

Abstract
Resveratrol is a polyphenol produced by plants that has multiple beneficial activities similar to those associated with caloric restriction (CR), such as increased life span and delay in the onset of diseases associated with aging. CR improves neuronal health, and the global beneficial effects of CR have been postulated to be mediated by the nervous system. One key enzyme thought to be activated during CR is the AMP-activated kinase (AMPK), a sensor of cellular energy levels. AMPK is activated by increases in the cellular AMP:ATP ratio, whereupon it functions to help preserve cellular energy. In this regard, the regulation of dietary food intake by hypothalamic neurons is mediated by AMPK. The suppression of nonessential energy expenditure by activated AMPK along with the CR mimetic and neuroprotective properties of resveratrol led us to hypothesize that neuronal activation of AMPK could be an important component of resveratrol activity. Here, we show that resveratrol activated AMPK in Neuro2a cells and primary neurons in vitro as well as in the brain. Resveratrol and the AMPK-activating compound 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) promoted robust neurite outgrowth in Neuro2a cells, which was blocked by genetic and pharmacologic inhibition of AMPK. Resveratrol also stimulated mitochondrial biogenesis in an AMPK-dependent manner. Resveratrol-stimulated AMPK activity in neurons depended on LKB1 activity but did not require the NAD-dependent protein deacetylase SIRT1 during this time frame. These findings suggest that neuronal activation of AMPK by resveratrol could affect neuronal energy homeostasis and contribute to the neuroprotective effects of resveratrol.
Keywords: caloric restriction, neuronal energy, neuronal protection
Resveratrol is a polyphenol that is present at high levels in grapes, nuts, pomegranates, and Polygonum cuspidatum, a component of Chinese herbal medicines. Resveratrol has potent antioxidant and antitumorigenic activities as well as important protective effects on the nervous system (1). For example, resveratrol blocks the accumulation of mutant protein aggregates and improves survival in nematode models of Parkinson's and Huntington's diseases (2). In the short-lived fish Nothobranchius furzeri, resveratrol delays age-dependent declines in locomotor activity and memory and reduces the neurofibrillary degeneration that occurs with normal aging (3). In mammalian neurons, resveratrol delays axonal degeneration after injury (4), blocks accumulation of Aβ peptide in vitro (5), and provides protection from brain ischemia in both adult and neonatal rodents (6). Because of these promising neuroprotective effects, resveratrol is currently being evaluated in clinical trials of patients with Alzheimer's disease. Interestingly, many of the activities of resveratrol are similar to the beneficial effects offered by caloric restriction (CR), including slowed aging and delaying the onset of chronic diseases (7, 8,).
Despite these protective effects on neurons, the mechanism of action of resveratrol is not fully understood. Resveratrol has been reported to alter expression of enzymes such as COX2 and ODC, inhibit cytochrome P450 enzymes, and activate the silent information regulator 2 (Sir2) protein, an NAD-dependent protein deacetylase (1). The activation of Sir2 was an exciting discovery because it provided a molecular link to the effects of resveratrol on longevity. Indeed, increased longevity due to resveratrol in nematodes and Drosophila depends on the presence of functional Sir2 (7). Resveratrol also consistently mimics the protective effects of SIRT1 (a mammalian Sir2 protein) overexpression in cell culture, suggesting that its neuroprotective effects are also mediated through this pathway.
Resveratrol and CR also cause metabolic changes such as decreased insulin/IGF signaling and increased mitochondrial biogenesis (1, 8). Interestingly, alterations in insulin signaling and mitochondrial activity also result from activation of AMP-activated kinase (AMPK), the central energy sensor in the cell (9–11). AMPK exists as a heterotrimeric complex containing a catalytic α subunit (α1 or α2), a regulatory β subunit (β1 or β2), and a γ subunit (γ1, γ2, or γ3) (12). AMPK is activated by alterations in the AMP:ATP ratio that occur in response to energetic stress and requires phosphorylation of Thr172 in the activation loop of the catalytic α subunit (13). Two upstream kinases have been identified as activators of AMPK, the tumor suppressor LKB1 (14, 15) and calcium/calmodulin-dependent protein kinase β (CaMKKβ) (16, 17).
