

Abstract
Androgens provide survival signals to prostate epithelial cells, and androgen ablation induces apoptosis in the prostate gland. However, the molecular mechanisms of actions of the androgen-signaling pathway in these processes are not fully understood. Here, we report that androgens induced expression of the cellular Fas/FasL-associated death domain protein-like inhibitory protein (c-FLIP) gene, which is a potent inhibitor of Fas/FasL-mediated apoptosis. The androgen receptor was recruited to the promoter of the c-FLIP gene in the presence of androgens. We found that c-FLIP promoter contained multiple functional androgen response elements. In addition, we show that c-FLIP overexpression accelerated progression to androgen independence by inhibiting apoptosis in LNCaP prostate tumors implanted in nude mice. Our results suggest that the androgen receptor affects survival and apoptosis of prostate cells through regulation of the c-FLIP gene in response to androgens.
ANDROGENS PLAY CRITICAL roles in cell survival, growth, and differentiation in the prostate gland (1). Prostate cancer cells are also androgen dependent, and the common treatment for advanced prostate cancer is androgen withdrawal (2, 3). Androgen withdrawal therapy, although effective in early androgen-dependent stages, nonetheless fails in the androgen-independent stages of advanced prostate cancer (4, 5). Although the mechanism for the clinical response to androgen withdrawal therapy is not clear, androgen-independent progression has been associated with mutations or amplification of the androgen receptor (AR) gene and activation of intracellular signaling pathways that stimulate AR function (6, 7). AR mediates the functions of androgens and is a ligand-inducible transcription factor that regulates the transcription of specific target genes by binding to specific DNA response elements in their promoters, referred to as androgen response elements (AREs) (8–10). Androgen-targeted genes that play key regulatory roles in development and maintenance of the prostate gland, as well as in the response of malignant prostate cells to androgen deprivation, are poorly defined.
A search for these genes would logically include prostate apoptotic pathways. The growth of prostate tumors is determined by cell proliferation and cell death. Indeed, a high cell proliferation activity is associated with advanced clinical stage of prostate cancer (11), and androgen withdrawal inhibits prostatic cell proliferation and induces apoptosis of both normal and malignant prostate epithelial cells (12). In hormone-refractory prostate cancer the apoptosis rate decreases, and expression of the apoptotic inhibitor bcl-2 increases (13, 14).
In this study, we identified multiple functional AREs in the cellular Fas/FasL-associated death domain protein-like inhibitory protein (c-FLIP) gene and found that they were directly regulated by AR in the presence of androgens. Overexpression of c-FLIP enhanced the androgen-independent growth of the LNCaP tumor in the nude mouse.
RESULTS
AR Directly Targets the c-FLIP Gene
The androgen pathway exerts a protective effect in the prostate gland (10) and is required for growth of androgen-sensitive human prostate cancer LNCaP cells (15). Although the mechanisms underlying these effects have not been clearly defined, androgen’s effects on both pro- and antiapoptotic gene expression have been demonstrated (15). c-FLIP was shown to prevent Fas/FasL-mediated apoptosis by inhibiting caspase-8 activation at the death-inducing signaling complex (16). We investigated whether the AR pathway could regulate the c-FLIP gene expression in prostate cancer cells. Two mRNA species of c-FLIP have been described, the major species of 6 kb and another species of 1.3 kb (17). Northern blot analysis demonstrated that the androgen up-regulated c-FLIP gene expression in LNCaP cells (Fig. 1A).
Fig. 1. AR Directly Targets the c-FLIP Gene.
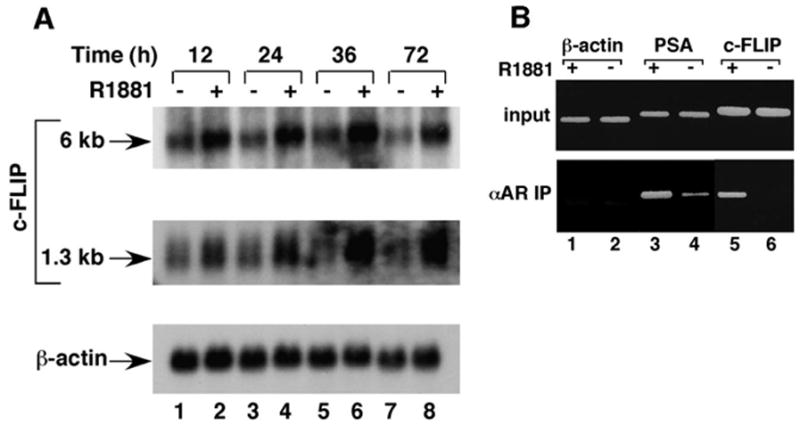
A, Northern blot analysis of c-FLIP expression in LNCaP cells. LNCaP cells were grown in the absence (lanes 1, 3, 5, and 7) or presence (lanes 2, 4, 6, and 8) of 10 nM R1881 for different times as indicated. mRNA was isolated, fractionated by electrophoresis, and transferred to a Hybond N+ membrane. The membrane was hybridized with c-FLIP probe (top two panels) or β-actin probe (bottom panel). B, ChIP assay to identify the c-FLIP gene as a direct target gene of AR. LNCaP cells were grown in the absence (lanes 2, 4, and 6) or presence (lanes 1, 3, and 5) of 10 nM R1881. Immunoprecipitation (IP) was performed with antigen-purified anti-AR antibody. The cross-links in the immunoprecipitated protein-DNA complexes were reversed, and the resulting DNA was amplified by a PCR with two specific primers derived from promoter regions of β-actin (lanes 1 and 2), PSA (lanes 3 and 4), or c-FLIP (lanes 5 and 6). The same set of PCR was performed with chromatin DNA used for immunoprecipitation (top panel).
In the chromatin immunoprecipitation (ChIP) assay, AR bound to the prostate-specific antigen (PSA) and c-FLIP promoter regions in the presence of androgen (Fig. 1B, lane 3 vs. lane 4 and lane 5 vs. lane 6). The products amplified by PCR at the same time from the β-actin promoter, which served as negative controls, were not changed in response to the addition of androgen (Fig. 1B, lanes 1 and 2). Because AR directly targets the PSA promoter (18), these results indicated that AR might directly target the c-FLIP promoter to enhance its gene expression in response to androgens.
