

Summary
Under ambient air conditions, NO inhibits NMDA-sensitive glutamate receptor responses by reacting with C399 of the NR2A subunit as well as possibly with two pairs of cysteine residues if their disulfide bonds are reduced to free thiol (C744, C798 in NR1 and C87, C320 in NR2). Here we demonstrate that relative hypoxia enhances S-nitrosylation of NMDA receptors by a unique mechanism involving NR1(C744,C798), which constitutes a novel ‘NO-reactive oxygen sensor motif.’ These two critical thiol groups sensitize other sites on the NMDA receptor to S-nitrosylation and consequent inhibition of receptor activity, while manifesting little if any effect on NMDA receptor activity by their own nitrosylation. The crystal structure of NR1 reveals a uniquely flexible (C744-C798) disulfide bond, which may account for the susceptibility of these two cysteines to reduction and subsequent facile oxidation by NO, as observed with biochemical techniques. Our findings have important implications for the treatment of hypoxia/stroke because these thiol groups can be nitrosylated preferentially during relative hypoxia, thus abrogating excessive activity associated with cytotoxicity while avoiding side effects caused by blockade of normal NMDA receptors.
Introduction
Glutamate is the major excitatory neurotransmitter of the central nervous system. NMDA-type glutamate receptors have been implicated in multiple physiological processes including, neuronal development (Akazawa et al., 1994; Bliss and Collingridge, 1993; Lu et al., 2001; Monyer et al., 1994), long-term potentiation (Collingridge et al., 1983), and long-term depression (Mulkey and Malenka, 1992). However, overstimulation of NMDA receptors can cause excessive Ca2+ influx, free radical generation, abnormal enzymatic activity, and thus contribute to a number of neurodegenerative disesases (Lipton and Rosenberg, 1994). To date, NMDA receptor antagonists have failed in clinical trials for stroke because of lack of clinical tolerability, arising from the blockade of normal NMDA receptor activity (Lipton, 2004; Lipton and Chen, 2004). Here, in an effort to overcome this problem, we present a mechanism whereby excessive NMDA receptor activity can be preferentially abated during hypoxic/stroke insults, but not under normal conditions.
A variety of chemical modifications that modulate NMDA receptor activity have been reported, including phosphorylation of tyrosine (Kohr and Seeburg, 1996; Wang and Salter, 1994) and serine/threonine residues (Omkumar et al., 1996; Tingley et al., 1997), redox modulation of disulfide bonds, and S-nitrosylation of free thiol groups on critical cysteine residues (Choi et al., 2000; Lei et al., 1992; Lipton et al., 1993; Lipton et al., 1996; Sucher et al., 1996; Lipton et al., 2002). Similarly, cysteine residues play an important role in redox modulation of a variety of other proteins, including Zn2+ finger transcription factors (Choi et al., 2001a; Kroncke and Carlberg, 2000), Zn2+ repository proteins (Aravindakumar et al., 1999; Kroncke et al., 1994; Misra et al., 1996; Pearce et al., 2000), hemoglobin (Stamler et al., 1997), and voltage- and ligand-gated ion channels (Lipton et al., 2002; Choi et al., 2001b; Choi et al., 2000; Kim et al., 1999; Lei et al., 1992). Redox modulators, including reducing (Aizenman et al., 1989; Aizenman, 1995; Aizenman et al., 1990; Aizenman et al., 1992; Scanlon et al., 1997; Tang and Aizenman, 1993a) and oxidizing agents (Gilbert et al., 1991; Kohr et al., 1994; Manzoni et al., 1992; Tang and Aizenman, 1993b), have been shown to alter NMDA receptor activity via reaction with cysteine residues (Choi et al., 2001b; Lipton et al., 2002). Reducing agents, such as dithiothreitol (DTT) or dihydrolipoic acid, potentiate NMDA-evoked currents (Kohr and Seeburg, 1996) by promoting formation of free thiol groups. Conversely, oxidizing agents, such as 5-5’-dithio-bis(2-nitrobenzoic acid) (DTNB) or oxidized glutathione (GSSG) (Sucher and Lipton, 1991), inhibit NMDA-evoked currents by favoring disulfide formation (Aizenman et al., 1989). Moreover, oxidation of free thiol to disulfide prevents other covalent reactions of sulfhydryl groups, for example with NO-related species.
It is very important to note that physiological concentrations of O2 in the brain are relatively low compared to room air, in the range of 10-20 mm Hg (Erecinska and Silver, 2001), thus producing a less oxidizing environment than ambient conditions. This relative hypoxia favors free thiol over disulfide formation. Additionally, pathologically hypoxic conditions, such as stroke, produce a state in vivo wherein many proteins are initially further reduced. In this pathological situation, disulfide bond formation is even less preferred. Regarding the NMDA receptor, we have reported evidence from electrophysiological experiments for a mixture of disulfide and free thiols of two pairs of cysteine residues on the NR1 and NR2A subunits of the receptor [NR1(C744,C798) and NR2A(C87,C322); the ratio of disulfide to free thiol varies depending on the chemical conditions (Aizenman et al., 1989; Choi et al., 2001b; Choi et al., 2000; Lei et al., 1992; Sullivan et al., 1994; Lipton et al., 2002).