AMPK is activated by a number of pathological stresses, including hypoxia, oxidative stress, glucose deprivation, as well as exercise and dietary hormones, such as leptin and adiponectin (12). AMPK activation plays a protective role against stress, in particular ischemia, where it decreases infarct size (11, 18–20). AMPK is also activated in the hypothalamic neurons under diet-restricted conditions (21). Because some of the metabolic changes caused by resveratrol mimic those observed in response to AMPK activation, we hypothesized that AMPK activation might be an important mediator of resveratrol actions in neurons. Our results show that resveratrol is a potent activator of AMPK in neuronal cell lines, primary neurons, and the brain. Furthermore, many of the actions of resveratrol, including mitochondrial biogenesis and neurite outgrowth, depended on the presence of a functional AMPK complex and its upstream regulator LKB1. However, resveratrol-mediated AMPK activation during this time period was independent of SIRT1. These results indicate that AMPK influences neuronal differentiation and that at least some of the actions of resveratrol in neurons are mediated by AMPK activation.
Results
Resveratrol Activates AMPK in Neuro2a Cells.
Polyphenols, in particular resveratrol, have been touted as CR mimetics and are neuroprotective in a number of paradigms. They are thought to act by stimulating the SIRT protein deacetylases; however, CR is also likely to influence other pathways, including energy sensing pathways, although this mode of action is not well characterized. To explore its role in modulating these pathways, we tested whether resveratrol altered the activity of AMPK in neuronal cells. Neuro2a neuroblastoma cells were treated with 10 μM resveratrol, and AMPK activation was examined by using phospho-AMPK-specific antibodies. Resveratrol treatment resulted in a robust increase in AMPK Thr172 phosphorylation within 2 h that persisted for up to 72 h (Fig. 1A). Interestingly, resveratrol activated AMPK to an extent similar to that of AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside), a well characterized activator of AMPK that is converted to ZMP, an AMP mimetic (18, 19). To confirm that AMPK activation by resveratrol results in typical AMPK-mediated downstream responses, we monitored phosphorylation of acetyl-CoA carboxylase (ACC), a primary target of activated AMPK (12). Using a phospho-ACC-specific antibody, we found that resveratrol stimulation led to robust phosphorylation of ACC both acutely and chronically to a degree similar to that observed with AICAR stimulation (Fig. 1B).
Fig. 1.

Resveratrol activates AMPK in Neuro2a cells. Neuro2a cells were switched to serum-starvation medium (medium containing 0.2% FCS) and treated with DMSO (vehicle control), 10 μM resveratrol (Resv), or 1 mM AICAR. (A) Both resveratrol and AICAR stimulated AMPK phosphorylation by 2 h and maintained the phosphorylated state through the 72-h test period. (B) Both resveratrol and AICAR promoted increased phosphorylation of the AMPK downstream effector ACC at all time points tested. Low levels of phosphorylated AMPK (pAMPK) and ACC (pACC) were detected in DMSO-treated control cells at all time points tested. Levels of total AMPK and ACC are shown in A and B (Bottom). (C and D) Densitometric analysis of changes in phosphorylated AMPK (C) and phosphorylated ACC (D) under different conditions is shown. AU, arbitrary units.
A well characterized mechanism of AMPK activation is a decrease in cellular energy levels reflected by an increased AMP:ATP ratio. To determine whether resveratrol-mediated AMPK activation occurs by this mechanism, we measured intracellular AMP and ATP levels in Neuro2a cells treated with DMSO (control) or resveratrol for 2 h. HPLC quantification of AMP and ATP levels failed to detect any increase in the AMP:ATP ratio in resveratrol-treated cells. In direct contrast, we observed a consistent increase in ATP and decrease in AMP levels in the resveratrol-treated cells compared with control cells. ATP and AMP concentrations in DMSO-treated cells were 34.66 ± 1.15 nmol/mg of protein and 6.17 ± 1.013 nmol/mg of protein, respectively, whereas in resveratrol-treated cells, the ATP level was 42 ± 8 nmol/mg of protein, and the AMP level was 4.16 ± 1.51 nmol/mg of protein. These results show that resveratrol treatment induced a statistically significant (P = 0.05) decrease in the AMP:ATP ratio (DMSO-treated cells, 0.178 ± 0.034; resveratrol-treated cells, 0.1 ± 0.036), thus demonstrating that resveratrol must stimulate AMPK activation in Neuro2a cells by mechanisms other than an increased AMP:ATP ratio.
AMPK Activation by Resveratrol Stimulates Neurite Outgrowth in Neuro2a Cells.