The c-FLIP Gene Contains Multiple Functional AREs
We cloned a 1.9-kb genomic fragment (−1744 to +156) of the c-FLIP gene and inserted it into the luciferase reporter construct (pGL3). Transient transfection of this reporter plasmid into PC3 cells resulted in an approximately 6-fold higher level of luciferase activity in the presence of AR than in the absence of AR (Fig. 2A, left panel). This promoter was also androgen inducible in the AR-positive LNCaP cells (Fig. 2A, right panel). These results suggest that the cloned c-FLIP genomic fragment contained functional AREs, which we then further mapped using a collection of deletions (Fig. 2A, left panel). The deletion from the 5′-end to −48 did not affect AR-induced luciferase activity, whereas deletion from the 3′-end to +20 resulted in its complete loss, indicating that the functional AREs localized within the region from +20 to +156 of the c-FLIP promoter. The alignment of the +20 to +156 region of the c-FLIP promoter with class I and class II ARE consensus sequences (19) revealed four putative AREs within this region (Fig. 2C). The point mutations on ARE-3, −2, −1, −2+3, or −1+3 decreased, but did not eliminate, AR-dependent lucif-erase activity (Fig. 2A, left panel), whereas the mutations on ARE-1+2 completely abolished it. These results suggest that there are multiple AREs in the +20 to +156 region of the c-FLIP gene. A gel shift assay confirmed that the identified AREs were functional. Three c-FLIP AREs interacted with the DNA-binding domain (DBD) of AR (Fig. 2B). The affinity of AR-DBD to ARE-2 and ARE-3 was similar (lanes 2–4 and lanes 6–8), but its affinity to ARE-1 was 3-fold lower (lanes 10–12 vs. lanes 2–4 and lanes 6–8). The mutations on the conserved bases in the ARE probes abrogated the DNA-AR-DBD complexes (lanes 13–21), confirming the specificity of the observed interactions of AR-DBD with the c-FLIP AREs. We have not yet tested the putative c-FLIP ARE-4 by the above analyses. Thus, the identified functional c-FLIP AREs lie within the region of +50 to +150, and this region and the region (−321 to −32) amplified by PCR in the ChIP assay could be in the same DNA fragments (average size of 1 kb) immunopurified by anti-AR antibody (Fig. 1B, lanes 5 and 6).
Fig. 2. Activation of the c-FLIP Promoter by AR.
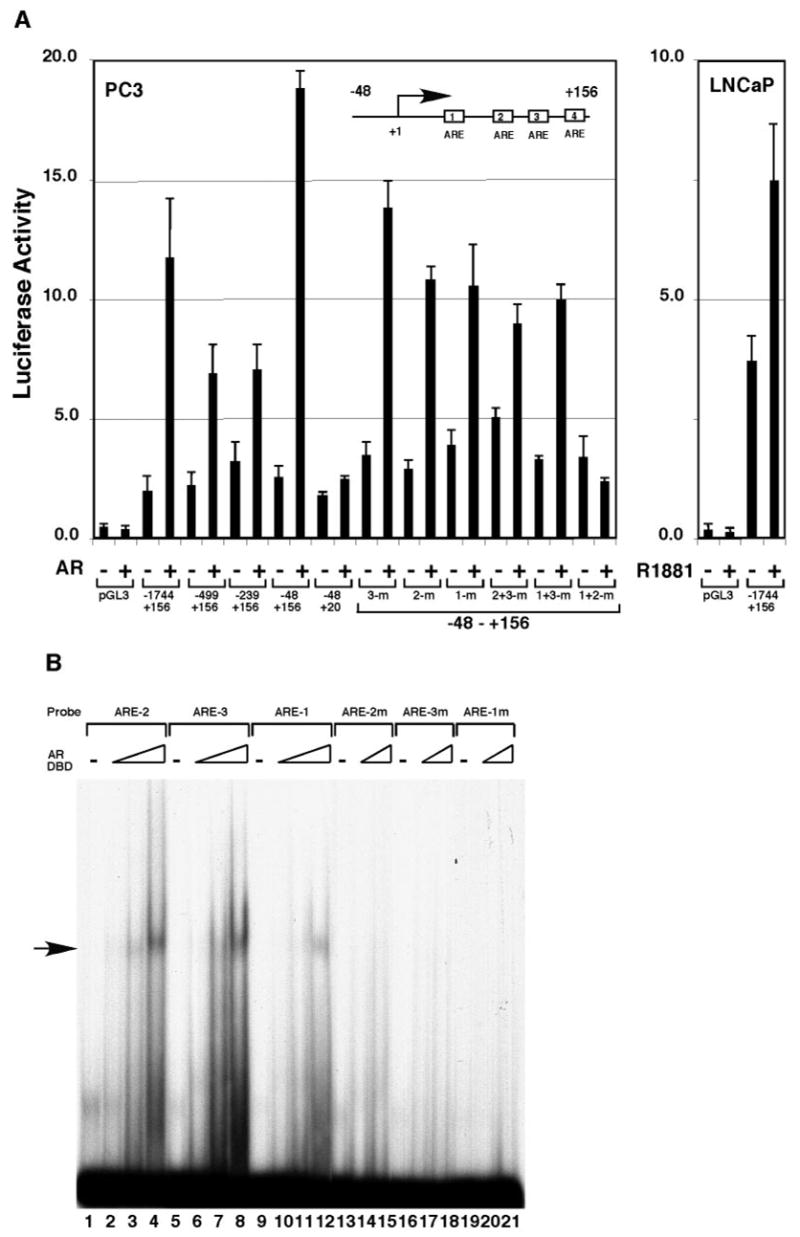

A, PC3 cells were transfected with 100 ng of the reporter plasmids containing various truncations and point mutations of the c-FLIP promoter and with or without 150 ng pcDNA-AR. LNCaP cells were transfected with 100 ng of the reporter plasmid containing the c-FLIP promoter (−1744 to +156) and grown in the absence or presence of 10 nM R1881. B, Binding of c-FLIP AREs to the DBD of AR. The different 32P-labeled ARE sites in the c-FLIP promoter were incubated with 0 ng (lanes 1, 5, 9, 13, 16, and 19), 20 ng (lanes 2, 6, and 10), 60 ng (lanes 3, 7, 11, 14, 17, and 20), or 180 ng (lanes 4, 8, 12, 15, 18, and 21) ng of the recombinant DBD of AR. The complexes of AR-DBD-ARE are indicated by an arrow at the left. C, The nucleotide sequence of the ARE-containing region in the c-FLIP promoter and the alignment of the c-FLIP AREs with the consensus AR binding sequences. The point mutations used in transient transfection and gel shift assays are indicated in lowercase letters.