Previously, we reported that the endogenous NO donor, S-nitrosocysteine (SNOC), inhibits NMDA-evoked currents via S-nitrosylation and possible disulfide bond formation of critical cysteine residues of NMDA receptors on primary cortical neurons (Lei et al., 1992; Lipton et al., 1993; Lipton and Rosenberg, 1994) as well as on NR1/NR2A recombinant receptors (Choi et al., 2000; Lipton et al., 2002). NO inhibition of NMDA current was abolished by pre-treatment with methanethiosulfonate derivatives such as 2-aminoethylmethanethio-sulfonate (MTSEA) and [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET), which react specifically with thiol groups to form mixed disulfides, thus rendering thiol groups unavailable to react with NO (Kim et al., 1999). These experiments showed that thiol groups of cysteine residues are crucial for S-nitrosylation of the NMDA receptor. Furthermore, from site-directed mutagenesis studies, we found that the cysteine residue at position #399 on the NR2A subunit accounted for the predominant effect of S-nitrosylation. Additionally, we found that the aforementioned two pairs of cysteines on the NR1 and NR2A subunits could possibly undergo S-nitrosylation or further oxidation and thus downregulate NMDA receptor activity if they were in the free thiol form (Choi et al., 2001b; Choi et al., 2000; Lipton et al., 2002). In the present study, we report that increasing hypoxia renders the receptor exquisitely sensitive to S-nitrosylation. This finding is reminiscent of previous observations on the ryanodine receptor-Ca2+ release channel (RyR) (Eu et al., 2000), showing that thiol groups are involved in O2 sensing to produce allosteric regulation of S-nitrosylation at physiological levels of pO2. Moreover, the mechanism reported here suggests that S-nitrosylation of the receptor may be used to curtail the excessive NMDA activity that occurs during hypoxia/stroke with relatively less effect on normal function. Importantly, we also report here that the C744,C798 pair of cysteine residues found on the NR1subunit constitute a unique ‘molecular oxygen sensor’ in the brain, controlling the degree of downregulation of NMDA receptor function by S-nitrosylation in the presence of low pO2 levels, and thus dictating the pathological effects of hypoxia that are mediated via the receptor.
Results
Relative Hypoxia Potentiates Endogenous NO-Induced Downregulation of NMDA Receptor Responses in Primary Cerebrocortical Neurons
Initially, we studied whether lowering oxygen tension would affect the attenuation of NMDA receptor responses by endogenous neuronal nitric oxide synthase (nNOS), which we had previously observed under room air conditions (Lei et al., 1992; Lipton et al., 1993; Kim et al., 1999; Choi et al., 2000; Lipton et al., 2002). It should be noted that, in fact, room air conditions actually represent a form of oxidative stress (pO2 150-160 mm Hg), but the term ambient conditions is used here as a matter of convention. In these experiments, to permit relatively long-term monitoring of NMDA responses without rundown, we performed digital [Ca2+]i imaging on cultured cerebrocortical neurons stimulated with consecutive pulses of 50 μM NMDA under control (ambient) and relatively hypoxic conditions. As we previously reported (Kim et al., 1999; Choi et al., 2000), the addition of L-arginine to the medium, as a substrate for NOS, led to a decrease in successive NMDA responses under room air conditions (Figure 1). This effect reflects downregulation of NMDA receptor activity by endogenous NO generated by nNOS in response to Ca2+ entry via NMDA receptor-operated channels. The effect was abrogated by the NOS inhibitor, L-nitro-arginine. Under hypoxic conditions (pO2 as low as 5 mm Hg; see Experimental Procedures), this attenuation of NMDA responses was greatly enhanced (Figure 1). In fact, L-arginine was twice as effective in decreasing NMDA receptor responses (60.0% decrement) as it was under control room air conditions (29.8%), determined as the difference in response between L-arginine—treated and L-nitro-arginine—treated cells. This phenomenon is pathophysiologically relevant since we observed it at oxygen tensions of 5 mm Hg or less, but it is also physiologically relevant because we could see a smaller but reproducible effect at oxygen tensions of 10-20 mm Hg, which approach those encountered in the brain.
Figure 1.

Hypoxia Enhances NO Inhibition of NMDA Responses in Primary Cerebrocortical Neurons
The mean of repetitive neuronal [Ca2+]i responses to 50 μM NMDA in each of 3 experiments were arbitrarily designated 100%. Addition of the nNOS substrate, L-arginine (1 mM for 30 min), resulted in a small but statistically significant decrement in the NMDA response under ambient conditions (p < 0.05). Concomitant incubation with the nNOS inhibitor L-nitro-arginine (1 mM) not only reversed this effect but also resulted in increased NMDA responses compared to the control condition (p < 0.001), probably because of the presence of endogenous levels of NO under baseline conditions. Under hypoxic conditions, the effect of L-arginine in decreasing the NMDA response was significantly greater than under ambient conditions (p < 0.001). Note that even under control hypoxic conditions, the effect of endogenous NO was greater than under control ambient conditions (p < 0.001). L-Nitro-arginine again reversed the effect under hypoxic conditions. Values are mean ± SEM for n = 13-21 microscopic fields with 8-15 cortical neurons/field in 3 separate cultures.
Biotin-Switch Assay Reveals that NR1 Thiols Are Increasingly S-Nitrosylated Under Relatively Hypoxic Conditions
We next asked if relative hypoxia could increase the degree of S-nitrosylation of the NMDA receptor. Previously, using the biotin-switch technique, Solomon Snyder’s group had shown that cysteine thiols on the NR1 subunit of the NMDA receptor are S-nitrosylated (Jaffrey et al., 2001). Here, using a similar protocol, we demonstrated that relative hypoxia enhanced S-nitrosylation of NR1 (Figure 2). For this purpose, we activated endogenous nNOS under room air vs. relatively hypoxic conditions and performed a biotin-swtich assay to monitor S-nitrosylation of NR1. We were able to compare the levels of NR1 S-nitrosylation using a technique we recently reported that normalizes the amount of S-nitrosylated protein to total protein (Uehara et al., 2006). We found that relative hypoxia increased S-nitrosylation of the NR1 subunit by approximately 2-fold (Figure 2).
Figure 2.

S-Nitrosylation of the NR1 Subunit of the NMDA Receptor Under Hypoxic Conditions
(A) Expression constructs for NR1 and NR2A were transfected into nNOS-HEK cells. Twenty-four hours later, nNOS-HEK cells were treated with 5 mM A23187 for 2 h to allow Ca2+ entry to stimulate endogenous nNOS under ambient (room air) or hypoxic conditions (pO2 = 5-10 mm Hg). Cell extracts were subjected to the biotin-switch assay. SNO-NR1: S-nitrosylated NR1.
(B) Quantitative analysis of SNO-NR1 formation. Blots from the biotin-switch assay and Western analyses were quantified by densitometry, and the relative ratio of SNO-NR1 to total NR1 was calculated. Error bars represent SEM (*p <0.05 by ANOVA).
To further investigate the mechanism of the enhanced downregulation of NMDA responses by NO in the presence of relative hypoxia, we next turned to electrophysiological recordings from recombinant receptors.
