

Abstract
Prostate-specific membrane antigen (PSMA) is a relatively omnipresent, but unique Type II dimeric transmembrane protein with a multiplicity of functions and has been shown to be a reasonable target for immunological approaches such as vaccines or more directed therapy with radioactively labelled monoclonal antibodies against PSMA. Given the abundance of various glycoprotein and carbohydrate antigens expressed on the surface of prostate cancer cells and cell lines, PSMA stands out as another ‘self’ antigen which is not only expressed on cancer cells, but on neovasculature. Although vaccines are varied in their design and target goal, recent technology has afforded researchers the opportunity to induce recruitment of multiple effector cell populations, cytokines and factors which can elicit both cellular and humoral responses. This review serves to present unique approaches in vaccine development which can induce immunological responsiveness with potential impact on disease progression and to introduce PSMA as a potential target for multimodality therapies.
Keywords: antigen, immunotherapy, prostate-specific antigen (PSA), prostate-specific membrane antigen (PSMA), vaccine, vector
1. Introduction
1.1 Treatment in the minimal disease state: is there a standard?
The last decade has revealed the evolution of a unique population of men who have failed primary therapies and have as the sole manifestation of disease relapse, a rising biomarker, prostate-specific antigen (PSA). Unlike other solid tumour malignancies which are not favoured by a validated marker which can be monitored for screening, disease relapse and progression, the patient with a rising PSA following primary treatments such as surgery or radiation often has no other recourse than to pursue hormone ablative therapy in an effort to control relapse. However, many patients are unwilling to pursue this avenue and are seeking ways to enhance their immune systems to actively, rather than passively, fight their cancer. Therefore, a distinct rationale exists for pursuing immunotherapeutic approaches in patients with prostate cancer, particularly for those in a minimal disease state as manifested by a rising PSA following primary therapies such as surgery or radiation. Among the reasons to pursue a vaccine approach are that, 1) there are an abundance of well-defined glycoprotein and carbohydrate antigens overexpressed on the surface of prostate cancer cell lines as well as prostate cancer cells which may serve as targets for immune recognition and attack; 2) the population under study is healthy, has minimal symptoms and will most likely have a long natural history; 3) the population is often younger with a poor tolerance of androgen ablation should they wish to pursue this treatment in the face of a rising PSA postsurgery or radiation; and 4) deferring hormonal therapy may not compromise survival.
Many of the molecules identified on prostate cancer cells and cell lines are ‘self’ antigens, meaning that they appear on the normal counterparts of the tumour cells derived from the organ in question, yet change in very subtle ways with malignant transformation. This is particularly true of the mucin family (i.e., MUC-1 and MUC-2) which are present normally within glandular elements and become overexpressed and underglycosylated with malignancy. It is unclear as to why the immune system does not see these altered ‘self’ antigens expressed on the new tumour cells as foreign and allows them to grow; however, strategies have focused on means of educating the immune system to recognise these molecules and break ‘immunological tolerance’, thereby allowing the body to respond to what is now perceived as a ‘foreign’, rather than ‘self ’, antigen.
1.2 Castrate metastatic disease - is there a role for immuotherapy in the presence of a new standard?
Patients with overt metastatic disease which progresses despite androgen ablation have limited treatment options. The combination of mitoxantrone and a glucocorticoid in patients with castrate metastatic prostate cancer (hormone refractory prostate cancer) and bone metastases demonstrated a palliative response in two large randomised trials; however, in neither trial did the combination result in a survival advantage over glucocorticoid alone [1,2]. A multi-centre Phase III study compared docetaxel plus prednisone with mitoxantrone plus prednisone, was recently reported [3]. A schedule of docetaxel (75 mg/m2 every 3 weeks) plus prednisone (5 mg orally, b.i.d) significantly improved overall survival, improved pain response and impacted favourably on PSA, compared with mitoxantrone plus prednisone. The regimen was well-tolerated, but with more grade III/IV neutropenia. Similarly, a second trial using docetaxel with estramustine also demonstrated a survival advantage over mitoxantrone and prednisone [4]. However, estramustine was not felt to contribute significantly to the regimen and the FDA approved docetaxel in combination with prednisone for treatment of patients with advanced metastatic prostate cancer [4]. These are the first Phase III studies to show a significant survival benefit in castrate metastatic disease. Thus, for patients who relapse following androgen ablation, there is a significant unmet medical need for additional targeted therapies. One approach addressing this need involves enhancing the body’s immune system as a means to both treat recurrent disease and delay or prevent disease progression.
1.3 Immunotherapeutic approaches: passive immunotherapies
In recent years, there has been an increased interest in the use of immunotherapy as an alternative or adjunct to chemotherapy and radiotherapy for the treatment of cancer. Immunotherapy encompasses a broad array of active and passive approaches that can be applied in many disease settings. Passive immunity implies the administration of an immune product, thereby bypassing the body’s ability to respond to an immunogenic challenge such as bacterial or viral species. No cellular or humoral response is generated by the body and the exogenously administered immune product serves to act on the body’s behalf. The most successful application of passive immunotherapy has been the administration of monoclonal antibodies to defined tumour-associated antigens, and FDA-approved monoclonal antibody products have provided new standards of care for breast cancer, colorectal cancer, non-Hodgkin’s lymphoma and B cell chronic lymphocytic leukaemia.