Overexpression of c-FLIP Accelerated Progression to Androgen-Independence in the Human Prostate LNCaP Tumor Model
Of the currently available human prostate cancer cell lines, LNCaP cells are androgen responsive, PSA secreting, and immortalized in vitro (20). The growth of LNCaP tumors in nude mice is regulated by androgen and is inhibited by castration. After a prolonged period of growth in castrated hosts, LNCaP tumors become androgen independent (21, 22). The LNCaP tumor model is, therefore, particularly useful in studying mechanisms controlling growth and progression of prostate cancer. To investigate the functions of c-FLIP in prostate cancer in vivo, we infected LNCaP cells with a retrovirus harboring the c-FLIP gene (Fig. 3A), leading to increased levels of c-FLIP protein (Fig. 3B, lane 2 vs. lane 1). The growth rates of green fluorescent protein (GFP)-LNCaP and c-FLIP-LNCaP cells were similar in tissue culture (data not shown). GFP-LNCaP and c-FLIP-LNCaP cells injected sc into male nude mice produced tumors of similar size (Fig. 3C). However, c-FLIP-LNCaP tumors progressed to androgen independence more rapidly than GFP-LNCaP tumors after the mice were castrated (Fig. 3C). By 50 d after castration, the c-FLIP-LNCaP tumors weighed twice as much as the GFP-LNCaP tumors (Fig. 3D). In intact mice, GFP-LNCaP and c-FLIP-LNCaP tumors progressed at a similar rate (Fig. 3C) and weighed about the same (Fig. 3D). These results indicate that c-FLIP overexpression accelerated progression to androgen independence in the human prostate LNCaP tumor model.
Fig. 3. Overexpression of c-FLIP Accelerated Progression to Androgen Independence in the Human Prostate LNCaP Tumor Model.
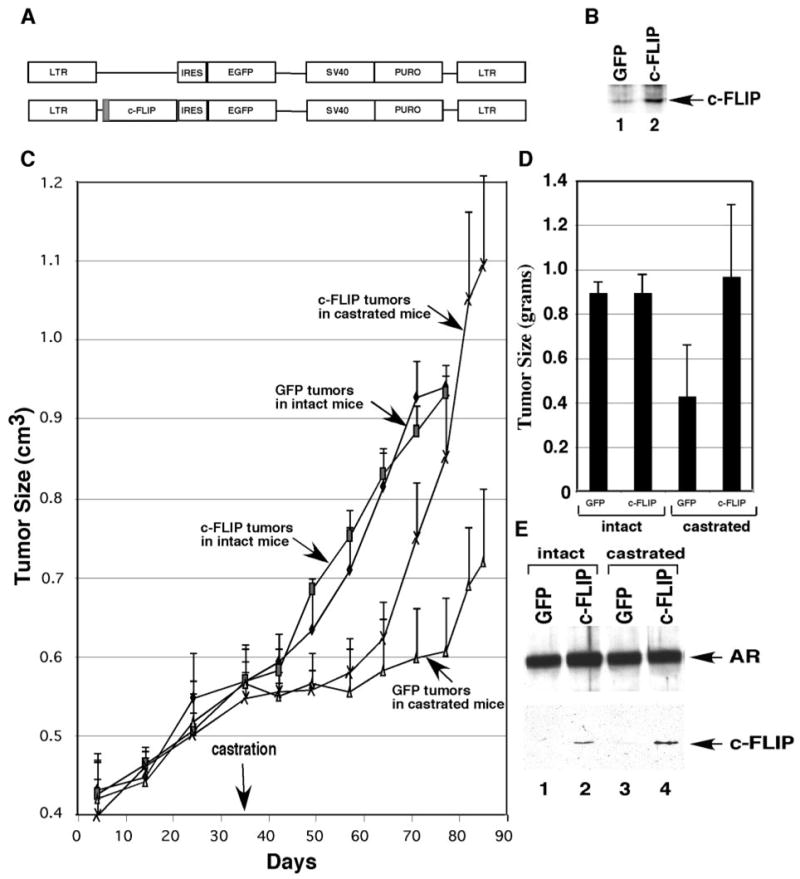
A, Schematic illustration of retrovirus constructs for expression of GFP and c-FLIP plus GFP. IRES, Internal ribosomal entry site. B, Western blot analysis with anti-c-FLIP antibody was performed with the whole-cell lysates made from cells infected with GFP-virus (lane 1) or with c-FLIP-virus (lane 2). C, Tumor growth curves in the intact and castrated male nude mice. D, The intact and castrated mice were killed by euthanasia 77 d and 85 d after implantation, respectively, and the weights of tumors were measured. E, Western blot analysis with anti-AR and anti-FLAG antibodies (to detect the FLAG-tagged c-FLIP) was performed with the LNCaP tumors. EGFP, Enhanced GFP; LTR, long terminal repeat; SV40, Simian virus 40 promoter; PURO, puromycin.
Overexpression of c-FLIP Inhibited Apoptosis in LNCaP Tumors
We next investigated the expression of AR and c-FLIP in the LNCaP tumors by Western blot and immunohistochemical analyses. AR expression in GFP-LNCaP and c-FLIP-LNCaP tumors in intact (Fig. 3E, lanes 1 and 2; Fig. 4, E and F) and castrated (Fig. 3E, lanes 3 and 4; Fig. 4, G and H) mice was similar. The Western blot and immunohistochemical analyses with anti-c-FLIP antibodies were unsuccessful because the antic-FLIP antibodies reacted nonspecifically with many proteins in LNCaP tumors. Immunoprecipitation of tumor extracts with the anti-FLAG antibody followed by Western blot analysis revealed that the FLAG-tagged c-FLIP was expressed in c-FLIP-LNCaP tumors in intact and castrated mice (Fig. 3E, lanes 2 and 4).
Fig. 4. The Overexpression of c-FLIP Inhibited Apoptosis in LNCaP Tumors.