Hypoxia Sensitizes Recombinant NR1/NR2A Receptor Responses to Downregulation by S-Nitrosylation
We used the oocyte expression system to examine the mechanism of the effect of hypoxia on responses of recombinant NMDA receptors. Under room air conditions, application of the endogenously-occurring NO donor, SNOC (500 μM), inhibited NMDA-evoked currents in oocytes expressing NR1/NR2A subunits (by 18.8 ± 1.1%, mean ± SEM, n = 10; Figure 3A), consistent with our previous findings (Choi et al., 2000; Kim et al., 1999). We found that lowering the ambient O2 tension to ∼10 mm Hg resulted in increased inhibition of NMDA-evoked currents by NO (to 29.7 ± 6.7 %, n = 10; p < 0.05 by Mann-Whitney; Figures 3B and 3C). This finding is consistent with the notion that the increased number of free thiol groups on critical cysteine residues, which are known to be present under hypoxic conditions, may react with NO to enhance the inhibitory effect of S-nitrosylation on the NMDA receptor. Additionally, DTT not only mimicked this effect of hypoxia but also occluded further effects of hypoxia (see Figure S1 in the Supplemental Data), suggesting hypoxia-sensitivity occurs at critical cysteines responsible for DTT modulation (Choi et al., 2001b) and that the hypoxic condition itself was chemically reducing.
Figure 3.

Hypoxia Enhances NO Inhibition of NMDA-Evoked Currents
(A) NMDA-evoked currents (200 μM NMDA and 10 μM glycine) in oocytes expressing NR1/NR2A subunits were inhibited by application of the NO donor, SNOC (500 μM), under ambient conditions. (B) Hypoxia enhanced SNOC-mediated inhibition of NMDA current. Holding potential -80 mV, pH 7.2. (C) Summary of the effect of hypoxia on SNOC-mediated inhibition of NMDA-evoked currents. Suppression of NMDA currents by SNOC is expressed as the normalized response to NMDA prior to application of SNOC. Each column represents mean ± SEM (*p < 0.05; Mann-Whitney U test).
Similar to the NMDA receptor, accumulating evidence suggests that the ryanodine receptor (RyR) can be poly-nitrosylated (Xu et al., 1998); relatively hypoxic conditions can increase the sensitivity of this reaction in an allosteric manner, allowing lower levels of NO to be effective (Eu et al., 2000; Stamler et al., 2001). To test this possibility for the NMDA receptor, we performed dose-response curves for NO. Under ambient air conditions, a ten-fold lower concentration of SNOC (50 μM) only slightly attenuated NMDA-evoked currents (Figure 4A), compared to inhibition with 500 μM. Intriguingly, however, during hypoxia, 50 μM SNOC inhibited the NMDA-evoked currents to a greater extent than the same concentration of SNOC under ambient conditions (Figure 4B). At an even lower concentration, 5 μM, SNOC did not significantly inhibit NMDA-evoked currents under ambient conditions (Figure 4A). Nonetheless, at a pO2 of ∼10 mm Hg, 5 μM SNOC manifested a significant inhibitory effect (Figures 4B and 4C). These results suggest that the NMDA receptor becomes more sensitive to inhibition by NO under relatively hypoxic conditions; in fact, physiologically relevant levels of NO inhibit NMDA receptor activity at physiological oxygen tensions.
Figure 4.

Hypoxia Shifts the Sensitivity of NMDA Receptor Inhibition Towards Lower Concentrations of NO
(A) Inhibition of NMDA responses by SNOC (5-500 μM) under ambient conditions. (B) Inhibition of NMDA responses by SNOC (5-500 μM) under hypoxic conditions. At each concentration of SNOC, hypoxic conditions augmented inhibition of NMDA-evoked currents greater than ambient air conditions. Remarkably, at lower concentrations of SNOC, inhibition of NMDA-evoked current was evident only under hypoxic conditions. (C) Potentiation of NMDA-evoked currents expressed as normalized responses to NMDA before SNOC application. Values are mean ± SEM (n = 7 in each case). SNOC inhibition of NMDA currents was significantly greater under hypoxic compared to ambient conditions (*p < 0.05, **p < 0.01; Mann-Whitney U test).
Cysteine 399 on the NR2A Subunit Contributes to Enhanced NO Sensitivity Under Relatively Hypoxic Conditions
From previous electrophysiological and chemical studies, we knew that, among other cysteine residues, Cys 399 of NR2A was a primary site of S-nitrosylation (Choi et al., 2000; Kim et al., 1999; Lipton et al., 2002). We found in the present study that under ambient air conditions, NO manifested virtually no effect on NMDA-evoked currents in oocytes expressing NR1/NR2A(C399A) mutant receptors (Figure 5A). Additionally, we found that hypoxia-mediated enhancement of NO inhibition of NMDA-evoked currents was virtually eliminated in this mutant compared to wild-type NR1/NR2A receptors (Figures 5B and 5C versus Figures 3 and 4).
Figure 5.
