mechanism of TGFβ1 induction was unknown. Here, we have found the location of the TGFβ1 response element to be within the AP-1 region of the vimentin promoter (Figs. 2D and 3C). Mutation of the tandem AP-1 elements drastically reduces TGFβ1 induction via reporter gene assays in both myogenic C2C12 and COS-1 cells. However, not only are the AP-1 elements themselves important, but the surrounding GC-boxes are also involved as Smad3 binding could still be detected in DAPA assays with dsDNA containing mutant AP-1 sites (Fig. 4) plus low levels of TGFβ1-induced reporter gene activity are still detectable with a -775CAT construct containing mutant AP-1 sites, but retaining the surrounding GC-boxes (Figs. 2D and 3C). Smad3 binding was finally eliminated with mutation of these GC-boxes. ChIP assays further support the interaction of c-Jun, c-Fos, Smad3 and Sp1/Sp3 in mediating vimentin transcriptional activity (Fig. 5). Finally, p300 is involved in TGFβ1 induction of vimentin transcriptional activity, which can be overcome by expression of JDP2 or HDAC3 (Figs. 6 and 7).
Although Smad3, c-Jun, c-Fos and Sp1/Sp3 are all located in a multiprotein complex over the AP-1 region, their relative composition is reported to change during myogenesis. Thus, it is not just the proximity of these binding sites, but the relative interaction of their cognate binding factors that modulates transcription of the vimentin gene. c-Jun (Fig. 1B) and JunD levels have been shown to decrease during differentiation [31,43]. Likewise, DNA binding activities as well as the level of c-Fos and Sp1 decline [31,42,43]. It has been proposed that c-Fos may be the preferred c-Jun binding partner during the myoblast stage [43]. Moreover, heterodimers of c-Jun and c-Fos have been shown to activate transcription to a greater extent than either protein co-expressed with Smad3 alone [45]. In contrast, c-Jun levels have been shown to decrease during myogenesis (Fig. 1B) and it has been postulated that c-Jun or JunD may prefer to complex with Fra2 in the myotube stage, an interaction which maybe be enhanced by post-translational modifications such as phosphorylation [43]. Yet, the inadequacy of decent Fos antibodies excluded a complete analysis of the c-Fos contribution to myotube gene expression. Our ChIP data supports a decline in the c-Jun/Fos heterodimer binding to the endogenous vimentin promoter in myotubes (Fig. 5). We propose this change in AP-1 element binding coupled with the presence of the transcriptional repressor ZBP-89 binding to the two PS elements could account for the observed decline in vimentin expression during myogenesis (Figs. 2B and 2C).
However, upon TGFβ1 treatment of C2C12 cells, ChIP assays detect a dramatic increase in binding of c-Jun, c-Fos and Sp3 with some increase in Sp1 binding to the endogenous vimentin gene promoter (Fig. 5). DAPAs suggest that even upon mutation of the AP-1 element, Sp1/Sp3 binding to the surrounding GC-boxes could retain Smad3 binding to the vimentin promoter at least in vitro (Fig. 4). While there have been reported cases where Smad3 and c-Jun or Smad3 and Sp1 can interact on a Smad3 binding site enhanced by upstream or downstream AP-1 elements and/or GC-boxes, to our knowledge this is the first example where TGFβ1 induction required both such elements in lieu of a Smad3 binding site [47]. Past reports for both collagenase and c-Jun promoters indicated that Smad and AP-1 family members can interact synergistically [45,54]. However, there are examples where Smad3 and c-Jun expression can lead to gene repression in which case there are often binding sites for both Smad3 and AP-1 family members [44]. In these examples, the role of c-Fos and Sp3 is unknown and for the vimentin promoter there is no known Smad binding site. From our studies, we propose that Sp1 binding to the GC-boxes surrounding the tandem AP-1 elements could serve as the initial scaffold for DNA-protein interaction, hence binding remains fairly constant in myoblasts and myotubes (Fig. 5). It is only upon TGFβ1 induction that Sp3 and somewhat Sp1 binding is enhanced, which in turn supports the subsequent binding of the Smad3/2/4 complex along with c-Jun/c-Fos thereby contributing to enhanced vimentin gene expression. In fact, this heterocomplex may have already assembled as suggested by IP data (Fig. 3E) and thus join the DNA as a preformed complex [44–46]. Thus, Smad3 binding is only completely obliterated when the surrounding GC-boxes are mutated, yet in this case c-Jun binding is still quite apparent at least in vitro (Fig. 4C).