1.4 Active immunotherapies
Active augmentation of the immune response to cancer can be attempted by two basic approaches: non-specific immunopotentiation which constitutes the bulk of past and current efforts at cancer immunotherapy, and specific immunisation, which has contributed much to the control of infectious diseases. It is the knowledge of microbial antigens which has permitted the development of successful specific immunisation against infections. The efficacy of antigen-presenting cells (APCs) loaded with PA2024, a recombinant fusion protein containing prostatic acid phosphatase (PAP) and granulocyte-macrophage colony stimulating factor (GM-CSF) was further evaluated by Burch et al. and the Dendreon Corporation [5]. This cellular product vaccine was based on the initial idea that specific APCs, dendritic cells (DCs), could process specific molecules and directly present them in an efficient manner to specialised lymphocytes so that they may be sensitised and effect an antitumour response to the tumour cells. Patients were leukophoresed, their white cells co-cultivated with GM-CSF and the PAP fusion protein (PA2024) and within a 48 hour turn around period, the same cells were reintroduced into the patient. Preliminary data from this Phase II trial indicated clinical responses with median time to progression was 118 days. Two patients exhibited a transient 25 - 50% decrease in PSA and a third patient had a significant PSA decline from 221 ng/ml at baseline to undetectable levels by week 24 and has remained as such for > 4 years. In addition, this patient’s metastatic retroperitoneal and pelvic adenopathy has resolved. Peripheral blood mononuclear cells (PBMC) collected from patients for at least 16 weeks proliferated upon in vitro stimulation by PA2024. For the patient with responsive disease as determined radiographically or by PSA declines, PBMC could be stimulated for 96 weeks.
A completed Phase III trial examined a vaccine based on autologous DCs pulsed with a recombinant protein, PAP fused with GM-CSF [6]. A total of 127 patients with asymptomatic metastatic hormone refractory prostate cancer were randomised (2:1 ratio) to receive antigen-loaded DCs or unloaded DCs given every 2 weeks for 3 cycles. A trend towards increased time to progression (TTP) was observed, but the data failed to reach statistical significance. In a post hoc subgroup analysis of patients with Gleason score (GS) < 7, a significantly delayed TTP and time to onset of disease related pain (TDRP) was suggested for treatment versus placebo. A Phase III trial (n = 275) to confirm these results in patients with GS ≤ 7 has been initiated [6].
GVAX® (Cell Genesys) is a prostate cancer vaccine that is in clinical-stage development for patients with advanced-stage castrate metastatic prostate cancer. The vaccine is comprised of two prostate cancer cell lines that have been genetically modified to secrete GM-CSF, a hormone which plays a key role in stimulating the body’s immune response to vaccines, and then irradiated for safety. Two Phase II multi-centre trials of GVAX have been conducted in hormone-refractory patients. The initial trial demonstrated improved survival of patients receiving the vaccine (median survival of 31 months in the high-dose group), and these results compare favourably to the reported median survival for castrate metastatic prostate cancer patients with bone metastases who are treated with taxane chemotherapy, the current standard of care for this patient group. The follow-up Phase II trial demonstrated improvements in bone cell loss and PSA values; however, median survival had reportedly not yet been reached for any treatment group. Phase III clinical trials of GVAX prostate cancer vaccine are planned to compare the vaccine to taxane-based chemotherapy in castrate metastatic prostate cancer patients with radiographic evidence of metastatic disease [7,8].
Fully synthetic carbohydrate-based vaccines have completed Phase I and 2 testing in biochemically relapsed prostate cancer [9,10]. The antigen was a trimeric form or cluster of the mucin-related O-linked glycopeptide, α-N-acetylgalactosamine-O-serine/threonine [Tn(c)]. When conjugated to the carrier protein keyhole limpet haemocyanin (KLH) and combined with QS21 adjuvant, the Tn(c) vaccine stimulated the production of high-titer antibody responses. A decline in PSA slopes post-treatment was also observed, although the clinical significance of this treatment effect is unknown at present. Similar findings were made in another trimeric vaccine with Thomsen-Friedenreich antigen also in a clustered formation conjugated to KLH and administered with QS-21 [11].
A recent ECOG (East Coast Oncology Group consortium) trial involving a vaccinia construct expressing PSA followed by a fowlpox construct expressing PSA in a ‘prime-boost strategy’ in men with a rising PSA following definitive local therapy, but without overt metastatic disease did not result in significant changes in PSA values. The best overall response was stabilisation of PSA, although 52% of patients remained free of PSA progression 2 years after entering the trial [12].
1.5 Summary
Preclinical studies have strongly supported the role of adoptive immunotherapeutic approaches and indicated that antitumour responses can be favourably induced by using a variety of effector cell populations. Each of the antigens studied by clinical trials has been able to generate immune responses as garnered by the development of high titer IgM and/or IgG responses to the antigens used for immunisation. However, there have been few antigens which have induced cellular immune responses which may prove to be more durable. The current technologies and the concurrent use of cytokines such as IL-2 or GM-CSF may favourably enhance response, but whether it is the cytokine, the vaccine or both which imparts enhanced immunogenicity or antitumour effects remains to be determined.
2. Treatment impact: post-therapy changes in prostate-specific antigen
Two main issues remain in determining response to vaccines and other biological therapies. The first, is that there is no clear standard of treatment response for these therapies and the ramifications of antibody titers and their role in complement dependent lysis or antibody-dependent cytolysis versus T cell responses as measured by effector functions using enzyme-linked immunoSPOT (ELISPOT) or cell proliferation assays remains unclear. Second, changes in a biomarker such as PSA may not truly reflect the biology of the disease, and third, changes in PSA doubling time or slope may be more effective gauges in deciding when therapy should be initiated. Guidelines for interpreting post-therapy changes in PSA values were recently provided by the PSA Working Group [13]. Serial serum PSA determinations may provide a valuable intermediate marker for screening new therapies [4]. Using this paradigm, agents that have no effect on the rate of rise in PSA following administration should be abandoned. In contrast, the pattern of change in PSA following an intervention that is associated with a favourable change in the natural history of the disease varies as a function of the specific therapy administered. Agents that reduce tumour burdens may produce declines in PSA. On the other hand, differentiation type approaches might result in an initial rise and later a fall in the measured values. In prior studies with conjugate vaccines, changes in PSA did not occur for several months after immunisations were begun [9]. In general, this was associated with the development of antibody titers after vaccination. In the present study, associations between the pattern of change in PSA, immunological responses, and changes in clinical status after vaccination will be examined. As the specific pattern of change in PSA that will be observed in this trial is unknown, this analysis is considered exploratory.