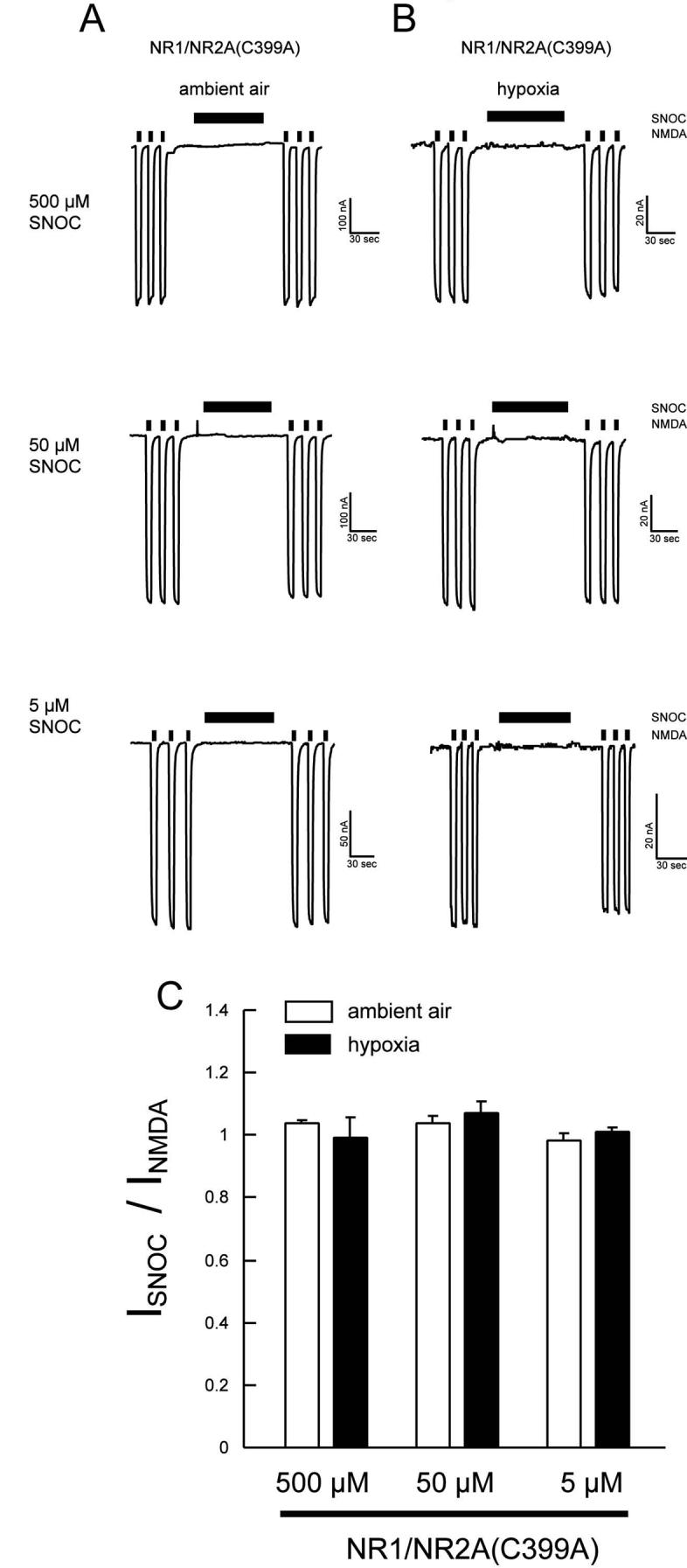
Cysteine Residues of the NR2A Subunit Involved in Hypoxic Enhancement of SNOC Inhibition
(A and B) SNOC-mediated NMDA current inhibition was abolished in oocytes expressing NR1/NR2A(C399A). The mutant also abrogated the enhancement of NO inhibition by hypoxia. (C) Summary histogram of the effect of NR2A cysteine mutation at residue 399 on NO-mediated NMDA receptor inhibition. Each bars represents mean ± SEM of responses obtained from 7 oocytes.
Cysteines 744 and 798 on the NR1 Subunit Constitute an Oxygen Sensor
We also knew from previous work (Sullivan et al., 1994; Choi et al., 2000; Choi et al., 2001a; Kim et al., 1999; Lipton et al., 2002) that the NR1 subunit harbors a pair of redox-active cysteine residues that can form a disulfide bond but can apparently be nitrosylated if in the free thiol state. In fact, by mutational studies these were the only cysteine thiols found to be redox active and S-nitrosylated on the NR1 subunit (Sullivan et al., 1994; Choi et al., 2000; Choi et al., 2001b). When we expressed mutant NR1(C744A,C798A)/NR2A receptors, NO could still significantly inhibit NMDA responses under ambient air conditions. Remarkably, however, hypoxic enhancement of the inhibitory effect of NO was totally abrogated (Figure 6). Therefore, we concluded that, under hypoxic conditions, while not directly inhibiting NMDA receptor activity itself, S-nitrosylation of NR1(C744,C798) influenced the overall sensitivity of receptor function to inhibition by S-nitrosylation at other sites.
Figure 6.

Mutation of Cysteines 744, 798 of the NR1 Subunit Abrogates Hypoxic Enhancement of NO-Mediated Inhibition NMDA-Evoked Currents
(A and B) Voltage-clamp traces from oocytes expressing NR1(C744A, C798A)/NR2A subunits under ambient air (A) and hypoxic (B) condition. SNOC produced dose-dependent inhibition of NMDA-evoked currents (200 μM NMDA plus 10 μM glycine) under ambient air conditions but hypoxia did not enhance this inhibition. (C) Inhibition of NMDA-evoked currents by NO, expressed as normalized responses to NMDA before SNOC application. Values are mean ± SEM (n = 4-5 oocytes).
Putative Structure of the Thiol Oxygen Sensor on NR1
The NR1(C744A,C798A) mutant abrogated NO inhibition of NMDA receptor activity under relatively hypoxic conditions. This observation was consistent with the notion that an oxygen sensing motif, consisting of cysteine thiol groups located at NR1 residues Cys744 and Cys798, preferentially reacts with NO under hypoxic conditions. Therefore, we investigated the crystal structure of this region of the NR1 subunit (Furukawa and Gouaux, 2003) before and after exposure to NO.
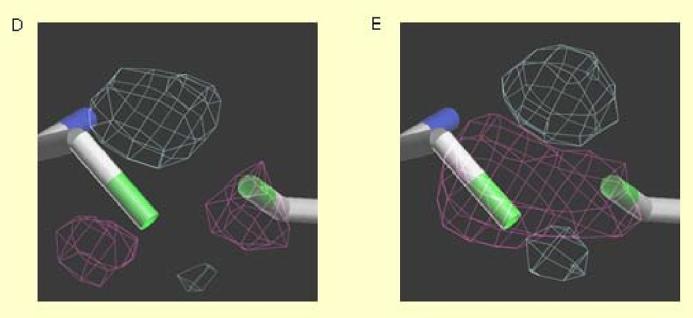
Figures 7A-C display the electron density (2mFo-DFc, representing the sigma A weighted omit minus map) in the vicinity of the three disulfide bridges within the structure of NR1-S1S2. Two of the disulfides (C420-C454 and C436-C455) have a well-defined density, indicative of a single conformation. For the third disulfide (C744-C798), the electron density is well defined for the main chain and Cβ atom, but more diffuse for the Sγ atoms comprising the disulfide bridge. Figures 7D and 7E show a Fo-Fc difference map that is the average of 4 or 5 diffraction sets, each collected from a distinct single crystal. The only strong features (>3 σ) in the map are those flanking the Sγ atoms of the C744-C798 disulfide (peak heights +5σ and -4σ). We attempted to model the structure as alternate conformations of Cys744 or Cys798, either as distinct disulfide bridges or as a mixture of free thiols and disulfides (Heras et al., 2004). However, we could not account for the density satisfactorily without imposing unfavorable geometry, raising the possibility that covalent modification of the cysteines was present in the crystals.