Interestingly, overexpression of c-Jun in COS-1 cells blocked Smad3 induction of vimentin reporter gene activity whereas c-Fos synergized expression (Fig. 6A). While Smad3 has been shown to interact directly with both c-Jun and Sp1 attenuated by TGFβ1 treatment, c-Fos has not been shown to directly interact with Smad3 [36,45,46,55]. In addition, it has been documented that c-Jun and Smad3 can interact off DNA [44–46]. Thus, overexpression of c-Jun could effectively lower the amount of Smad3 available for DNA binding resulting in the noted decreases in gene expression (Fig. 5A). In support of this hypothesis, c-Jun and Smad3 have been shown to interact via the leucine-zipper and N-terminal domains, respectively [56]. Mutation of the leucine zipper domain of c-Jun partially diminished repression both in our studies (Fig. 6B) and others, and deletion of any Smad3 domain completely abrogated activation (Figs. 2F and 3D). Inclusion of the c-Jun inhibitor, JPD2, completely repressed Smad3 activation (Fig. 6B). On the other hand, c-Fos does not interact with Smad3 off DNA and thus can synergize Smad3 activation as seen here (Fig. 6A).
Previously, Smad3 and CBP have been shown to directly interact only after TGFβ1 treatment [35,51]. This interaction has been linked to amino acids 210–342 of Smad3. The inclusion of either the N- or C-terminal domains of Smad3 represses its interaction with p300 possibly by restricting the binding of Smad3 and CBP. Furthermore, Smad3, Smad4 and p300/CBP have been shown to interact together on the PAI-1 promoter following TGFβ1 treatment in the presence of a Smad binding site [35]. In the case of vimentin, there is no Smad binding site, yet we have shown via co-transfection assays that p300 can still contribute to Smad3-dependent activation of the vimentin promoter negating the requirement for an actual Smad DNA-binding element (Fig. 7). Activation can only be eliminated by co-expression of an E1A mutant (Δ2–36), which retains the p300 interaction domain.
In contrast, the Jun dimerization protein (JDP2) is a potent repressor of AP-1 transcriptional activity. Ectopic expression of JDP2 by itself has been shown to induce myogenesis in C2C12 myoblasts [50]. JDP2 is an ubiquitously expressed protein that can form stable heterodimers with c-Jun, JunB or JunD and act as a repressor of c-Jun and the c-Jun/c-Fos heterodimers [49]. In addition, JDP2 has been shown to inhibit the recruitment of p300 to AP-1 elements by recruiting the histone deacetylase, HDAC3 [53]. It has been proposed that JDP2 requires HDAC activity in order to function as a repressor forming a multi-protein complex with the known co-repressor mSin3A. In agreement, we find that ectopic expression of JDP2 inhibits a vimentin promoter CAT construct containing the tandem AP-1 elements (Fig. 6). Altogether this result coupled with the fact that JDP2 levels naturally increase during myogenesis and overexpression accelerates differentiation suggests that JDP2 may play a similar role in the down-regulation of vimentin via the recruitment of HDAC3 [50]. This could be facilitated by the repressor function of ZBP-89. Although interesting, additional experiments understanding the putative interaction of c-Jun, JPD2 and ZBP-89 in repressing vimentin gene expression during muscle differentiation is beyond the scope of this initial study.
In summary, we present a model for how TGFβ1 and its downstream effector Smad3 might regulate vimentin gene expression. In the absence of a Smad binding site, Sp1/3 assists in recruiting the “activated” Smad complex to vimentin’s AP-1 elements as promoted by AP-1 family members. By interacting with c-Jun and its preferred binding partner c-Fos, Smad3 in turn recruits p300, which could act either as a co-activator linking the bound transcriptional factors to the basal transcriptional machinery or exert its acetylation activity to enhance regulatory factor activity and/or further “open up” chromatin structure. In converse, the recruitment of HDAC3 by JDP2 may remove acetyl groups from the basal transcription machinery and/or an AP-1 family member thereby resulting in repression of vimentin gene expression during myogenesis.