PSA as an end point for clinical trials is still under intense scrutiny. It remains unclear as to whether PSA stabilisation or decline correlates with a change in the biology of the tumour or whether there is any direct correlation between a biomarker and disease progression. There are no standard parameters for evaluating biological therapies and each trial may mandate a separate end point to assess a ‘response’. There is the danger of being too eager about Phase I or Phase II data which may lead to a misinterpretation of the data and false claims. Response criteria which can appropriately interpret the trial are stronglyneeded. Enthusiasm with Phase I and Phase II trials results must be tempered by the need to invest time and due consideration of how the best trial design must be incorporated for Phase III investigations.
3. Prostate-specific membrane antigen
The most thoroughly studied molecules in prostate cancer immunology have been PSA and prostate-specific membrane antigen (PSMA), the latter on which a diagnostic radiographic test has been previously marketed [14]. PSMA was originally thought to be a unique molecule whose expression, like PSA, was limited to the prostate gland. However, it is an extraordinary molecule in that it appears to be omnipresent, highly expressed on normal and malignant prostate cells, the brain, salivary glands, biliary tree and on neovasculature in renal cell carcinoma [15-17]. In prostate cancer cells, PSMA is expressed at 1000-fold higher levels compared with normal prostate epithelium, where it is expressed in the cytoplasm as the product of an RNA splice variant (PSM’) [18]. As a dimeric Type II integral membrane glycoprotein (Figure 1), it is a large, extracellular globular-like molecule with an intracellular segment and is expressed at levels > 1000-fold greater than the minimal expression seen in the kidney, proximal small intestine and salivary gland [15,16]. It has multiple domains including an intracellular segment (amino acids 1 - 18); a transmembrane domain (amino acids 19 - 43); and an extracellular domain (amino acids 44 - 750). PSMA is alternatively spliced to produce at least three variants, the most interesting, PSM’, which is located in the cytoplasm and is of unclear importance. It is also upregulated in the androgen-resistant state. Its ubiquitousness, rather than hindering its usefulness, ends up being its strength as it is a fascinating target for treatment schemes which include radiopharmaceutical targeting using a monoclonal antibody directed to the external domain, as well as its being widely expressed on renal tubular epithelium [19-21] suggesting that alternative forms of therapy may be directed along a similar venue for a disease which has relied heavily on surgical and investigational approaches often with limited success. In prostate cancer, expression of PSMA increases with disease progression and is highest in metastatic disease, hormone refractory cases, and higher-grade lesions. Important clinical proof-of-concept for PSMA tumour targeting is provided by ProstaScint® (Cytogen), which is a 111In-labelled anti-PSMA monoclonal antibody that is licensed by the FDA for imaging and staging of prostate cancer in patients with both newly diagnosed and recurrent disease [14]. In addition, PSMA is abundantly expressed on the neovasculature of a variety of other solid tumours, including bladder, pancreas, melanoma, lung and kidney cancers, but not on normal neovasculature [19,22]. Second generation humanised forms of a monoclonal antibody against the external domain of PSMA (J591) developed by Bander et al. [23], have been conjugated to iodine as well as yttrium with additional plans to conjugate to lutecium in an effort to image all sites of metastases, especially in bone. These studies have shown 100% specificity and sensitivity for sites in bone and have been extended to other malignancies including renal cell cancer. Antitumour activity was seen, with two patients experiencing 85 and 70% declines in PSA levels lasting 8 and 8.6 months, respectively, before returning to baseline [25]. These patients had objective measurable disease responses. An additional 6 patients (21%) experienced PSA stabilisation. Particularly exciting is the expression of PSMA protein in the neovasculature of solid tumours (except prostate), but not in normal vasculature facilitating additional imaging and therapeutic target development [23-26]. Several clinical trials and preclinical studies [27-36] have used PSMA peptides and whole length proteins in an effort to determine immunogenicity and clinical response as determine biochemically and radiographically. Horiguchi et al. [37] have identified HLA-restricted peptides capable of inducting robust T cell responses which may be applied to clinical trial. A Phase II trial by Murphy [38] involved infusions of autologous DCs with two HLA-A2.1+-restricted PSMA peptides in patient with androgen-independent disease and found cellular responses and impact on serum PSA.
Figure 1.

Globular nature of PSMA and the amino acids (aa) which make up the various domains. Note the distinct transmembrane domain (B), the intracellular cytoplasmic domain (A), the catalytic binding site (E) and extracellular domains C, D, E, F. [Domain A: aa 1 - 19; Domain B: aa 20 - 39; Domain C: aa 40 - 144; domain D: aa 173 - 248; Domain E: aa 275 - 596; Domain F: aa 597 - 756].
PSMA: Prostate-specific membrane antigen; PSM’: RNA splice variant of PSMA.
PSMA subserves several behaviours and functions. It can function in glutamatergic neurotransmission and folate absorption. As a NAALADase [14-16], it metabolises a brain neurotransmitter, N-acetyl-aspartyl-glutamate, and can remove gamma-linked glutamates in the small intestine from poly-γ-glutamated folates as a folate hydrolase [14-16,39-42] and as a carboxypeptidase in the form of carboxypeptidase II. In a manner similar to other cell surface receptors, PSMA can be internalised constitutively and spontaneously in a dose-dependent manner when bound to monoclonal antibody J591 [23,24,43]. The PSMA-antibody complex undergoes internalisation through clathrin-coated pits and finally ends up in the lysosomes [15,43]. Its internalisation scheme bears marked resemblance to the epidermal growth factor receptor (EGFR) and its ligand as it is well known that many ligands and their transmembrane receptors are internalised through clathrin-coated pits as part of receptor-mediated endocytosis [15]. The implication of this observation is that PSMA may have a transport function to another unknown ligand in a manner resembling other receptor-mediated endocytosis such as the peptide hormones, or growth factors.