Neuro2a cells are a widely used as an in vitro model of neuronal differentiation. These cells cease to proliferate and begin to differentiate, as evidenced by neurite outgrowth, in response to serum starvation, retinoic acid, or growth factors such as neurotrophins and glial cell-derived neurotrophic factor family ligands. Because AMPK activation inhibits proliferation of a number of cell types (22), we first tested whether AMPK activation also inhibits Neuro2a cell proliferation. Neuro2a cells grown under serum starvation conditions (0.2% FCS) were treated with 1 mM AICAR or 10 μM resveratrol. Twenty-four hours after plating, the number of proliferating cells identified by using Ki67 immunocytochemistry was decreased dramatically (12.8% and 11%) by AMPK activation with AICAR or resveratrol compared with 50% proliferating cells in DMSO controls (data not shown). Both AICAR and resveratrol also induced differentiation of Neuro2a cells as evidenced by increased neurite outgrowth compared with serum starvation alone (Fig. 2).
Fig. 2.

Resveratrol and AICAR stimulate neurite outgrowth in Neuro2a cells. Neuro2a cells were switched to serum-starvation medium (medium containing 0.2% FCS) and treated with DMSO (vehicle control), 10 μM resveratrol (Resv), or 1 mM AICAR. (A) Serum deprivation of Neuro2a cells results in growth of short neurites that increase in length over 72 h. (B) Resveratrol induced rapid neurite outgrowth resulting in elaborate neurite network formation by 48 h. (C) AICAR stimulated extensive neurite outgrowth similar to that observed with resveratrol. (D) Quantification of neurite length showed significantly longer neurites in resveratrol- or AICAR-treated cells compared with Neuro2a cells grown in 0.2% serum alone (P < 0.001).
Because resveratrol activated AMPK and altered the differentiation of Neuro2a cells, we next asked whether AMPK activity was required for resveratrol-induced neurite outgrowth. To address this issue, we infected Neuro2a cells with lentivirus expressing either GFP alone (FUGW-control) or dominant-negative AMPK (dnAMPK). After 3 days of growth to allow robust lentiviral transgene expression, the medium was replaced with serum starvation medium. Resveratrol promoted robust neurite outgrowth in cells expressing GFP alone, whereas resveratrol-stimulated neurite outgrowth was severely diminished in Neuro2a cells expressing dnAMPK (Fig. 3). Resveratrol-induced neurite outgrowth was reduced in the presence of 10 μM AMPK pharmacological inhibitor Compound C (CC), further supporting the importance of AMPK activity for resveratrol-induced neurite outgrowth. We also infected Neuro2a cells with lentivirus expressing constitutively active AMPK (caAMPK) and found that constitutive AMPK activity significantly enhanced neurite outgrowth (Fig. 3E). Notably, CC (Fig. 3H) and dnAMPK (Fig. 3F) by themselves did not cause any significant inhibition of neurite growth, indicating that AMPK inhibition specifically reversed resveratrol-stimulated neurite growth. Taken together, these results indicate that AMPK activation is necessary and sufficient to inhibit Neuro2a proliferation and promote neuronal differentiation.
Fig. 3.

Resveratrol-induced neurite outgrowth and mitochondrial biogenesis are dependent on AMPK. Neuro2a cells were infected with lentivirus expressing GFP only (FUGW, control), dnAMPK, or caAMPK. Three days later, cells were shifted to serum-starvation medium (0.2% FCS) containing DMSO (control) or 10 μM resveratrol (Resv). In addition, uninfected Neuro2a cells in serum-starvation medium were treated with resveratrol alone, 10 μM AMPK inhibitor CC alone, or with resveratrol and CC together. Images of the cultures captured in bright-field and green fluorescence were overlaid to visualize neurite outgrowth. Resveratrol-treated cells demonstrated robust neurite outgrowth (B) that was blocked by inhibition of AMPK by dnAMPK (C) or CC (D). Conversely, cells infected with caAMPK demonstrated increased neurite outgrowth in the absence of resveratrol (compare E with A). dnAMPK alone (F) or CC alone (H) did not inhibit neurite outgrowth by themselves. (G) Neurite length in cells treated with caAMPK + resveratrol. (I) Quantitative analysis of average neurite length showed significant (denoted by asterisks) neurite outgrowth inhibition by AMPK inhibition (P < 0.001) and neurite outgrowth promotion by caAMPK (P < 0.005). (J and K) Quantitative RT-PCR analysis of markers of mitochondrial biogenesis demonstrated that resveratrol treatment resulted in an 18-fold increase in Tfam (K) and 2-fold increases in PGC-1α and mitofusin 2 (MFN2) mRNA levels (J), which were inhibited by CC. (L and M) Similar results were observed by genetic inhibition of AMPK with dnAMPK. Values were normalized to the 18S transcript. Data shown are representative of two independent experiments.