Figure 7.
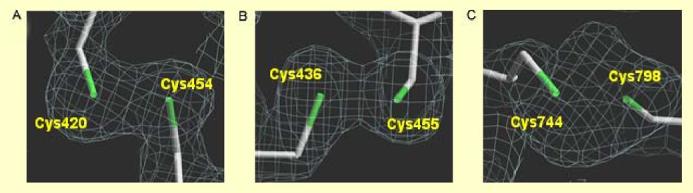
Crystal Structure of Critical Cysteine Residues on the NR1 Subunit of the NMDA Receptor
(A-C) Sigma A weighted omit map (2mFo-DFc at +1.0 σ contour) of the three disulfide bonds in the NR1-S1S2 ligand-binding domain: C420-C454 (A), C436-C455 (B), C744-C798 (C). (D, E) Electronic density of the oxygen sensor motif consisting of cysteine Thiol groups located at NR1 residues C744 and C798 that appear to react with NO: Mean of 4 untreated crystals (D); mean of 5 SNOC-treated crystals (E). Fo-Fc averaged electron density map of C744-C798 contoured at ±3.0 σ (+gray -pink).
In NR1 crystals treated with SNOC (4 independent crystal structures), the only significant features in the averaged Fo-Fc difference map occur in the vicinity of Cys744,Cys798. The peaks are in similar positions to but higher than (+7σ, -7σ) those of the untreated crystals. The larger difference peaks suggest that a fraction of disulfides are reduced in the crystal and that the free cysteine sulfurs reacted with NO. This change is reflected in the altered crystal structure, accounting for the more positive peaks in the difference map. The inherent disorder and/or redox mixture at these bonds precludes further description at the atomic level. However, we can infer that SNOC treatment specifically affects the Cys744/Cys798 pair and that the data are consistent with NO-induced covalent modification in the form of facile S-nitrosylation of at least a fraction of the free thiols. This notion is further supported by our chemical evidence of S-nitrosylation of the NR1 subunit using the biotin-switch technique (Figure 2, above).
We also analyzed NO-treated NR1-S1S2 peptide by Mass Spectrometry using a modification of our previously published methods (Gu et al., 2002; Yao et al., 2004; Uehara et al., 2006). The predominant species detected were free thiol at Cys 744 and 1/2 of a disulfide on Cys 798 (data not shown). Interestingly, this kind of ‘intermediate result’ might have been predicted from the unusual flexible disulfide bond that we saw between Cys 744 and Cys 798 in the crystal structure of NR1-S1S2. However, we did not observe NO on either thiol, possibly because, chemically-speaking, NO is a good leaving group. As such, NO may not be expected to be seen by this Mass Spectrometry method since the SNO-intermediate on NR1-S1S2 may be rather ephemeral. Nonetheless, as mentioned above, the biotin-switch assay on native NMDA receptors revealed increasing S-nitrosylation of the NR1 subunit with relative hypoxia.
There are several possible reasons that we might be able to detect NO (or in this case, its biotin replacement) on the NR1 subunit by the standard biotin-switch assay but not by Mass Spectrometry. For example, salt content and detergents must be kept to a minimum during Mass Spectrometry analysis in order to obtain interpretable spectra, while this is not as critical for the biotin-switch assay. There are also various potential explanations why we can observe S-nitrosylation of other peptides but not the NMDA receptor by Mass Spectrometry (Gu et al., 2002; Yao et al., 2004; Uehara et al., 2006), such as the differential ionization efficiency for disparate peptides during Mass Spectrometry analysis. These various features could contribute to the failure of Mass Spectrometry to detect sites of S-nitrosylation compared to the biotin-switch assay in this instance.
Taken together with prior work, however, we have very solid chemical data for redox modulation of by S-nitrosylation of Cys744 or Cys798 on NR1 using the biotin-switch assay in conjunction with site-directed mutagenesis experiments (Sullivan et al., 1994; Kim et al., 1999; Choi et al., 2000; Choi et al. 2001; Jaffrey et al., 2001).
Discussion
In this study, we have established that hypoxia enhances NO modulation of the NMDA receptor, resulting in increased attenuation of NMDA-evoked currents. Since cysteine residues are the primary target for S-nitrosylation, the redox state of the NMDA receptor is critical to the efficacy of NO modulation. NMDA receptors normally function in the brain where pO2 levels are substantially lower than in ambient air. Thus, our findings highlight the physiological relevance of S-nitrosylation of the NMDA receptor. Moreover, these results suggest that under pathologically hypoxic conditions, S-nitrosylation of the NMDA receptor becomes even more important because progressively lower concentrations of NO inhibit excessive NMDA receptor activity. A likely explanation for the enhancement of NO inhibition of NMDA-evoked currents during hypoxia is the increase in free thiol groups that are available to react with NO under these conditions (Figure 8). In a reminiscent manner, relatively hypoxic conditions favor poly-nitrosylation of the RyR (Eu et al., 2000; Stamler et al., 2001).
Figure 8.

Schematic Representation of Hypoxic Enhancement of NO-Mediated Inhibition of NMDA Receptor Activity
(A) Ambient O2 levels are significantly higher than in living tissues or during hypoxic insults, favoring disulfide formation (red line). (B) In the presence of disulfide, NO cannot be transferred to these cysteine residues since no free thiol exists for reaction. S-Nitrosylation can occur on NR2A C399 (cysteine residue at left) but only in the presence of a high concentration of NO donor. (C) Under physiological or even more hypoxic conditions, the relatively reducing state favors free thiol groups on the NMDA receptor. (D) Under relatively hypoxic conditions, the free thiols groups are more readily available to react with NO to form S-nitrosothiol, and low concentrations of an NO donor can effect this reaction. The reaction route that possibly leads from S-nitrosylation to disulfide formation of vicinal thiols (NR1 C744/C798) remains unknown, although it is likely that in this scenario only one of these thiols is S-nitrosylated. During relative hypoxia, our hypothesis is that NO is more likely to react with both C744 and C798 to S-nitrosylate their thiol groups, and under these conditions disulfide formation would be blocked. Such dual S-nitrosylation of C744 and C798 would then lead to the increased NO sensitivity that we have observed experimentally at other NMDAR sites, such as C399 on NR2A. Therefore, NO itself may not be regulatory at NR1 C744/C798, but the redox status of these cysteine residues would exert an allosteric influence on S-nitrosylation of other thiol groups on the NMDAR such as C399 on NR2A.