4. DNA vaccines
Multiple strategies have been used toward breaking immunological tolerance as manifested by the development of antibodies and/or T cell responses to the immunising antigen(s). Significant experience with synthetic conjugate vaccines given with the carrier keyhole limpet haemocyanin and the immunological saponin adjuvant, QS-21, have shown that high titer IgM and IgG antibodies can be generated with specificity for the immunising glycoprotein or glycolipid antigen suggesting that immunological tolerance can be overcome in these patients [9-11,44,45]. The justification for using a DNA vaccine [46,47] is seen in Box 1 and is based on the fact that it is relatively inexpensive and simple to purify in large quantities and avoids using an ex vivo approach to expand patient’s cells. The plan encompasses presenting the antigen, in this case PSMA, to specialised APCs which can then take the antigen, digest it into smaller components or peptides and fold it within a complex which can be presented to the T cells which can then activate and initiate factors which can cause tumour cell death (Figure 2). However, there are other mechanisms at play which help to ensure that the immune system can recognise the immunogen. This includes the idea of cross-priming whereby transcription and translation of antigens) by non-APCS such as myocytes or keratinocytes can then release mature antigen via secretion or their death. These preformed antigen(s) can be captured by APCs and presented to naive T cells in lymph nodes which are stimulated to recruit additional immune cells to the sites of disease to effect an antitumour response. Immunisation with plasmid DNA may allow direct transfection of APCs, or facilitate cross-priming.
Box 1. Rationale for a DNA vaccine.
Relatively inexpensive; efficient purification in large quantity
Avoids complex ex vivo expansion and manipulation of patients’ cells
Antigen is cloned into a bacterial expression plasmid with a constitutively active promoter
Bacterial plasmid DNA contains immunostimulatory sequences (CpG motifs) that may serve as an immunological adjuvant
Direct entry of the antigen into the intracellular MHC class I processing pathway
MHC: Major histocompatibility complex.
Figure 2.

Immunogenicity of rsPSMA vaccine. Compared with other adjuvants, such as the saponin QS-21, a much more vigorous response was seen using alum [48].
ELISA: Enzyme-linked immunosorbent assay; FACS: Fluorescence-activated cell sorting; PSMA: Prostate-specific membrane antigen.
As part of developing a DNA vaccine, it is necessary to clone the antigen of interest into a bacterial expression plasmid with a constitutively active promoter. The plasmid can then be introduced into the skin or muscle with an intradermal or intramuscular injection. The skin and muscle host APCs, particularly DCs, which can then present the transcribed and translated antigen into the context of the major histocompatibility complex and co-stimulatory molecules which can then elicit an immune response. Recent preclinical work by Gregor et al. [46,47], using a melanoma model, demonstrated that immunisation of mice with xenogeneic (foreign) human DNA coding for the melanosomal differentiation antigen gp75/translocation protein 1 (TRP-1), gp100 and TRP-2 via Biojector (a carbon dioxide-driven delivery system which delivers the vaccine at high pressure into muscle without benefit of gold particles or other adjuvants), resulted in antibody production and cytotoxic T cell responses. This is in contradistinction to immunisation of the mouse with self-DNA to which there was no immune response. In preclinical studies of PSMA vaccines, similar results were obtained demonstrating that xenogeneic human PSMA DNA was able to elicit an antibody response against recombinant and cell-associated murine PSMA in immunised BALB/c mice while syngenetic (murine) PSMA DNA was not immunogenic. This was consistent with other published reports [48] suggesting that prostatic acid phosphatase could be a good target, particularly when xenogeneic PAP was used and was able to elicit both antibody and T cell responses. The platform for the DNA immunisations in man by Gregor et al. [49] was developed at the author’s institution and contained DNA which had been inserted into the pING plasmid vector which contained a CMV promoter and kanamycin resistance selection marker [46,47]. The CMV promoter and intron A assured that high levels of constitutive expression were maintained. Of note, the kanamycin gene was incorporated into the vector design as a means to place the kanamycin gene under the control of a prokaryotic promoter as a means to eliminate the risk of generation of antibiotic resistance. Along similar lines, investigators have translated this observation into the clinic where patients with advanced prostate cancer who are HLA-A2.1+ have been randomised to receive either murine or human DNA encoded for PSMA intramuscularly for several immunisations followed by cross-over to the respective other species. This randomisation allows a determination of the necessity of using xenogeneic DNA for initial priming to break immunological tolerance. Preliminary data documents the safety of the approach although immune study results are still pending.
5. A protein vaccine approach
The enthusiasm for PSMA as a target continues to be recognised for immune approaches. Recently, a recombinant soluble (rs) PSMA vaccine was studied which comprised the entire extracellular domain of PSMA [49]. It was produced in secreted form in recombinant Chinese hamster ovary cells to a purity of > 95% homogeneity. Preclinical studies in BALB/c mice immunised subcutaneously at two week intervals with rsPSMA vaccine (protein 25 μg) given with the adjuvant Alhydrogel® (Superfos) (325 μg aluminum) showed the generation of PSMA-specific antibodies with titers of > 1/100,000 by enzyme-linked immunsorbent assay. The response was directed to the cell surface native PSMA as indicated by reactivity by FACS analysis using cells expressing PSMA. Splenocytes were also evaluated by ELISPOT assay for activated T cells secreting IFN-γ (Figure 2).
Preliminary results of a Phase I clinical trial in patients with biochemically relapsed prostate cancer following prostatectomy or radiation therapy were recently presented [49]. The primary objective was to investigate the safety and tolerability of this treatment. Secondary end points included evaluation of the PSMA-specific immune responses and treatment effect manifest by change in serum PSA levels. The vaccine was administered with a dose-escalating design in cohorts of 6 patients receiving 4 immunisations of 50, 250 or 1000 μg of rsPSMA vaccine by subcutaneous injection. Recent data shows that the immunisations are safe with minimal reactivity at the injection sites. Patients at the 250 μg cohort generated antibodies although flow cytometry data has yet to show reactivity of patients’ sera against PSMA-expressing cell lines. The outcomes of this study will guide development of the rsPSMA protein and VRP-PSMA vaccines used alone and/or in prime-boost combinations and demonstrates the effectiveness of different adjuvants.