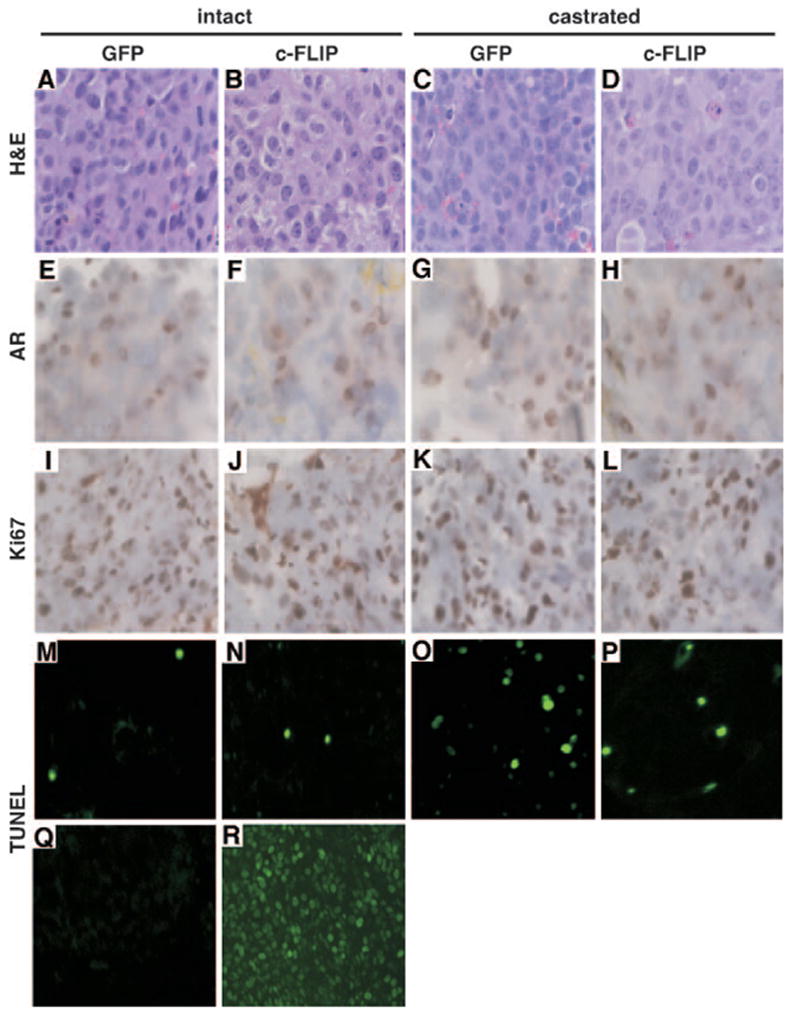
The GFP-LNCaP (A, E, I, M, C, G, K, O, Q, and R) and c-FLIP-LNCaP (B, F, J, N, D, H, L, and P) tumors were obtained from intact (A, B, E, F, I, J, M, N, Q, and R) or castrated (C, D, G, H, K, L, O, and P) nude mice. The tumors were stained with hematoxylin and eosin (H&E) (A–D), immunostained with anti-AR (E–H), or anti-Ki67 (I–L), or stained with TUNEL (M–R). Q, The negative control (without TdT); R, the positive control (DNase I treated) for the TUNEL assay. Original magnification is ×200 for A–L and ×100 for M–R.
The proliferation of LNCaP tumor cells was measured by immunohistochemistry staining with anti-Ki67 antibody. High percentages (>90%) of Ki67-positive cells were observed in GFP-LNCaP and c-FLIP-LNCaP tumors in both intact and castrated mice (Fig. 4, I–L). These results suggested that c-FLIP overexpression did not change the growth rate of LNCaP tumors in nude mice. Because c-FLIP inhibits Fas/FasL-mediated apoptosis (23), we also evaluated the effect of c-FLIP overexpression on apoptosis in LNCaP tumors by using the in situ transferase-mediated dUTP nick end labeling (TUNEL) method. Few TUNEL-positive cells were detectable in GFP-LNCaP (9 ± 1 apoptotic cells per 1500 tumor cells) and c-FLIP-LNCaP (11 ± 1 apoptotic cells per 1500 tumor cells) tumors obtained from intact mice (Fig. 4, M and N). However, more TUNEL-positive cells (55 ± 5 apoptotic cells per 1500 tumor cells) were found in GFP-LNCaP tumors from castrated mice (Fig. 4O). Overexpression of c-FLIP reduced the TUNEL-positive cells in c-FLIP-LNCaP tumors (25 ± 3 apoptotic cells per 1500 tumor cells) obtained from the castrated mice (Fig. 4P). The negative (without terminal transferase) and positive (DNase I-treated) controls for the TUNEL assay are shown in panels Q and R (Fig. 4), respectively. Taken together, these results indicate that c-FLIP overexpression promoted the androgen-independent growth of LN-CaP tumors through alterations in apoptosis.
C-FLIP Overexpressed in Prostate Cancer by cDNA Microarray and in Situ Hybridization
We have previously reported global cancer-specific gene expression in prostate cancer using Affymetrix oligonucleotide DNA microarrays (24). We extracted the levels of the transcript corresponding to c-FLIP to examine its individual levels of expression in normal (n = 11) and cancerous (n = 16) prostate. The average levels of c-FLIP expression in prostate cancer cases increased compared with normal prostate tissues (Fig. 5A). Using nonparametric analysis, Wald-Wolfowitz test showed the differences among the normal and primary cancers are statistically significant (P = 0.026).
Fig. 5. Overexpression of the c-FLIP Gene in Prostate Cancer.
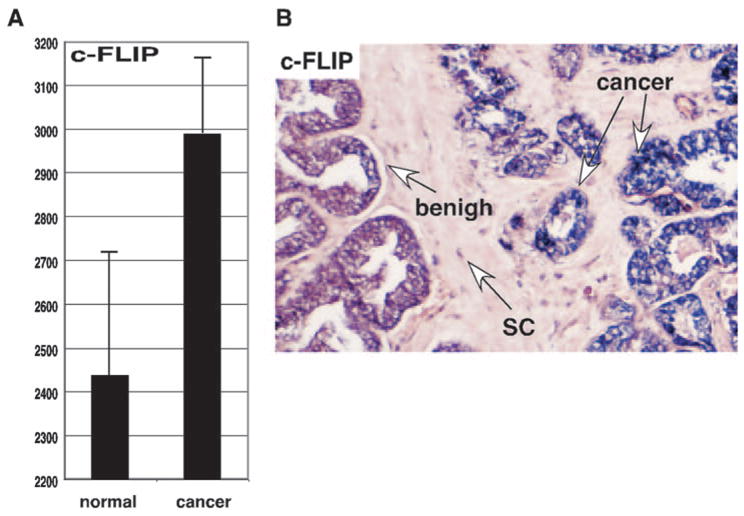
A, DNA microarray analysis of expression of the c-FLIP gene in normal prostate and cancerous tissue. y-Axis represents gene expression values (means ± SD) determined by default settings of Affymetrix Microarray Suite 4.0. B, The slides were hybridized with the digoxigenin-labeled RNA probes of c-FLIP. Positive signal is purple/blue, and the negative cells are colorless/light pink. SC, Prostatic stromal cells.
Because samples used in the DNA microarray analysis were macrodissected they contained prostate cancer cells as well as normal prostate and other cells. To overcome this problem, we further examined the expression of c-FLIP in 20 prostate cancer tissues derived from archival prostectomy specimens by in situ hybridization analysis. Of 20 prostate cancer cases examined, 12 cases (60%) showed higher expression levels for c-FLIP in cancer tissues (Fig. 5B). c-FLIP expression was not increased in high-grade prostatic intraepithelial neoplasia, a precursor of prostate cancer. Thus, these results suggest that the up-regulation of c-FLIP expression might contribute to prostate cancer growth.