Intriguingly, mutation of a pair of cysteines on the NR1 subunit (C744, C798), that are also known to form a disulfide bond under oxidizing conditions (Choi et al., 2001b; Choi et al., 2000; Lipton et al., 2002; Furukawa and Gouaux, 2003), had little if any effect on inhibition of the NMDA response by NO under ambient conditions. However, this mutation completely abrogated enhancement of the effect of NO during hypoxia. Thus, this pair of cysteines functions as an ‘oxygen sensor motif,’ in that it sensitizes the NMDA receptor to inhibition by NO under hypoxic conditions but has little direct effect on inhibition of receptor activity by its own nitrosylation. One possible explanation for this effect is that under hypoxic conditions, reaction of the free thiols at C744 or C798 with NO exerts an allosteric effect on the receptor that facilitates further nitrosylation at another critical cysteine site; we know that such a site exists at NR2A(C399) because mutation of this site abrogated NO inhibition of receptor activity under either ambient air or hypoxic conditions.
Structural studies are needed to prove this premise, and we have therefore examined the electronic structure of C744/C798 in crystals of this region of the NR1 subunit. We found that exposure to an endogenous NO donor changes the electronic density adjacent to the cysteine sulfur atoms, consistent with S-nitrosylation. Furthermore, our electrophysiological experiments suggest that such nitrosylation is facilitated in an hypoxic environment. Hence, taken together with the chemical evidence for enhanced S-nitrosylation under relatively hypoxic conditions obtained via the biotin-switch technique, our electrophysiological results suggest that the NR1 thiol not only sense oxygen tension but can also be S-nitrosylated themselves. It is intriguing that the same thiol(s) that are S-nitrosylated may constitute the molecular O2 sensor that makes additional thiol groups more easily nitrosylated.
Whatever the exact mechanism, the present study shows that C744/C798 can sense the oxygen environment to render the receptor exquisitely sensitive to inhibition by S-nitrosylation under hypoxic conditions. The data thus provide mechanistic insight into downregulation of NMDA receptor activity by the physiological environment. Evolutionary pressure may have allowed the brain to develop this mechanism as a means to curtail excitotoxic insults caused by NMDA receptor hyperactivity (Lipton and Rosenberg, 1994). Furthermore under pathological conditions such as stroke, the levels of O2 are further depressed, making S-nitrosylation even more favorable, and thus this reaction can contribute to neuroprotection by limiting the influx of excessive Ca2+ through NMDA receptor-associated channels (Choi et al., 2000; Lei et al., 1992; Lipton et al., 1993). In fact, since the receptor is more sensitive to inhibitory control by S-nitrosylation during hypoxia, future therapeutic modalities could take advantage of this mechanism. By targeting nitrosylatable cysteine residues during stroke to prevent NMDA receptor-mediated neurotoxicity, possible side effects would be avoided because the effect of S-nitrosylation on NMDA activity under physiological conditions is potentially less than in pathologically hypoxic areas of the brain.
Experimental Procedures
Fluorescence Imaging of Neuronal [Ca2+]i in Primary Cerebrocortical Cultures
Cerebrocortical cultures were prepared and digital [Ca2+]i imaging was performed with standard methods, as previously described (Lei et al., 1992; Lipton et al.,. 1993; Kim et al., 1999; Choi et al., 2000). The cortical cultures were loaded for 30 min at 37°C with 5 μM Fura-2 acetoxymethylester (Fura-2-AM, Molecular Probes) in Hanks solution buffered with HEPES (138 mM NaCl, 1 mM NaHCO3, 0.34 mM Na2HPO4, 5.4 mM KCl, 0.44 mM KH2PO4, 2.5 mM CaCl2, 10 mM HEPES, 0.01 mM glycine, and 22.2 mM glucose, adjusted to pH 7.2 with NaOH) and transferred to the recording chamber of an inverted microscope (Zeiss Axiovert 35; 20X, 0.5 NA, objective) equipped with a Sutter DG-4 fast wavelength switcher. Pairs of images from cells stimulated at 350 nm and 380 nm with output measured at 510 nm were acquired every 250 ms with a cooled CCD camera (Cooke Sensicam, PCO, Germany) controlled by SlideBook software (Intelligent Imaging Innovations, Santa Monica, CA). Signals were averaged over regions of interest (cell soma), background subtracted, and then converted into [Ca2+]i using standard methods (Grynkiewicz et al., 1985). Fura-2 loading capacity was assessed by monitoring the fluorescence output at 360 nm (isosbestic point).
Biotin-Switch Assay to Detect S-Nitrosylation of the NR1 Subunit of the NMDA Receptor
HEK293 cells stably expressing nNOS (nNOS-HEK) were cultured in DMEM supplemented with 10% FBS, 4 mM glutamine, 200 mg/ml G418, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. nNOS-HEK cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The biotin-switch assay was performed as described (Jaffrey et al., 2001; Uehara et al., 2006; Yao et al., 2004). In brief, cells were lysed, and free thiols were blocked with 4 mM methyl methanethiosulfonate at 50 °C for 20 min. After blocking, cell extracts were precipitated with two volumes of acetone, and resuspended in 0.15 ml of HENS buffer (250 mM HEPES, pH 7.4/1 mM EDTA/0.1 mM neocuproine/1% SDS). Subsequently, S-nitrosylated cysteines were reduced with 1mM ascorbate and biotinylated with 1 mM Biotin-HPDP (Pierce, Rockford, IL) at room temperature for 1 h. After incubation, proteins were precipitated with acetone as described above. The biotinylated proteins were pulled down with streptavidin-agarose and analyzed by Western blotting.
cRNA Synthesis
Templates were prepared from circular plasmid cDNA by linearizing the 3’ untranslated region with NheI (for NR1-1a), or EcoRV (for NR2A). cRNA, which incorporates the 5’ cap analog [m7G(5’)P(5’)G], was transcribed from 1 μg of linearized template in vitro by T7 (for NR1) or T3 (for NR2A) RNA polymerase according to the mMessage mMachine protocol (Ambion). cRNA concentrations were determined by measuring the optical density at 260 nm and by agarose gel electrophoresis.