6. Recombinant viral vaccines: the concept
There have been several vaccines using viral vectors which have been examples of proof-of-principle as they have been used as intraprostatic immunotherapy for the treatment of newly diagnosed localised disease. Toward this end, concerns have been entertained as to how one evaluates a ‘response’ to localised therapy. Traditionally, inflammatory infiltrates comprised of lymphocytes have been used as an indication of the reactivity of the organ, although flagrant tumour necrosis is more desirable treatment goal. A unique vaccine replicon particle (VRP) system is currently under investigation as a potential approach [50,51]. PSMA-VRP is a propagation-defective, viral replicon vector system derived from an attenuated strain of Venezuelan equine encephalitis virus (VEE) [22] encoding PSMA for use as a vaccine for prostate cancer. VEE is part of the Alphavirus group that is comprised of enveloped viruses containing plus-sense RNA genomes within icosahedral capsids that replicate in the cytoplasm of infected cells. The VRP system is derived from a VEE virus that has been genetically modified to be avirulent, and consists of a self-amplifying RNA (replicon) in which the structural protein genes of VEE are replaced by the full-length human PSMA gene [50]. The replicon RNA is packaged into VRPs by supplying the structural proteins in trans during the production phase. After VRP enters cells, the replicon RNA is first translated to produce the viral replicase proteins necessary to initiate self-amplification and expression (Figure 3). The PSMA gene is encoded by a subgenomic mRNA, leading to high level of expression of PSMA protein and presentation to the immune system. Attenuating mutations in the VEE structural proteins used to package VRP limit dissemination of the vaccine in vivo. The encoded gene is preferentially expressed in DCs in the draining lymph nodes, an ideal location for induction of immune responses. This approach has a number of benefits in its plan including: 1) mediates high-level full-length PSMA expression; 2) maintains innate adjuvanticity via dsRNA intermediates and induction of cellular apoptosis; 3) VRP infects DCs in lymph nodes; 4) elicits antigen-specific immunity; and 5) confers protective immunity in infectious disease and tumour models. Of note, the genes for the VEE structural proteins are not encoded by the replicon RNA, therefore, progeny viral particles are not assembled and replication is limited to a single cycle within the infected cell. Therefore, there is no DNA present within the vaccine, and the risk of incorporation of genetic material into the host chromosome is minimal. A similar vaccine has been studied in other virus systems and is currently under clinical development for HIV [52].
Figure 3.
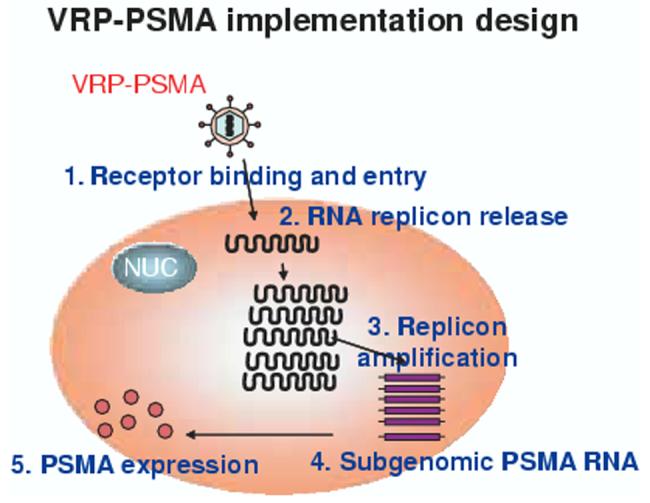
Strategy for the VRP-PSMA vaccine currently under clinical development.
PSMA: Prostate-specific membrane antigen; VRP: Vaccine replicon particle.
6.1 Preclinical studies in the PSMA-VRP replicon system
Gardner et al. [50] prepared PSMA-VRP constructs encoding full-length human PSMA and used them to immunise normal BALB/c mice subcutaneously with various doses (up to 108) units of vaccine. PSMA-specific immune responses were measured with a panel of cellular and humoral immunoassays, including ELISPOT and intracellular cytokine staining for IFN-γ and IL-4 in T cell subsets. Potent, durable TH1-based cellular and humoral responses were elicited after a single dose of vaccine, and these responses increased with subsequent immunisations in all mice tested (Figure 4). The immune response was directed to cell surface, native PSMA as indicated by sera reactivity in the flow cytometry assay using cells expressing PSMA. Splenocytes were also examined in an ELISPOT assay, and cells secreting IFN-γ were detected after PSMA stimulation in all groups of immunised mice. Intracellular cytokine fluorescence-activated cell sorting analysis was performed to measure IFN-γ levels in CD8+ T cells in response to PSMA stimulation. Between 10 - 20% of total CD8+ T cells were specific for PSMA after immunisations with PSMA VRP and these cells responded to antigen by producing IFN-γ. This cell phenotype was associated with cytotoxic killer T cells, and helper CD4+ T cells that recognised PSMA were detected using similar techniques. Unlike other vaccines, humoral and cellular immune responses were maintained for the course of the studies (up to 1 year) after the final immunisation. This durability has been highly sought after in multiple approaches and is the rationale for similar approaches in man. Interestingly, dose increments of 102, 104, 105 and 106, respectively, of PSMA-VRP were used in preclinical studies in mice with immune responses observed at all doses tested which were maintained at bi-weekly intervals with immunisation. Safety and toxicity studies showed no evidence of systemic, haematopoietic, and autoimmune reactivity related to PSMA+VRP at any dose or time in the study. Physical examination of the injection site showed minimal erythema that resolved within 1 - 2 days and was not related to the PSMA-VRP vaccine. Histopathological analysis of the injection site two days following the last immunisation showed a mild local inflammatory response in a subset of animals in all groups which resolved within two weeks and is thought to be related to injection trauma.