Acknowledgments
This work was supported by NHLBI, National Institutes of Health (NIH) Grant HL-45422 to Z.E.Z., DK073932 to X.L. and an American Heart Association Mid-Atlantic Affiliate pre-doctoral fellowship (0415464U) to M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Glick AB. TGFβ1, back to the future. Cancer Biol Therapy. 2004;3:276–283. doi: 10.4161/cbt.3.3.849. [DOI] [PubMed] [Google Scholar]
- 2.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Cheifetz S, Endo T, Nadal-Ginard B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci U S A. 1986;83:8206–8210. doi: 10.1073/pnas.83.21.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olson EN, Sternberg E, Hu JS, Spizz G, Wilcox C. Regulation of myogenic differentiation by type beta transforming growth factor. J Cell Biol. 1986;103:1799–1805. doi: 10.1083/jcb.103.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan TJ, Edmondson DG, Li L, Olson EN. Transforming growth factor beta represses the actions of myogenin through a mechanism independent of DNA binding. Proc Natl Acad Sci U S A. 1991;88:3822–3826. doi: 10.1073/pnas.88.9.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaidya TB, Rhodes SJ, Taparowsky EJ, Konieczny SF. Fibroblast growth factor and transforming growth factor beta repress transcription of the myogenic regulatory gene MyoD1. Mol Cell Biol. 1989;9:3576–3579. doi: 10.1128/mcb.9.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu D, Black BL, Derynck R. TGF-β inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–2966. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu D, Kang JS, Derynck R. TGF-β activated Smad represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004;23:1557–1566. doi: 10.1038/sj.emboj.7600179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinert PM, Liem RK. Intermediate filament dynamics. Cell. 1990;60:521–523. doi: 10.1016/0092-8674(90)90651-t. [DOI] [PubMed] [Google Scholar]
- 10.Parry DA, Steinert PM. Intermediate filament structure. Curr Opin Cell Biol. 1992;4:94–98. doi: 10.1016/0955-0674(92)90064-j. [DOI] [PubMed] [Google Scholar]
- 11.Duprey P, Paulin D. What can be learned from intermediate filament gene regulation in the mouse embryo. Int J Dev Biol. 1995;39:443–457. [PubMed] [Google Scholar]
- 12.Sax CM, Farrell FX, Zehner ZE. Down-regulation of vimentin gene expression during myogenesis is controlled by a 5′-flanking sequence. Gene. 1989;78:235–242. doi: 10.1016/0378-1119(89)90226-6. [DOI] [PubMed] [Google Scholar]
- 13.Moura-Neto V, Kryszke MH, Li Z, Vicart P, Lilienbaum A, Paulin D. A 28-bp negative element with multiple factor-binding activity controls expression of the vimentin-encoding gene. Gene. 1996;168:261–266. doi: 10.1016/0378-1119(95)00789-x. [DOI] [PubMed] [Google Scholar]
- 14.Wieczorek E, Lin Z, Perkins EB, Law DJ, Merchant JL, Zehner ZE. The zinc finger repressor, ZBP-89, binds to the silencer element of the human vimentin gene and complexes with the transcriptional activator, Sp1. J Biol Chem. 2000;275:12879–12888. doi: 10.1074/jbc.275.17.12879. [DOI] [PubMed] [Google Scholar]
- 15.Passantino R, Antona V, Barbieri G, Rubino P, Melchionna R, Cossu G, Feo S, Giallongo A. Negative regulation of β-enolase gene transcription in embryonic muscle is dependent upon a zinc finger factor that binds to the G-rich box within the muscle-specific enhancer. J Biol Chem. 1998;273:484–494. doi: 10.1074/jbc.273.1.484. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari S, Battini R, Kaczmarek L, Rittling SR, Calabretta B, de Riel JK, Philiponis V, Wei JF, Baserga R. Coding sequence and growth regulation of the human vimentin gene. Mol Cell Biol. 1986;6:3614–3620. doi: 10.1128/mcb.6.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson CW, Rittling SR, Hirschhorn RR, Kaczmarek L, Calabretta B, Stiles CD, Baserga R. Cell cycle dependent genes inducible by different mitogens in cells from different species. Mol Cell Biochem. 1986;71:61–69. doi: 10.1007/BF00219329. [DOI] [PubMed] [Google Scholar]
- 18.Carey I, Zehner ZE. Regulation of chicken vimentin gene expression by serum, phorbol ester, and growth factors: identification of a novel fibroblast growth factor-inducible element. Cell Growth Differ. 1995;6:899–908. [PubMed] [Google Scholar]
- 19.Izmailova ES, Snyder SR, Zehner ZE. A Stat1a factor regulated the expression of the human vimentin gene by IFN-γ. J Inter Cytokine Res. 2000;20:13–20. doi: 10.1089/107999000312694. [DOI] [PubMed] [Google Scholar]