Resveratrol Induces Mitochondrial Biogenesis Through AMPK Activation.
CR induces mitochondrial biogenesis and bioenergetic efficiency in cultured cells (23) and animals (24). Because AMPK activation also induces mitochondrial biogenesis (10, 11, 25) and resveratrol mimics CR, we tested whether AMPK activation by resveratrol could promote mitochondrial biogenesis. We treated Neuro2a cells for 3 days with either DMSO (control) or 10 μM resveratrol in the presence or absence of 10 μM CC. We assessed mitochondrial biogenesis by monitoring mRNA levels of mitofusin 2, a mitochondrial protein and marker of mitochondrial mass and two key regulators of mitochondrial biogenesis, peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and mitochondrial transcription factor A (Tfam) (26). Quantitative RT-PCR analysis revealed that resveratrol treatment increased Tfam mRNA ≈18-fold, whereas PGC-1α and mitofusin 2 mRNA levels were increased 2-fold. Interestingly, the resveratrol-induced up-regulation of these mitochondrial markers was severely diminished when cells were treated with resveratrol in the presence of CC (Fig. 3 J and K) or were expressing dnAMPK (Fig. 3 L and M). These results suggest that one mechanism by which resveratrol exerts its protective effects is through promotion of mitochondrial biogenesis resulting from AMPK activation.
Resveratrol-Stimulated AMPK Activation Is Independent of SIRT1.
SIRT1 participates in many crucial functions, including protection from stress, aging, and cell cycle regulation (1, 7). Because a number of biological effects of resveratrol and other polyphenols depend on SIRT1 function, we explored whether AMPK activation and neurite outgrowth by resveratrol depend on SIRT1. First, we confirmed by Western blot analysis and immunocytochemistry that SIRT1 is expressed in Neuro2a cells (data not shown). Next, we stimulated Neuro2a cells with 10 μM resveratrol for 2 h in the presence or absence of three inhibitors of SIRT1 (10 μM sirtinol, 10 μM splitomycin, and 10 mM nicotinamide). None of the SIRT1 inhibitors attenuated the robust activation of AMPK by resveratrol as judged by the increased phosphorylation of AMPK and its downstream target ACC (Fig. 4 A and B). Similarly, SIRT1 inhibitors had no effect on the ability of resveratrol to stimulate Neuro2a neurite outgrowth (Fig. 4C). These results suggested that resveratrol effects on AMPK are independent of SIRT1 activity.
Fig. 4.

Resveratrol-mediated AMPK activation and neurite outgrowth are independent of SIRT1 and CaMKKβ in Neuro2a cells. Phospho-specific antibodies were used to assess activation of AMPK and ACC in lysates of Neuro2a cells treated with DMSO (control) or 10 μM resveratrol (Resv) in the presence or absence of three SIRT1 inhibitors [sirtinol, splitomycin (Splito), or nicotinamide (NAM)] or the CaMKKβ inhibitor STO 609 for 2 h. Resveratrol induces rapid activation of AMPK (A) that occurs in concurrence with phosphorylation of ACC (B) and is not prevented by SIRT1 or CaMKKβ inhibitors. AICAR is included as a positive control for AMPK and ACC phosphorylation. Total AMPK and ACC are shown in the A and B (Bottom). (C) Neuro2a cells were allowed to differentiate in serum-starvation medium containing resveratrol in the absence or presence of SIRT1 inhibitors [10 μM splitomycin, 10 mM nicotinamide, or 10 μM sirtinol (data not shown)] or 2.5 μM STO 609. No inhibition of neurite outgrowth was observed in the presence of either SIRT1 or CaMKKβ inhibitors.
Two kinases, LKB1 and CaMKKβ (14–17), have been identified as upstream activators of AMPK. Although no pharmacological inhibitors of LKB1 are presently available, we were able to use a selective CaMKKβ inhibitor STO 609 (2.5 μM) to test whether resveratrol activates AMPK in Neuro2a cells through CaMKKβ. We found that inhibition of CaMKKβ had no effect on resveratrol-mediated AMPK activation or neurite outgrowth (Fig. 4). Together, these results suggest that resveratrol-stimulated AMPK activation in Neuro2a cells is independent of SIRT1 or CaMKKβ function, and we predict the involvement of other upstream activators of AMPK.