Figure 4.

A) VRP generate potent cellular immune responses to PSMA in vivo. BALB/c mice were inoculated s.c in the footpad three times at 2-week intervals with PSMA-VRPs. CD8+ T cells were isolated from splenocytes using Miltenyi MACS columns. T cells and syngeneic CT26-PSMA stimulator cells were added to ELISPOT wells coated with anti-murine IFN-γ capture antibody. After 20 hour incubation, cells were washed off and IFN-γ spots developed with detection antibody and HRP catalysis [49]. B) VRP generate potent humoral immune responses to PSMA in vivo. Groups of 5 BALB/c mice were inoculated subcutaneous in the footpad 3 times at 2-week intervals with PSMA-VRP. Two weeks after the final dose, serum was separated from blood, and used at 1:100 dilution in FACS analysis with 3T3 cells expressing human PSMA [49].
ELISPOT: Enzyme-linked immunoSPOT; FACS: Fluorescence-activated cell sorting; HRP: Horse radish peroxidase; IFN: Interferon; MFI: Mean fluorescence intensity; PSMA: Prostate-specific membrane antigen; VRP: Vaccine replicon particle.
7. Expert opinion
The case for PSMA as a target for vaccine development stands on its own. Its omnipresence, although originally thought to be a deterrent for clinical development, has been well-tempered by a variety of approaches which focus solely on the unique biological behaviour of the molecule. However, as is the case for many of the ‘self ’ antigens under evaluation, one needs to consider the potential, however remote, of developing autoimmunity. To date, there have been no reports of Hashimoto’s thyroiditis, rheumatoid arthritis, vitiligo, diabetes or anything phenomena suggestive of developing a response against self. Nevertheless, caution needs to be exercised, given the abundant expression of PSMA on brain and biliary trees. Efforts should be made to monitor blood parameters for the potential development of these medical problems.
The current technologies do not guarantee that immune responses will translate into a biological effect; however, each approach contributes to our understanding of the complex interaction of the cells of the immune system and how their functions may be enhanced. There is still a need to understand how to assess response in patients treated with biological therapies as an immune read-out may be insufficient in the setting of patients who have only a biomarker as the indication of their disease activity. It is gratifying that there remains continued interest in addressing this problem through multidisciplinary and multimodality strategies.
There remains a continued paucity of investigational trials for prostate cancer patients compared with other diseases such as breast cancer. For patients with continued good performance statuses and minimal tumour burden, a vaccine approach may be a reasonable option to explore other avenues of treatment prior to, or after, hormonal ablation or chemotherapy. Vaccines offer the opportunity to learn how immune responses can be maximised, whether alone or in combination with cytokines or chemotherapeutic drugs. Efforts in this direction should be continued and offer the potential to develop more rational approaches towards targeted therapy.
Acknowledgments
The author acknowledges the contributions of the following researchers: Polly Gregor, PhD, Jedd Wolchok, MD, Jason Gardner, PhD, William Goeckler, PhD, William Olson, PhD, Robert Israel, MD.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.TANNOCK IF, OSOBA D, STOCKLER MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisonealone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996;14:1756–1764. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 2.KANTOFF PW, HALABI S, CONAWAY M, et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. J. Clin. Oncol. 1999;17:2506–2513. doi: 10.1200/JCO.1999.17.8.2506. [DOI] [PubMed] [Google Scholar]
- 3.TANNOCK IF, DE WIT R, BERRY WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]; •• This reports the first chemotherapy regimen approved for the treatment of metastatic prostate cancer which also showed a survival benefit.
- 4.PETRYLAK DP, TANGEN CM, HUSSAIN MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004;351:1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 5.BURCH PA, CROGHAN GA, GASTINEAU DA, et al. Immunotherapy (APC8015, Provenge) targeting prostatic acid phosphatase can induce durable remission of metastatic androgen-independent prostate cancer: a Phase II trial. Prostate. 2004;60:197–204. doi: 10.1002/pros.20040. [DOI] [PubMed] [Google Scholar]
- 6.KYLSTRA JW, NEMUNAITIS J, SMALL EJ, JONES LA. A placebo-controlled Phase III trial of immunotherapy (APC8015, Provenge®) for androgen independent prostate cancer (AIPC): Evidence that Gleason Score predicts immunologic as well as clinical responses to therapy. Proc. Am. Assoc. Cancer Res. 2004;45 Abstract 1408. [Google Scholar]
- 7.SIMONS J, HIGANO C, CORMAN J, et al. A Phase I/II study of high dose allogeneic GM-CSF gene-transduced prostate cancer cell line vaccine in patients with metastatic hormone-refractory prostate cancer. Proc. Am. Soc. Clin. Oncol. 2003;22:166. Abstract 667. [Google Scholar]; •• An interesting study of GM-CSF used in concert with a prostate cancer cell line vaccine.
- 8.SIMONS J, NELSON W, NEMUNAITIS J, et al. Phase II trials of a GM-CSF gene-transduced prostate cancer cell line vaccine (GVAX) in hormone refractory prostate cancer. Proc. Am. Soc. Clin. Oncol. 2002;21:183a. Abstract 729. [Google Scholar]; • Results of a phase II trial of a prostate cancer cell line vaccine demonstrating an antitumor effect.