- 20.Bussemakers MJ, Verhaegh GW, van Bokhoven A, Debruyne FM, Schalken JA. Differential expression of vimentin in rat prostatic tumors. Biochem Biophys Res Commun. 1992;182:1254–1259. doi: 10.1016/0006-291x(92)91866-o. [DOI] [PubMed] [Google Scholar]
- 21.Gilles C, Thompson EW. The epithelial to mesenchymal transition and metastatic progression in carcinoma. The Breast J. 1996;2:83–96. [Google Scholar]
- 22.Rittling SR, Baserga R. Functional analysis and growth factor regulation of the human vimentin promoter. Mol Cell Biol. 1987;7:3908–3915. doi: 10.1128/mcb.7.11.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lilienbaum A, Paulin D. Activation of the human vimentin gene by the Tax human T-cell leukemia virus I: Mechanisms of regulation by the NF-kB transcription Factor. J Biol Chem. 1993;268:2180–2188. [PubMed] [Google Scholar]
- 24.Chen JH, Vercamer C, Li Z, Paulin D, Vandenbunder B, Stehelin D. PEA3 transactivates vimentin promoter in mammary epithelial and tumor cells. Oncogene. 1996;13:1667–1675. [PubMed] [Google Scholar]
- 25.Salvetti A, Lilienbaum A, Li Z, Paulin D, Gazzolo L. Identification of a negative element in the human vimentin promoter: modulation by the human T-cell leukemia virus type I Tax protein. Mol Cell Biol. 1993;13:89–97. doi: 10.1128/mcb.13.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rittling SR, Coutinho L, Amram T, Kolbe M. AP-1/jun binding sites mediate serum inducibility of the human vimentin promoter. Nucl Acids Res. 1989;17:1619–1633. doi: 10.1093/nar/17.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izmailova ES, Zehner ZE. An antisilencer element is involved in the transcriptional regulation of the human vimentin gene. Gene. 1999;230:111–120. doi: 10.1016/s0378-1119(99)00046-3. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Zhang X, Zehner ZE. c-Jun and the dominant-negative mutant, TAM67, induce vimentin gene expression by interacting with the activator Sp1. Oncogene. 2003;22:8891–8901. doi: 10.1038/sj.onc.1206898. [DOI] [PubMed] [Google Scholar]
- 29.Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, Verma IM. Functional antagonism between c-jun and myoD proteins: A direct physical association. Cell. 1992;68:507–519. doi: 10.1016/0092-8674(92)90187-h. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Zhou J, James G, Hellier-Harrison R, Czech MP, Olson EN. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell. 1992;71:1182–1194. doi: 10.1016/s0092-8674(05)80066-2. [DOI] [PubMed] [Google Scholar]
- 31.Lehtinen SK, Rahkila P, Helenius M, Korhonen P, Salminen A. Down-regulation of transcription factors AP-1, Sp-1, and NF- B precedes myocyte differentiation. Biochem Biophys Res Commun. 1996;229:36–43. doi: 10.1006/bbrc.1996.1754. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Wu Y, Zehner ZE, Jackson-Cook CK, Ware JL. Proteomic analysis of the tumorigenic human prostate cell line M12 after microcell-mediated transfer of chromosome 19 demonstrates reduction of vimentin. Electrophoresis. 2004;24:3445–3453. doi: 10.1002/elps.200305574. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Diab IH, Zehner ZE. ZBP-89 represses vimentin gene transcription by interacting with the transcriptional activator, Sp1. Nucl Acids Res. 2003;31:2900–2914. doi: 10.1093/nar/gkg380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Y, Diab I, Zhang X, Izmailova ES, Zehner ZE. Stat3 enhances vimentin gene expression by binding to the antisilencer element and interacting with the repressor protein, ZBP-89. Oncogene. 2004;23:168–178. doi: 10.1038/sj.onc.1207003. [DOI] [PubMed] [Google Scholar]
- 35.Feng XH, Zhang Y, Wu RY, Derynck R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-βinduced transcriptional activation. Genes Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng XH, Lin X, Derynck R. Smad2, Samd3, and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J. 2000;19:5178–5193. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang HGH, Moran E, Yaciuk P. E1A promotes association between p300 and pRB in multimeric complexes required for normal biological activity. J Virol. 1995;69:7917–7924. doi: 10.1128/jvi.69.12.7917-7924.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farrell FX, Sax CM, Zehner ZE. A negative element involved in vimentin gene expression. Mol Cell Biol. 1990;10:2349–2358. doi: 10.1128/mcb.10.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J. Transforming growth factor-β1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle. Amer J Path. 2004;164:1007–1019. doi: 10.1016/s0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kryszke MH, Moura-Neto V, Lilienbaum A, Paulin D, Auclair C. Involvement of histone H4 gene transcription factor 1 in downregulation of vimentin gene expression during skeletal muscle differentiation. FEBS Lett. 2001;491:30–34. doi: 10.1016/s0014-5793(01)02142-1. [DOI] [PubMed] [Google Scholar]