LKB1 but Not SIRT1 Is Required for Resveratrol-Stimulated AMPK Activation in Cortical and Dorsal Root Ganglia Sensory Neurons.
Resveratrol activation of AMPK in Neuro2a cells along with the crucial role of AMPK in promoting neurite outgrowth in these cells encouraged us to examine this pathway in primary neurons. We treated E13.5 mouse dorsal root ganglia (DRG) sensory and cortical neuron cultures with 10 μM resveratrol or 1 mM AICAR. Western blotting demonstrated that resveratrol stimulated phosphorylation of AMPK and ACC in neurons from both peripheral and central nervous systems [Fig. 5C and supporting information (SI) Fig. 7C].
Fig. 5.

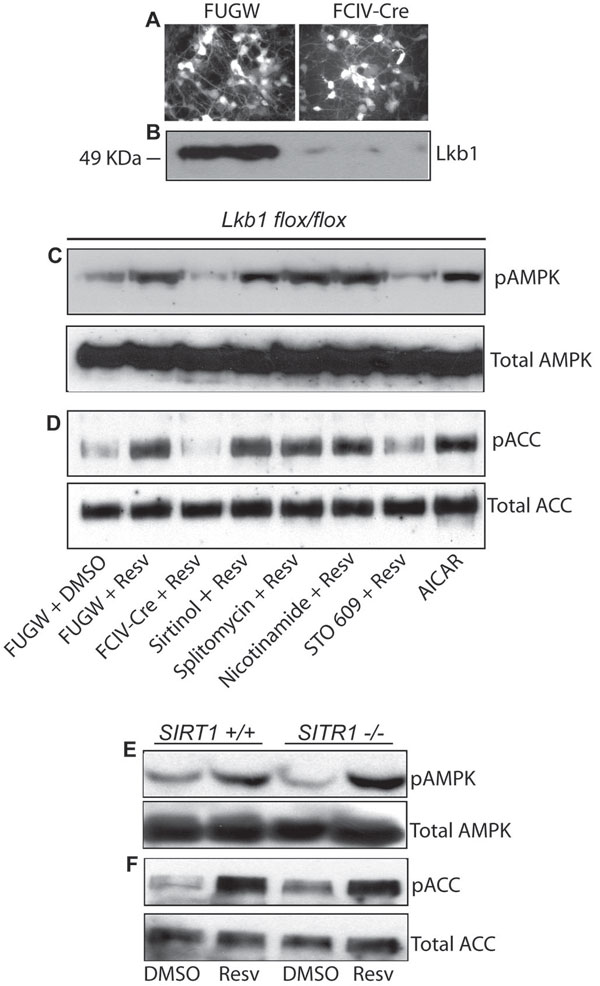
AMPK activation by resveratrol in DRG sensory neurons requires Lkb1 but is independent of SIRT1. (A) Embryonic DRG neurons from Lkb1flox/flox mice were infected with lentivirus expressing Cre recombinase (FCIV-Cre) or GFP only (FUGW control). Lentiviral infection was monitored by GFP fluorescence. (B) A Western blot with LKB1 antibody demonstrated complete loss of Lkb1 in Cre-expressing neurons. (C and D) Lkb1flox/flox DRG neurons were infected with FUGW or FCIV-Cre and treated as indicated. AMPK or ACC was immunoprecipitated from neuronal lysates, and Western blots were probed with the respective phospho-specific antibodies. Resveratrol-mediated AMPK (C) and ACC phosphorylation (D) were significantly reduced upon Lkb1 excision by FCIV-Cre. No inhibition of AMPK or ACC phosphorylation was observed by treatment with SIRT1 or CaMKKβ inhibitors. Lysate from AICAR-treated DRG neurons was included as a positive control. (E and F) Embryonic DRG neurons were cultured from wild-type and SIRT1-deficient littermates derived from SIRT1 heterozygous matings. Western blot analysis with phospho-specific antibodies revealed that resveratrol stimulated AMPK (E) and ACC phosphorylation (F) equivalently in wild-type and SIRT1-deficient neurons. Levels of total AMPK and ACC are shown in C–F (Bottom). Densitometric analysis of changes in levels of phospho-AMPK (G) and phospho-ACC (H) in SIRT1+/+ and SIRT1−/− DRG neurons in the presence and absence of resveratrol is shown. Resv, resveratrol; Splito, splitomycin; NAM, nicotinamide; AU, arbitrary units.