DISCUSSION
The role of the androgen-signaling pathway in survival of the prostate epithelial and prostate cancer cells has been appreciated for some time, but the mechanisms underlying this process have not been addressed fully. In this study, we found that androgens up-regulated c-FLIP gene expression through multiple AREs within the c-FLIP gene. ChIP analysis indicated that AR directly bound to the promoter region of the c-FLIP gene in response to androgen treatment, suggesting that it directly targeted the c-FLIP gene. The ectopic expression of c-FLIP promoted androgen-independent growth of LNCaP tumors in nude mice. Our data shed light on a transcription-mediated mechanism for the effects of AR on cell survival and apoptosis in the prostate gland.
The identification and characterization of androgen-responsive genes have advanced our understanding of androgen action and prostate cancer in a variety of ways. One of the best-characterized androgen-responsive genes, PSA, is widely used as a diagnostic marker for prostate cancer. Other androgen-responsive genes such as hKLK2 (25) and PSCA (26) have been proposed as potential biomarkers for prostate cancer or for prostate cancer progression. The promoters of the androgen-responsive genes have been successfully used to generate mouse transgenic models for prostate cancer (27). Our results show androgen directly activated c-FLIP gene expression. Supporting this view was the discovery of multiple AREs in the c-FLIP promoter that complexed with AR-DBD and activated the c-FLIP promoter reporter plasmids in the presence of androgens. The c-FLIP AREs are similar to those found in the androgen-responsive enhancers in PSA (28), pro-basin (19), kKH2 (29), and PSCA (26) genes. These enhancers contain multiple low-affinity AREs that activate transcription in response to androgens. The pivotal role of c-FLIP in the control of Fas/FasL-mediated apoptosis suggests that it may be involved in androgen-mediated prostate cell survival.
Although the mechanisms underlying androgen functions in survival of the prostate gland as well as in promoting prostate tumor cell growth have not been clearly defined, the effects of androgen on both pro-and antiapoptotic gene expression have been demonstrated (15). Our present data provide additional information about how androgen can determine survival and apoptosis in the prostate gland. Specifically, in the presence of androgens, AR in the nucleus acts as a transcriptional activator for expression of the c-FLIP gene the function of which is to inhibit apoptosis. If androgens are depleted by chemical or surgical castration, however, this protective function is abolished, resulting in apoptosis in the prostate gland.
Whereas prostate cancer begins as an androgen-dependent tumor that regresses in response to androgen ablation, tumors eventually reappear and progress in an androgen-independent manner (4, 5). Altered gene expression patterns in prostate cancer cells may contribute to the androgen-independent growth of prostate cancer cells (30, 31). Consistent with this hypothesis, ectopic expression of some genes promotes androgen-independent growth in human prostate cancer cells (32, 33). c-FLIP expression is increased in human melanoma, and overexpression of c-FLIP contributed to tumor escape from the Fas/FasL-dependent apoptosis (34). Consistent with these observations, the c-FLIP gene was overexpressed in most prostate cancer specimens we surveyed. Loss or down-regulation of c-FLIP expression could sensitize various cells to the Fas/FasL-mediated apoptosis (35–39). Our finding that LNCaP tumors expressing high c-FLIP grew more quickly in castrated nude mice supports the relevance of the increased c-FLIP expression to prostate cancer. Moreover, androgen-independent growth correlated with the inhibition of apoptosis in the LNCaP tumors expressing high levels of c-FLIP. Whether dysfunction of the AR-c-FLIP signaling pathway might contribute to the survival of the androgen-independent prostate tumors in prostate cancer patients is currently being examined in our laboratory.
MATERIALS AND METHODS
Northern Blot Analysis
LNCaP cells (American Type Culture Collection, Manassas, VA) were maintained in RPMI 1640 medium supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) (HyClone Laboratories, Inc., Logan, UT). Cells were seeded into 15-cm dishes and cultured with phenol red-free RPMI 1640 supplemented with 10% charcoal/dextran (Sigma Chemical Co., St. Louis, MO)-stripped FBS for 3 d. The medium was then changed to phenol red-free RPMI 1640 with 10% charcoal/dextran-striped FBS with or without the synthetic androgen, R1881 (10 nM) (NEN Life Science Products, Boston, MA). Cells were harvested for mRNA isolation with oligo (dT) cellulose (Bio-Rad Laboratories, Inc., Hercules, CA) at 12, 24, 48, and 72 h after the medium change (40). For Northern blot analysis, 4 μg mRNA was fractionated with a 1% denatured formaldehyde-agarose gel (41), transferred to a Hybond N+ membrane (Amersham Biosciences, Piscataway, NJ), and cross-linked with UV light (GS Gene Linker, Bio-Rad Laboratories). The 771-bp fragment (729–1499) of human c-FLIP cDNA (encoding the long form of c-FLIP protein) (GenBank accession no. U97074) and the human β-actin cDNA were labeled with α-32P-dCTP using a random primer labeling kit (Amersham Biosciences) and purified over a 1-ml Sephadex G-50 column (Amersham Biosciences). Membranes were hybridized with probes (0.5 × 106 cpm/ml) in 5 ml of Express Hyb hybridization solution (CLONTECH Laboratories, Inc., Palo Alto, CA) with continuous shaking at 60 C overnight. The membrane was washed with 0.3 M NaCl-0.03 M sodium citrate-0.1% sodium dodecyl sulfate (SDS) at room temperature and with 0.03 M NaCl-0.003 M sodium citrate-0.1% SDS at 55 C, and then the membrane was exposed to x-ray film.
Transient Transfection Assay
PC3 cells was purchased from American Type Culture Collection and maintained in RPMI 1640 medium supplemented with 10% FBS. Cell transfection and the Due-Glo Luciferase Assay System (Promega, Madison, WI) were performed as described previously (42).
Gel Shift Assay
The gel shift assay was performed as described (43). Briefly, six pairs of oligos derived from the c-FLIP wild-type and mutant AREs (Fig. 2C) were synthesized and annealed and were labeled with γ-32P-ATP (Amersham Biosciences) by T4 polynucleotide kinase (New England Biolabs, Beverly, MA). In gel shift assays, the 20 μl reaction contained 20 mM HEPES (pH 7.9), 70 mM KCl, 1 μg poly(dI-dC), 1 mM dithiothreitol, 0.1% Nonidet P-40, 100 μg/ml BSA, and various recombinant AR-DBD protein (43). The reaction mixture was incubated for 30 min at 37 C and loaded directly onto a 4% (37.5:1, acrylamide-bisacrylamide) nondenaturing polyacryl-amide gel with 0.25 × Tris borate-EDTA buffer and run at 150 V for 2 h at room temperature.