Preparation of Oocytes and Injection of cRNA
Xenopus oocytes were prepared as previously described (Choi and Lipton, 1999; Sullivan et al., 1994). Frog oocytes at stage V or VI were surgically removed from the ovaries of Xenopus laevis. Lumps of ∼100 oocytes were incubated with 580 U/ml (2 mg/ml) collagenase type I (Sigma) for 2 hr in Ca2+-free solution (in mM: 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, adjusted to pH 7.5 with NaOH). After slow agitation to remove the follicular cell layer, the oocytes were washed extensively with Ca2+-free frog Ringer’s solution. Oocytes were maintained at 18 °C in frog Ringer’s solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 mM HEPES, adjusted to pH 7.5 with NaOH) supplemented with 550 mg/l sodium pyruvate as a carbon source and 100 μg/ml gentamicin. Twenty-four hours later, oocytes were injected with up to 10 ng of cRNA of each NMDA receptor subunit using a 10 μl Drummond microdispenser under visual control with a dissecting microscope.
Two-Electrode Voltage-Clamp Recording in the Oocyte Expression System
Two to seven days after injection, oocytes were recorded in frog Ringer’s solution under two-electrode voltage clamp at -80 mV. Recordings were performed at room temperature with an Oocyte Clamp OC-725b amplifier (Warner Instrument Corporation) using Mac Lab version 3.5 software (AD Instruments). Voltage-sensing electrodes had a resistance of 1-4 MΩ, and current-injecting electrodes had a resistance of 0.5-1 MΩ. Both were filled with 3 M KCl. Oocytes were continuously superfused with a solution containing (in mM): 90 NaCl, 1 KCl, 10 HEPES, 1.5 BaCl2,10 μM glycine, adjusted to pH 7.2-7.5 with NaOH (the lower pH was used in some cases to prolong the life of S-nitrosothiol donors). Ba2+ was used as the divalent cation. Drugs dissolved in frog Ringer’s solution were applied by superfusion at a flow rate of ∼2 ml/min in a 100-μl chamber with an array of pipettes similar to the ‘sewer pipe’ system used in patch-clamp recording to achieve relatively rapid solution exchange. Using a concentration jump paradigm with Mg2+, we measured a time constant of less than 3 s for complete solution exchange in the recording chamber (Choi et al., 2001b).
Hypoxic Conditions
During experiments involving digital Ca2+ imaging and electrophysiological recording, relative hypoxia was maintained by continuously bubbling a solution of 100% nitrogen gas into the recording chamber. To ensure hypoxic conditions throughout the solution, diffusion stones (Fisher Scientific) were used. After 15 min of hypoxic equilibration, the O2 content of the solution was measured digitally as partial pressure of oxygen (pO2) with an Arterial Blood Gas Analyzer, Stat profile pHOX plus (NOVA Medical); values as low as 5 mm Hg could be maintained in this manner and could be increased to 10-20 mm Hg by shortening the duration of bubbling in nitrogen. The osmolarity and pH of the solution remained unaffected by the bubbling procedure. For the experiments involving the biotin-switch assay, all procedures were performed in a hypoxia chamber (Biotrace).
Atomic Resolution Structures
NR1-S1S2 recombinant protein was expressed and purified in E. coli as described41. Briefly, Origami B (DE3) cells harboring pET-22b(+)-NR1-S1S2 were grown and induced by 0.5 mM isopropyl-β-D-thiogalactoside. All purifications steps were performed at 4 °C. The protein was purified by metal-chelating column followed ion exchange column. Crystals were grown at 4 °C by the hanging drop vapor diffusion method by mixing NR1-S1S2 at 5 mg/ml with equal volume drops from a reservoir consisting of 20-23% polyethylene glycol 1000, 100 mM sodium cacodylate (pH 6.0), and 100 mM lithium sulfate, 10 mM glycine. For freezing, the crystals were transferred to a cryo-buffer consisting of 25-28% polyethylene glycol 1000, 100 mM lithium sulfate, 10 mM glycine, 100 mM sodium cacodylate (pH 6.0) with 15-20% glycerol for 5 minutes, mounted on cryo-loops and plunged into liquid nitrogen.
S-Nitrosylation of crystals was performed for 30-60 min in the cryo-buffer supplemented with 10 mM SNOC. The crystals were cryo-cooled. Diffraction data were collected at 110 K using a Rigaku FRE X-ray source equipped with an RAXIS-IV image plate detector. Diffraction images were processed and scaled with the HKL package (Otwinoski and Minor, 1997). The NR1 structure (pdb id: 1PB7) was used as the starting model for refinement using CNS (Brunger et. al., 1998) and program O (Jones and Kjeldgaard, 1997). Data collection and refinement statistics are given in Supplemental Tables S1 and S2. For difference map calculations, the B values of the Sγ atoms of Cys744 and Cys798 were assigned the average refined value of 31 Å2. For map averaging, refined models were overlaid (RMS main chain deviations of .1-.2 Å) in order to generate transformation matrices, and data sets were truncated to 2 Å resolution. The average B factors for protein atoms are very similar - 21.7 Å2 (untreated; 5 data sets) and 21.9 Å2 (SNOC-treated; 4 data sets).
Statistical Methods
Values are presented as mean ± SEM. To be conservative in our assessments, no underlying assumptions were made as to whether the data fit a normal distribution or not. Hence, significance was determined using high-stringency methods with a non-parametric test (Mann-Whitney U test).