- 9.SLOVIN SF, RAGUPATHI G, MUSSELLI C, et al. Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: clinical trial results with alpha-N-acetylgalactosamine-O-serine/threonine conjugate vaccine. J. Clin. Oncol. 2003;21:4292–4298. doi: 10.1200/JCO.2003.04.112. [DOI] [PubMed] [Google Scholar]
- 10.SLOVIN S, RAGUPATHI G, CLARKE T, et al. Multivalency in a Phase II prostate cancer (PC) vaccine trial: Are more antigens better? Proc. Am. Soc. Clin. Oncol. 2003;22:167. Abstract 671. [Google Scholar]
- 11.SLOVIN SF, RAGUPATHI G, MUSSELLI C, et al. Thomsen-Friedenreich. Cancer Immun. Immunother. doi: 10.1007/s00262-004-0598-5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.KAUFMAN HL, WANG W, MANOLA J, et al. Prime/boost vaccination using poxviruses expressing PSA in D0 prostate cancer: preliminary results of ECOG 7897, a randomized Phase II clinical trial. Proc. Am. Soc. Clin. Oncol. 2002;21 4a Abstract 12. [Google Scholar]
- 13.SCHER HI, EISENBERGER M, D’AMICO AV, et al. Eligibility and outcomes reporting guidelines for clinical trials for patients in the state of a rising prostate-specific antigen: recommendations from the Prostate-Specific Antigen Working Group. J. Clin. Oncol. 2004;22:537–556. doi: 10.1200/JCO.2004.07.099. [DOI] [PubMed] [Google Scholar]; •• This paper represents a consensus by authorities in prostate cancer clinical trial development which addresses the establishment of appropriate clinical trial end points.
- 14.HINKLE GH, BURGERS JK, NEAL CE, et al. Multicenter radioimmunoscintigraphic evaluation of patients with prostate carcinoma using indium-111 capromab pendetide. Cancer. 1998;83:739–747. [PubMed] [Google Scholar]
- 15.GHOSH H, HESTON WDW. Tumor target Prostate Specific Membrane Antigen (PSMA) and its regulation in prostate cancer. J. Cell. Biochem. 2004;91:528–539. doi: 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- 16.SCHULKE N, VARLAMOVA OA, DONOVAN GP, et al. The homodimer of prostate-specific membrane antigen is a functional target for cancer therapy. Proc. Natl. Acad. Sci. USA. 2003;100:12590–12595. doi: 10.1073/pnas.1735443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.SILVER DA, PELLICER I, FAIR WR, et al. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- 18.SU SL, HUANG IP, FAIR WR, et al. Alternatively spliced variants of prostate-specific membrane antigen RNA: ratio of expression as a potential measurement of progression. Cancer Res. 1995;55:1441–1443. [PubMed] [Google Scholar]
- 19.CHANG SS, REUTER VE, HESTON WD, GAUDIN PB. Metastatic renal cell carcinoma neovasculature expresses prostate specific membrane antigen. Urology. 2001;57:801–805. doi: 10.1016/s0090-4295(00)01094-3. [DOI] [PubMed] [Google Scholar]
- 20.CHANG SS, REUTER VE, HESTON WD, et al. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumorassociated neovasculature. Cancer Res. 1999;59:3192–3198. [PubMed] [Google Scholar]; • A study suggeting a pivotal role for PSMA in neovasculature.
- 21.DIVGI CR, BANDER NH, SCOTT AM, et al. Phase I/II radioimmunotherapy trial with iodine-131-labeled monoclonal antibody G250 in metastatic renal cell carcinoma. Clin. Cancer Res. 1998;4:2729–3279. [PubMed] [Google Scholar]
- 22.CHANG SS, O’KEEFE DS, BACICH DJ, et al. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin. Cancer Res. 1999;5:2674–2681. [PubMed] [Google Scholar]
- 23.BANDER NH, TRABULSI EJ, KOSTAKOGLU L, et al. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J. Urol. 2003;170:1717–1721. doi: 10.1097/01.ju.0000091655.77601.0c. [DOI] [PubMed] [Google Scholar]; •• This describes a new monoclonal antibody which targets the external domain of PSMA.
- 24.SMITH-JONES PM, VALLABAHAJOSULA S, GOLDSMITH SJ, et al. In vitro characterization of radiolabeled monoclonal antibodies specific for the extracllular domain of prostate-specific membrane antigen. Cancer Res. 2000;60:5237–5243. [PubMed] [Google Scholar]
- 25.MILWSKY MI, NANUS DM, KOSTAKOGLU L, et al. Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J. Clin. Oncol. 2004;22:2522–2531. doi: 10.1200/JCO.2004.09.154. [DOI] [PubMed] [Google Scholar]
- 26.NANS DM, MILOWSKY MI, KOSTAKOGLU L, et al. Clinical use of monoclonal antibody HuJ591 therapy: targeting prostate specific membrane antigen. J. Urol. 2003;170:S84–S88. doi: 10.1097/01.ju.0000095151.97404.7c. [DOI] [PubMed] [Google Scholar]
- 27.TJO BA, SIMMONS SJ, ELGAMA A, et al. Follow-up evaluation of a Phase II prostate cancer vaccine trial. Prostate. 1999;4:125–129. doi: 10.1002/(sici)1097-0045(19990701)40:2<125::aid-pros8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 28.GONG MC, LATOUCHE JB, KRAUSE A, et al. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1:123–127. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’KEEFE DS, UCHIDA A, BACICH D, et al. Prostate-specific suicide gene therapy using the prostate-specific membrane antigen promoter and enhancer. Prostate. 2000;45:149–157. doi: 10.1002/1097-0045(20001001)45:2<149::aid-pros9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 30.GONG MC, CHANG SS, WATT F, et al. Overview of evolving strategies incorporating prostate-specific membrane antigen as target for therapy. Mol. Urol. 2000;4:217–222. [PubMed] [Google Scholar]