ChIP
LNCaP cells were grown in phenol red-free RPMI 1640 supplemented with 10% charcoal/dextran-stripped FBS for 3 d and then treated with 10 nM R1881 for 36 h. Cells not treated with R1881 were used as the control. ChIP was performed as described elsewhere (44) with the following modifications: cross-linking was initiated with 1% formaldehyde at room temperature for 10 min and then stopped by addition of glycine to 0.125 M. The cross-linked chromatin was sonicated with Sonifer 450 microtip (Branson Ultrasonic Corp., Dan-bury, CT) at power setting 6 for 3 × 30 sec on ice. This treatment produced DNA fragments with an average size of 1 kb. For immunoprecipitation, 2 μg of antigen-purified anti-AR antibody was mixed with 200 μg of the purified cross-linked chromatin and incubated overnight at 4 C. Immunocomplexes were washed five times with a buffer containing 1% Triton X-100, 0.1% Na-deoxycholate, 0.05% SDS, 140 mM NaCl, and 1 mM phenylmethylsulfonylfluoride (Sigma); once with a buffer containing 0.25 M LiCl, 0.5% Nonidet P-40, 0.5% Na-deoxycholate, 1 mM EDTA, and 10 mM Tri-HCl (pH 8.0); and twice with 1 mM EDTA, 10 mM Tris-HCl (pH 8.0). After reversal and recovery of the immunoprecipitated chromatin DNA, the final DNA pellets were dissolved with 50 μl H2O. Immunopurified DNA (2.5 μl) was used for a PCR (30 cycles, annealing at 50 C and extension at 72 C for 0.5 min), with primers as follows: for c-FLIP, the forward primer sequence was GATCACGAGGTCAAGAGTTC, and the reverse primer sequence was GACACAAGTTCCACGCGTTAG, which amplifies a 290-bp product from −321 to −32 upstream of the c-FLIP gene. For PSA, the forward primer sequence was TCTGCCTTTGTCCGCTAGAT, and the reverse primer sequence was AACCTTCATTCCCCAGGACT, which amplifies a 212-bp product from −250 to −39 upstream of the PSA gene. For β-actin, the forward primer sequence was TCCTCCTCTTCCTCAATCTCG, and the reverse primer sequence was AAGGCAA CTTTCGGAACGG, which amplifies a 145-bp product from −1118 to −974 of the β-actin gene (the A of the ATG translation start codon was arbitrarily given the number +1).
Retrovirus-Mediated Expression of c-FLIP in LNCaP Cells
The retrovirus vector pBabe-GFP was prepared by inserting the EGFP cDNA from pEGFP-N1 and an internal ribosomal entry site from pCIN4 (45). The pBabe-c-FLIP-GFP was prepared by inserting the human c-FLIP cDNA (encoding the long form of c-FLIP protein) into pBabe-GFP. The human c-FLIP cDNA was modified to express the N-terminal Flag tag. Phoenix cells (46) were split at 1:4 in 10-cm dishes in DMEM-10% FBS. After 24 h, cells were transfected with 20 μg of retrovirus vector by lipofectAmine 2000 reagent (Invitrogen, Carlsbad, CA). The medium was changed, 48 h after transfection, to RPMI 1640–10% FBS. After an additional 12 h, virus-containing supernatant was collected and filtered through a 0.45-μm membrane, and the medium was supplemented with 8 μg/ml polybrene (Sigma). LNCaP cells were split at 1:3 in 10-cm dishes 1 d before infection. Cells were incubated with retrovirus for 36 h and split at 1:3 in 10-cm dishes. Cells were selected with 2 μg/ml puromycin (Sigma) for 6 d (~80–90% confluence). All the selected cells were GFP positive. The cells were split at 1:3 and grown in RPMI 1640–10% FBS supplemented with 2 μg/ml puromycin (Sigma) for 4–5 d.
Tumorigenicity Assays
LNCaP cells (3 × 106) cells expressing retrovirally transduced c-FLIP plus GFP, or GFP, were injected sc into each side of the back of 6-wk-old male nude mice (10 mice were injected). When the size of tumors reached about 1 cm in diameter, half of the nude mice were castrated, and the other half were kept intact as controls. Tumor volumes were calculated using the formula for hemiellipsoids: V = length (cm) × width (cm) × height (cm) × 0.5236. According to the protocol approved by the Institutional Animal Care and Use Committee at M.D. Anderson Cancer Center, the tumor-bearing mice must be euthanized when tumors reach 1.5 cm in diameter. Tumor xenografts grew more quickly in the intact mice than in the castrated mice. Therefore, the intact and castrated mice were killed on d 77 and d 85, respectively.
Expression of c-FLIP Using DNA Microarray Analysis
DNA microarray were performed as described (24). Briefly, total RNA was extracted from macrodissected benign and cancerous prostate tissue and evaluated for integrity by denaturing formaldehyde agarose gel. cDNA was synthesized from total RNA using a T7 promoter-tagged oligo-dT primer. RNA targets were synthesized by in vitro transcription and labeled with biotinylated nucleotides (Enzo Biochem, Farmingdale, NY). Gene expression analysis was performed using Affymetrix U95 human gene arrays (Affymetrix, Santa Clara, CA) consisting of five distinct microarrays (A–E), each containing probes for approximately 12,000 genes per expressed sequence tag transcript. The average difference was used as the primary measure of expression level, and absolute call was retained as a secondary measure. The data for the levels of transcripts of genes were extracted from the database, and the average of the levels of the c-FLIP transcript was subjected to a nonparametric Wald-Wolfowitz test to examine the statistical significance for the difference in the levels of the transcript.
Western Blot and Immunohistochemical Staining
Cells and tumor xenografts were washed with PBS and lysed with the passive lysis buffer (Promega, San Luis Obispo, CA) followed by centrifugation (10,000 × g, 5 min). Whole-cell or tumor lysates (30 μg per lane) were electrophoresed on 10% SDS-polyacrylamide gels at 250 V for 1.5 h (47). The resolved proteins were transferred to a nitrocellulose membrane using a Semi-Dry Transfer System (Bio-Rad Laboratories). Membranes were blocked for 30 min in 3% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.6; 150 mM NaCl; and 0.1% Tween 20). Blots were incubated with anti-AR (1:5000), anti-c-FLIP (1: 1000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), or anti-FLAG (1:1000) (Sigma) antibody for 2 h at room temperature, washed with TBST three times, and incubated for 1.5 h with the peroxidase-conjugated second antibody (1:5,000) (Amersham Biosciences). Antibodies were diluted with 2% BSA in TBST. The protein bands were detected by an enhanced chemiluminescence kit (Amersham Biosciences).