Supplementary Material
Acknowledgments
We thank E. Gouaux, J.M. Sullivan, S.F. Heinemann, and E. Liman for sharing cDNA clones, constructs or vectors, and B. Eckelman for discussion. This work was supported in part by NIH grants P01 HD29587, R01 EY05477, and R01 EY09024 (to S.A.L.), and American Heart Association grant SDG 9930256N (to H.-S.V. C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenman E. Modulation of N-methyl-D-aspartate receptors by hydroxyl radicals in rat cortical neurons in vitro. Neurosci. Lett. 1995;189:57–59. doi: 10.1016/0304-3940(95)11442-y. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Hartnett KA, Reynolds IJ. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron. 1990;5:841–846. doi: 10.1016/0896-6273(90)90343-e. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Hartnett KA, Zhong C, Gallop PM, Rosenberg PA. Interaction of the putative essential nutrient pyrroloquinoline quinone with the N-methyl-D-aspartate receptor redox modulatory site. J. Neurosci. 1992;12:2362–2369. doi: 10.1523/JNEUROSCI.12-06-02362.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman E, Lipton SA, Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–1263. doi: 10.1016/0896-6273(89)90310-3. [DOI] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- Aravindakumar CT, Ceulemans J, De Ley M. Nitric oxide induces Zn2+ release from metallothionein by destroying zinc-sulphur clusters without concomitant formation of S-nitrosothiol. Biochem. J. 1999;344(Pt 1):253–258. doi: 10.1042/0264-6021:3440253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Choi H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, Ryu S. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001a;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J. Neurosci. 2001b;21:392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Lipton SA. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- Choi Y-B, Tenneti L, Le DA, Ortiz J, Bai G, Chen H-SV, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. The antagonism of amino acid-induced excitations of rat hippocampal CA1 neurones in vitro. J. Physiol. (Lond) 1983;334:19–31. doi: 10.1113/jphysiol.1983.sp014477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 2001;128:263–276. doi: 10.1016/s0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert KR, Aizenman E, Reynolds IJ. Oxidized glutathione modulates N-methyl-D-aspartate- and depolarization-induced increases in intracellular Ca2+ in cultured rat forebrain neurons. Neurosci. Lett. 1991;133:11–14. doi: 10.1016/0304-3940(91)90045-u. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-Nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. USA. 2004;101:8876–8881. doi: 10.1073/pnas.0402769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jones TA, Kjeldgaard M. Electron-density map interpretation. Meth. Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- Kim WK, Choi Y-B, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO- Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994;12:1031–1040. doi: 10.1016/0896-6273(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Kohr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J. Physiol. (Lond.) 1996;492(Pt 2):445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroncke KD, Carlberg C. Inactivation of zinc finger transcription factors provides a mechanism for a gene regulatory role of nitric oxide. FASEB J. 2000;14:166–173. doi: 10.1096/fasebj.14.1.166. [DOI] [PubMed] [Google Scholar]
- Kroncke KD, Fehsel K, Schmidt T, Zenke FT, Dasting I, Wesener JR, Bettermann H, Breunig KD, Kolb-Bachofen V. Nitric oxide destroys zinc-sulfur clusters inducing zinc release from metallothionein and inhibition of the zinc finger-type yeast transcription activator LAC9. Biochem. Biophys. Res. Commun. 1994;200:1105–1110. doi: 10.1006/bbrc.1994.1564. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen H-SV, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Concepts: Turning down but not off—Neuroprotection requires a paradigm shift in drug development. Nature. 2004;428:473. [Google Scholar]
- Lipton SA, Chen H-SV. Paradigm shift in neuroprotective drug development: clinically tolerated NMDA receptor inhibition by memantine. Cell Death Differ. 2004;11:18–20. doi: 10.1038/sj.cdd.4401344. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi Y-B, Pan ZH, Lei SZ, Chen H-SV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi Y-B, Sucher NJ, Pan ZH, Stamler JS. Redox state, NMDA receptors and NO-related species. Trends Pharmacol. Sci. 1996;17:186–187. doi: 10.1016/0165-6147(96)20028-8. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi Y-B, Takahashi T, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Mechanisms of disease: Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-t. [DOI] [PubMed] [Google Scholar]
- Misra RR, Hochadel JF, Smith GT, Cook JC, Waalkes MP, Wink DA. Evidence that nitric oxide enhances cadmium toxicity by displacing the metal from metallothionein. Chem. Res. Toxicol. 1996;9:326–332. doi: 10.1021/tx950109y. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pearce LL, Gandley RE, Han W, Wasserloos K, Stitt M, Kanai AJ, McLaughlin MK, Pitt BR, Levitan ES. Role of metallothionein in nitric oxide signaling as revealed by a green fluorescent fusion protein. Proc. Natl. Acad. Sci. USA. 2000;97:477–482. doi: 10.1073/pnas.97.1.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon JM, Aizenman E, Reynolds IJ. Effects of pyrroloquinoline quinone on glutamate-induced production of reactive oxygen species in neurons. Eur. J. Pharmacol. 1997;326:67–74. doi: 10.1016/s0014-2999(97)00137-4. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC. Nitrosylation. The prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Sucher NJ, Awobuluyi M, Choi Y-B, Lipton SA. NMDA receptors: from genes to channels. Trends Pharmacol. Sci. 1996;17:348–355. [PubMed] [Google Scholar]
- Sucher NJ, Lipton SA. Redox modulatory site of the NMDA receptor-channel complex: regulation by oxidized glutathione. J. Neurosci. Res. 1991;30:582–591. doi: 10.1002/jnr.490300316. [DOI] [PubMed] [Google Scholar]
- Sullivan JM, Traynelis SF, Chen H-SV, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994;13:929–936. doi: 10.1016/0896-6273(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Tang LH, Aizenman E. Allosteric modulation of the NMDA receptor by dihydrolipoic and lipoic acid in rat cortical neurons in vitro. Neuron. 1993a;11:857–863. doi: 10.1016/0896-6273(93)90115-8. [DOI] [PubMed] [Google Scholar]
- Tang LH, Aizenman E. Long-lasting modification of the N-methyl-D-aspartate receptor channel by a voltage-dependent sulfhydryl redox process. Mol. Pharmacol. 1993b;44:473–478. [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi Z-Q, Ma Y, Gaston B, Plamer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson’s disease:S-Nitrosylation of parkin regulates it E3 ligase activity. Proc. Natl. Acad. Sci. USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.