- 31.UCHIDA A, O’KEEFE DS, BACICH DJ, et al. In vivo suicide gene therapy model using a newly discovered prostate-specific membrane antigen promoter/enhancer: a potential alternative approach to androgen deprivation therapy. Urology. 2001;85:132–139. doi: 10.1016/s0090-4295(01)01256-0. [DOI] [PubMed] [Google Scholar]
- 32.MINCHEFF M, ALTANKOVA I, ZOUBAK S, et al. In vivo transfection and/ or cross-priming of dendritic cells following DNA and adenoviral immunizations for immunotherapy of cancer - changes in peripheral mononuclear subsets and intracellular IL-4 and IFN-gamma lymphokine profile. Crit. Rev. Oncol. Hematol. 2001;39:125–132. doi: 10.1016/s1040-8428(01)00111-1. [DOI] [PubMed] [Google Scholar]
- 33.LU J, CELLIS E. Recognition of prostate tumor cell by cytotoxic T lymphocytes specific for prostate-specific membrane antigen. Cancer Res. 2002;62:5807–5812. [PubMed] [Google Scholar]
- 34.KURATSUKURI K, SONE T, WANG CT, et al. Inhibition of prostate specific membrane antigen (PSMA)-positive tumor growth by vaccination with either full-length or the C-terminal end of PSMA. Int. J. Cancer. 2002;102:244–249. doi: 10.1002/ijc.10700. [DOI] [PubMed] [Google Scholar]
- 35.MINCHEFF M, ZOUBAK S, ALTANKOVA I, et al. Human dendritic cells genetically engineered to express cytosolically retained fragment of prostate-specific membrane antigen prime cytotoxic T-cell responses to multiple epitopes. Cancer Gene Ther. 2003;10:907–917. doi: 10.1038/sj.cgt.7700647. [DOI] [PubMed] [Google Scholar]
- 36.MINCHEFF M, TCHAKAROV S, ZOUBAK S, et al. Naked DNA and adenoviral immunizations for immunotherapy of prostate cancer: a Phase I/II clinical trial. Eur. Urol. 2000;38:208–217. doi: 10.1159/000020281. [DOI] [PubMed] [Google Scholar]
- 37.ORIGUCHI Y, NUKAYA I, OKAZAWA K, et al. Screening of HLA-A24-restricted epitope peptides from prostate-specific membrane antigen that induce specific antitumor cytotoxic T lymphocytes. Clin. Cancer Res. 2002;8:3885–3892. [PubMed] [Google Scholar]
- 38.MURPHY GP, TJOA BA, SIMMONS SJ, et al. Higher-dose and less frequent dendritic cell infusions with PSMA peptides in hormone-refractory metastatic prostate cancer patients. Prostate. 2000;43:59–62. doi: 10.1002/(sici)1097-0045(20000401)43:1<59::aid-pros8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 39.NILKUMAR G, RAJASEKARAN SA, WANG S, et al. Prostate-specific membrane antigen association with filamin A modulates its internalization and NAALADase activity. Cancer Res. 2003;63:2645–2648. [PubMed] [Google Scholar]
- 40.ACICH DJ, PINTO JT, TONG WP, HESTON WD. Cloning, expression, genomic localization, and enzymatic activities of the mouse homolog of prostate-specific membrane antigen/NAALADase/folate hydrolase. Mamm. Genome. 2001;12:117–123. doi: 10.1007/s003350010240. [DOI] [PubMed] [Google Scholar]
- 41.YAO R, SCHNEIDER E, RYAN TJ, et al. Human gamma-glutamyl hydrolase: cloning and characterization of the enzyme expressed in vitro. Proc. Natl. Acad. Sci. USA. 1996;93:10134–10138. doi: 10.1073/pnas.93.19.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.TASCH J, GONG M, SADELAIN M, HESTON WD. A unique folate hydrolase, prostate-specific membrane antigen (PSMA): a target for immunotherapy? Crit. Rev. Immunol. 2001;21:249–261. [PubMed] [Google Scholar]
- 43.LIU H, RAJASEKARAN AK, MOY P, et al. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–4060. [PubMed] [Google Scholar]
- 44.SLOVIN SF. Vaccines as treatment strategies for relapsed prostate cancer: approaches for induction of immunity. Hematol. Oncol. Clin. North. Am. 2001;15:477–496. doi: 10.1016/s0889-8588(05)70227-6. [DOI] [PubMed] [Google Scholar]
- 45.LOVIN SF, SCHER HI. Peptide and carbohydrate vaccines in relapsed prostate cancer: immunogenicity of synthetic vaccines in man-clinical trials at Memorial Sloan-Kettering Cancer Center. Semin. Oncol. 1999;26:448–454. [PubMed] [Google Scholar]
- 46.WOLCHOK JD, GREGOR PD, NORDQUIST LT, et al. DNA vaccines: an active immunization strategy for prostate cancer. Sem. Oncol. 2003;30:659–666. doi: 10.1016/s0093-7754(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 47.GREGOR PD, WOLCHOK JD, FERRONE CR, et al. CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine. 2004;22:1700–1708. doi: 10.1016/j.vaccine.2003.10.048. [DOI] [PubMed] [Google Scholar]
- 48.FONG L, RUEGG CL, BRICKSTEDT D, et al. Induction of tissue specific autoimmune prostatitis with prostatic acid phosphatase immunization: implications for immunotherapy of prostate cancer. Cancer Res. 1999;59:3102–3198. [PubMed] [Google Scholar]
- 49.GARDNER JP, SLOVIN SF, MORRISSEY DM, et al. Recombinant soluble prostate-specific membrane antigen (rsPSMA) vaccine: preliminary findings of a Phase I safety/immunogenicity trial. Proc. Am. Soc. Clin. Onc. 2004;23:184. Abstract 2584. [Google Scholar]
- 50.GARDNER JP, DONOVAN GP, SCHUELKE N, et al. Active tumor immunotherapy using novel PSMA-based alphavirus and protein vaccines. Proc. Am. Assoc. Cancer Res. 2003;44:1088. Abstract 4747. [Google Scholar]
- 51.VELDERS MP, MCELHINEY S, CASSETTI MC, et al. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavurus 16 E7 RNA. Cancer Res. 2001;61:7861–7867. [PubMed] [Google Scholar]
- 52.AVIS NL, CALEY IJ, BROWN KW, et al. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine encephalitis virus replicon particles. J. Virol. 2000;74:371–378. doi: 10.1128/jvi.74.1.371-378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]