- 41.Izmailova ES, Wieczorek E, Perkins EB, Zehner ZE. A GC-box is required for expression of the human vimentin gene. Gene. 1999;235:69–75. doi: 10.1016/s0378-1119(99)00209-7. [DOI] [PubMed] [Google Scholar]
- 42.deLeon MB, Montanez C, Gomez P, Morales-Lazaro SL, Tapia-Ramirez V, Valadez-Graham V, Recillas-Targa F, Yaffe D, Nudel U, Cisneros B. Dystrophin Dp71 gene expression is down-regulated during myogenesis: Role of Sp1 and Sp3 on the Dp71 promoter activity. J Biol Chem. 2005;280:5290–5299. doi: 10.1074/jbc.M411571200. [DOI] [PubMed] [Google Scholar]
- 43.Andreucci JJ, Grant D, Cox DM, Tomc LK, Prywes R, Goldhamer DJ, Rodrigues N, Bedard PA, McDermott JC. Composition and function of AP-1 transcription complexes during muscle cell differentiation. J Biol Chem. 2002;277:16426–16432. doi: 10.1074/jbc.M110891200. [DOI] [PubMed] [Google Scholar]
- 44.Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-β in a promoter-specific manner. Oncogene. 2001;20:3332–3340. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 46.Liberati NT, Datto MB, Frederick JP, Shen X, Wong C, Rougier-Chapman EM, Wang XF. Smads bind directly to the Jun family of AP-1 transcription factors. Proc Natl Acad Sci U S A. 1999;96:4844–4849. doi: 10.1073/pnas.96.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brodin G, Ahgren A, Dijke P, Heldin CH, Heuchel R. Efficient TGF-β induction of the Smad7 gene requires cooperation between AP-1, Sp1, and Smad proteins on the mouse Smad7promoter. J Biol Chem. 2000;275:29023–29030. doi: 10.1074/jbc.M002815200. [DOI] [PubMed] [Google Scholar]
- 48.Thinakaran G, Ojala J, Bag J. Expression of c-jun/AP-1 during myogenic differentiation in mouse C2C12 Myoblasts. FEBS Lett. 1993;319:271–276. doi: 10.1016/0014-5793(93)80561-8. [DOI] [PubMed] [Google Scholar]
- 49.Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mole Cell Biol. 1997;17:3094–3102. doi: 10.1128/mcb.17.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ostrovsky O, Bengal E, Aronheim A. Induction of terminal differentiation by the c-Jun dimerization protein JPD2 in C2 myoblasts and rhabdomyosarcoma cells. J Biol Chem. 2002;277:40043–40054. doi: 10.1074/jbc.M205494200. [DOI] [PubMed] [Google Scholar]
- 51.Janknecht R, Wells NJ, Hunter T. TGF-β-stimulated cooperation of Smad proteins with the coactivators CBP/p300. Genes Dev. 1998;12:2114–2119. doi: 10.1101/gad.12.14.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang HGH, Rikitake Y, Corrigan M, Yaciuk P, Abraham SE, Zerler B, Moran E. Identification of specific adenovirus E1A N-Terminal residues critical to the binding of cellular proteins and to the control of cell growth. J Virol. 1993;67:476–488. doi: 10.1128/jvi.67.1.476-488.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin C, Li H, Murata T, Sun K, Horikoshi M, Chiu R, Yokoyama KK. JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol Cell Biol. 2002;22:4815–4826. doi: 10.1128/MCB.22.13.4815-4826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong C, Rougier-Chapman EM, Frederick JP, Datto MB, Liberati NT, Li JM, Wang XF. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor β. Mole Cell Biol. 1999;19:1821–1830. doi: 10.1128/mcb.19.3.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Datta PK, Blake MC, Moses HL. Regulation of plasminogen activator inhibitor-1 expression by Transforming Growth Factor-β-induced physical and functional interactions between Smads and Sp1. J Biol Chem. 2000;275:40014–40019. doi: 10.1074/jbc.C000508200. [DOI] [PubMed] [Google Scholar]
- 56.Qing J, Zhang Y, Derynck R. Structural and functional characterization of the transforming growth factor-beta-induced Smad3/c-Jun transcriptional cooperativity. J Biol Chem. 2000;275:38802–38812. doi: 10.1074/jbc.M004731200. [DOI] [PubMed] [Google Scholar]