Tumors were removed from mice. One fraction of tumors was fixed in 10% formalin overnight and embedded in paraffin. The other fraction of tumors was placed in OCT compound (Sakura Finetek USA, Inc., Torrance, CA) and snap frozen in liquid nitrogen. The slides were incubated with 3% BSA for 30 min and then incubated with the anti-AR (1:1000) or anti-Ki67 (1:20) (DAKO Corp., Carpinteria, CA) antibody overnight at 4 C. The slides were rinsed with PBS and incubated with the peroxidase-conjugated secondary antibody (1:500) (DAKO) for 30 min at room temperature. After being washed with PBS, slides were developed with stable 3,3′-diaminobenzidine (Phoenix BioTechnologies Ltd., Milpitas, CA) and counterstained with Hill’s hematoxylin no. 3 (Sigma). The TUNEL assay was performed with the paraffin sections of tumors using the apoptosis detection kit (Promega) according to the manufacturer’s instructions. Slides were evaluated under an Olympus IX71 microscope (Olympus Corp., Lake Success, NY) with a digital camera (Retiga 1300) interfaced to a computer with PCI software. The apoptotic cells were quantified as follows. The number of cells of the area of tumor xenograft was captured and counted. There were about 500 cells per area, and three areas were captured in each slide. The fluorescence-labeled cells (apoptotic cells) from the same area of the tumor xenograft were captured and counted. Three tumor xenografts derived from three mice were analyzed.
Prostate Tissue Specimens and Pathological Evaluation
Prostate cancer and normal control tissues were derived from the radical prostatectomy specimens of 20 prostate cancer patients treated at New York University Medical Center. The study protocol was approved by the Institutional Review Board of New York University Medical Center. Tissues were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections of the tissue (4 μm) were cut and mounted on Super-frost Plus adhesion slides (Fisher Scientific, Pittsburgh, PA) and used for histology and in situ hybridization. The histological features of each individual specimen were confirmed by the pathologist (P.L.). DNA fragments corresponding to the c-FLIP gene were subcloned into pBluescript II, and the digoxigenin-labeled RNA probes were synthesized by incubation of DNA with T7 or T3 RNA polymerase. After wax removal and rehydration, 4-μm sections of formalin-fixed tissue were hybridized with the digoxigenin-labeled RNA probes, washed, and developed by incubation with chromogenic substrate (positive signal is purple/blue, and the negative cells are colorless/light pink) (48).
Acknowledgments
We thank Mr. Walter Pagel for the critical editorial review. We also thank Dr. Corazon D. Bucana for help with immunohistochemistry staining and Meisheng Zhou for statistical analysis.
Abbreviations
- AR
Androgen receptor
- ARE
androgen response element
- c-FLIP
cellular Fas/FasL-associated death domain protein-like inhibitory protein
- ChIP
chromatin immunoprecipitation
- DBD
DNA-binding domain
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- PSA
prostate-specific antigen
- SDS
sodium dodecyl sulfate
- TBST
20 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.1% Tween 20
- TUNEL
transferase-mediated dUTP nick end labeling
Footnotes
This work was supported in part by National Institutes of Health Grant 1R01 DK065156 01 from National Institute of Diabetes and Digestive and Kidney Diseases (to Z.W.), and by a Cancer Center Support Core Grant (NIH Grant CA16672) to M.D. Anderson Cancer Center from the National Cancer Institute. This work is also supported by a New York University Pathology departmental start-up fund and Veterans Affairs Merit Review Award (to P.L.).
References
- 1.Denmeade SR, Lin XS, Isaacs JT. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate. 1996;28:251–265. doi: 10.1002/(SICI)1097-0045(199604)28:4<251::AID-PROS6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 2.Porkka KP, Visakorpi T. Molecular mechanisms of prostate cancer. Eur Urol. 2004;45:683–691. doi: 10.1016/j.eururo.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Santos AF, Huang H, Tindall DJ. The androgen receptor: a potential target for therapy of prostate cancer. Steroids. 2004;69:79–85. doi: 10.1016/j.steroids.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 5.Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, Said J, Reiter RE, Sawyers CL. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res. 1999;59:5030–5036. [PubMed] [Google Scholar]
- 6.Koivisto PA, Rantala I. Amplification of the androgen receptor gene is associated with P53 mutation in hormone-refractory recurrent prostate cancer. J Pathol. 1999;187:237–241. doi: 10.1002/(SICI)1096-9896(199901)187:2<237::AID-PATH224>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 7.Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, Hung MC. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60:6841–6845. [PubMed] [Google Scholar]
- 8.Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999;69:307–313. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- 9.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–3015. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Hakimi JM, Rondinelli RH, Schoenberg MP, Barrack ER. Androgen-receptor gene structure and function in prostate cancer. World J Urol. 1996;14:329–337. doi: 10.1007/BF00184606. [DOI] [PubMed] [Google Scholar]
- 11.Visakorpi T, Kallioniemi OP, Koivula T, Isola J. New prognostic factors in prostatic carcinoma. Eur Urol. 1993;24:438–449. doi: 10.1159/000474347. [DOI] [PubMed] [Google Scholar]
- 12.Westin P, Stattin P, Damber JE, Bergh A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am J Pathol. 1995;146:1368–1375. [PMC free article] [PubMed] [Google Scholar]
- 13.Bruckheimer EM, Brisbay S, Johnson DJ, Gingrich JR, Greenberg N, McDonnell TJ. Bcl-2 accelerates multistep prostate carcinogenesis in vivo. Oncogene. 2000;19:5251–5258. doi: 10.1038/sj.onc.1203881. [DOI] [PubMed] [Google Scholar]
- 14.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52:6940–6944. [PubMed] [Google Scholar]
- 15.Coffey RN, Watson RW, O’Neill AJ, Mc Eleny K, Fitzpatrick JM. Androgen-mediated resistance to apoptosis. Prostate. 2002;53:300–309. doi: 10.1002/pros.10159. [DOI] [PubMed] [Google Scholar]
- 16.Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Bamhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–288. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Zhang S, Murtha PE, Zhu W, Hou SS, Young CY. Identification of two novel cis-elements in the promoter of the prostate-specific antigen gene that are required to enhance androgen receptor-mediated transactivation. Nucleic Acids Res. 1997;25:3143–3150. doi: 10.1093/nar/25.15.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reid KJ, Hendy SC, Saito J, Sorensen P, Nelson CC. Two classes of androgen receptor elements mediate cooperativity through allosteric interactions. J Biol Chem. 2001;276:2943–2952. doi: 10.1074/jbc.M009170200. [DOI] [PubMed] [Google Scholar]
- 20.Tilley WD, Wilson CM, Marcelli M, McPhaul MJ. Androgen receptor gene expression in human prostate carcinoma cell lines. Cancer Res. 1990;50:5382–5386. [PubMed] [Google Scholar]
- 21.Chen Y, Martinez LA, LaCava M, Coghlan L, Conti CJ. Increased cell growth and tumorigenicity in human prostate LNCaP cells by overexpression to cyclin D1. Oncogene. 1998;16:1913–1920. doi: 10.1038/sj.onc.1201719. [DOI] [PubMed] [Google Scholar]
- 22.Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, Pathak S, Chung LW. LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate. 2000;44:91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 23.Roth W, Reed JC. FLIP protein and TRAIL-induced apoptosis. Vitam Horm. 2004;67:189–206. doi: 10.1016/S0083-6729(04)67011-7. [DOI] [PubMed] [Google Scholar]
- 24.LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, Gerald WL. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 25.Partin AW, Catalona WJ, Finlay JA, Darte C, Tindall DJ, Young CY, Klee GG, Chan DW, Rittenhouse HG, Wolfert RL, Woodrum DL. Use of human glandular kallikrein 2 for the detection of prostate cancer: preliminary analysis. Urology. 1999;54:839–845. doi: 10.1016/s0090-4295(99)00270-8. [DOI] [PubMed] [Google Scholar]
- 26.Jain A, Lam A, Vivanco I, Carey MF, Reiter RE. Identification of an androgen-dependent enhancer within the prostate stem cell antigen gene. Mol Endocrinol. 2002;16:2323–2337. doi: 10.1210/me.2002-0004. [DOI] [PubMed] [Google Scholar]
- 27.Abate-Shen C, Shen MM. Mouse models of prostate carcinogenesis. Trends Genet. 2002;18:S1–S5. doi: 10.1016/s0168-9525(02)02683-5. [DOI] [PubMed] [Google Scholar]
- 28.Farmer G, Connolly ES, Jr, Mocco J, Freedman LP. Molecular analysis of the prostate-specific antigen upstream gene enhancer. Prostate. 2001;46:76–85. doi: 10.1002/1097-0045(200101)46:1<76::aid-pros1011>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Xie X, Zhao X, Liu Y, Young CY, Tindall DJ, Slawin KM, Spencer DM. Robust prostate-specific expression for targeted gene therapy based on the human kallikrein 2 promoter. Hum Gene Ther. 2001;12:549–561. doi: 10.1089/104303401300042483. [DOI] [PubMed] [Google Scholar]
- 30.Chang GT, Blok LJ, Steenbeek M, Veldscholte J, van Weerden WM, van Steenbrugge GJ, Brinkmann AO. Differentially expressed genes in androgen-dependent and -independent prostate carcinomas. Cancer Res. 1997;57:4075–4081. [PubMed] [Google Scholar]
- 31.Amler LC, Agus DB, LeDuc C, Sapinoso ML, Fox WD, Kern S, Lee D, Wang V, Leysens M, Higgins B, Martin J, Gerald W, Dracopoli N, Cordon-Cardo C, Scher HI, Hampton GM. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22–R1. Cancer Res. 2000;60:6134–6141. [PubMed] [Google Scholar]
- 32.Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res. 2003;9:370–376. [PubMed] [Google Scholar]
- 33.Kiyama S, Morrison K, Zellweger T, Akbari M, Cox M, Yu D, Miyake H, Gleave ME. Castration-induced increases in insulin-like growth factor-binding protein 2 promotes proliferation of androgen-independent human prostate LNCaP tumors. Cancer Res. 2003;63:3575–3584. [PubMed] [Google Scholar]
- 34.Medema JP, de Jong J, van Hall T, Melief CJ, Offringa R. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez D, White E. E1A sensitizes cells to tumor necrosis factor α by downregulating c-FLIP(S) J Virol. 2003;77:2651–2662. doi: 10.1128/JVI.77.4.2651-2662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hietakangas V, Poukkula M, Heiskanen KM, Karvinen JT, Sistonen L, Eriksson JE. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol Cell Biol. 2003;23:1278–1291. doi: 10.1128/MCB.23.4.1278-1291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siegmund D, Hadwiger P, Pfizenmaier K, Vornlocher HP, Wajant H. Selective inhibition of FLICE-like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Mol Med. 2002;8:725–732. [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol Ther. 2002;1:520–527. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- 39.Hyer ML, Sudarshan S, Kim Y, Reed JC, Dong JY, Schwartz DA, Norris JS. Downregulation of c-FLIP sensitizes DU145 prostate cancer cells to Fas-mediated apoptosis. Cancer Biol Ther. 2002;1:401–406. doi: 10.4161/cbt.1.4.15. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Lu W, Dong Z. Tumor-infiltrating macrophages are involved in suppressing growth and metastasis of human prostate cancer cells by INF-β gene therapy in nude mice. Clin Cancer Res. 2002;8:2942–2951. [PubMed] [Google Scholar]
- 41.Rosen KM, Lamperti ED, Villa-Komaroff L. Optimizing the northern blot procedure. Biotechniques. 1990;8:398–403. [PubMed] [Google Scholar]
- 42.Yu X, Li P, Roeder RG, Wang Z. Inhibition of androgen receptor-mediated transcription by amino-terminal enhancer of split. Mol Cell Biol. 2001;21:4614–4625. doi: 10.1128/MCB.21.14.4614-4625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu GZ, Wang H, Wang Z. Identification of a highly conserved domain in the androgen receptor that suppresses the DNA-binding domain-DNA interactions. J Biol Chem. 2003;278:14956–14960. doi: 10.1074/jbc.M212229200. [DOI] [PubMed] [Google Scholar]
- 44.Orlando V, Paro R. Mapping polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell. 1993;75:1187–1198. doi: 10.1016/0092-8674(93)90328-n. [DOI] [PubMed] [Google Scholar]
- 45.Rees S, Coote J, Stables J, Goodson S, Harris S, Lee MG. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques. 1996;20:102–104. 106, 108–110. doi: 10.2144/96201st05. [DOI] [PubMed] [Google Scholar]
- 46.Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM, Roeder RG. Transcription coactivator TRAP220 is required for PPARγ 2-stimulated adipogenesis. Nature. 2002;417:563–567. doi: 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- 47.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Li P, Yu X, Ge K, Melamed J, Roeder RG, Wang Z. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am J Pathol. 2002;161:1467–1474. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]