The ineffectiveness of CaMKKβ inhibitors suggested that LKB1 is likely to be the major effector of AMPK activation in neurons. To test directly the role of LKB1 in the resveratrol-mediated activation of AMPK in primary neurons, we took advantage of genetic models of LKB1 deficiency in which LKB1 can be conditionally deleted by using Cre recombinase. We cultured DRG sensory neurons and cortical neurons from E13.5 Lkb1flox/flox mouse embryos. Neurons were infected with either FUGW lentivirus (GFP control) or lentivirus expressing Cre recombinase to excise the floxed LKB1 alleles (Fig. 5 A and B). Lkb1-positive (those infected with FUGW) and Lkb1-deficient (those infected with Cre) DRG and cortical neurons were treated with 10 μM resveratrol for 2 h, and AMPK phosphorylation was examined by Western blot analysis. Loss of LKB1 significantly reduced resveratrol-stimulated phosphorylation of AMPK and its downstream target ACC in both DRG and cortical neurons (Fig. 5C and SI Fig. 7). As observed in Neuro2a cells, the CaMKKβ inhibitor STO 609 had no effect on resveratrol-induced AMPK phosphorylation in DRG neurons. However, STO 609 did inhibit AMPK and ACC phosphorylation by resveratrol in cortical neurons (SI Fig. 7). These results indicate that the primary regulator of resveratrol-stimulated AMPK activation in DRG neurons is LKB1. However, in cortical neurons, CaMKKβ also plays a role in AMPK activation by resveratrol, in accord with previous results indicating that it is a crucial regulator of AMPK in the brain.
To confirm the SIRT-independence of resveratrol-mediated AMPK activation that we observed in Neuro2a cells and in primary neurons, we performed experiments with pharmacological inhibitors as well as genetic experiments with neurons from SIRT1-deficient mice. Similar to our observations in Neuro2a cells, none of the SIRT inhibitors (sirtinol, splitomycin, and nicotinamide) inhibited resveratrol-stimulated AMPK phosphorylation in DRG or cortical neurons (Fig. 5C and SI Fig. 7). We also treated embryonic DRG and cortical neurons from SIRT1-deficient mice with resveratrol. Western blot analysis demonstrated that resveratrol stimulated equivalent levels of AMPK and ACC phosphorylation in wild-type and SIRT1-deficient DRG and cortical neurons (Fig. 5 E and F and SI Fig. 7). Collectively, these results indicate that resveratrol activates AMPK through the LKB1 pathway and that SIRT1 is not involved in this process.
Resveratrol Acutely Activates AMPK in Vivo.
Resveratrol has a number of beneficial effects in whole organisms, including protection against cerebral ischemia (6, 27), oxidative stress, excitotoxic brain damage (28), and posttraumatic seizures and it can lead to an increased life span (7). To extend our in vitro results, we tested whether treating mice with resveratrol activates AMPK in the brain. We injected i.p. 2-month-old male mice with either resveratrol (20 mg/kg body weight) or DMSO (vehicle) (n = 3 for each group). Western blot analysis revealed that a single i.p. injection of resveratrol resulted in increased AMPK (2.5-fold) and ACC (2.1-fold) phosphorylation in the brain within 2 h (Fig. 6). Taken together, these results indicate that i.p. administration of resveratrol acutely activates AMPK in the brain and that this activation translates into the phosphorylation and presumed inhibition of ACC, an important downstream target.
Fig. 6.

Resveratrol treatment causes AMPK phosphorylation in the brain. Two-month-old male mice were injected i.p. with resveratrol (20 mg/kg body weight) or DMSO (vehicle) (n = 3 for each treatment). Two hours after treatment, the animals were killed, and brain lysates were prepared. Western analysis with AMPK and ACC phospho-specific antibodies showed increased levels of phosphorylated AMPK (A) and ACC (C) in brains of resveratrol (Resv)-treated animals. Total AMPK and ACC levels are shown as loading controls in B and D. (E) Densitometry was used to quantify the increased level of AMPK and ACC phosphorylation in the brain of resveratrol-treated animals (∗, P < 0.005). AU, arbitrary units.
Discussion
CR results in increased efficiency of energy utilization and promotes longevity and a decreased incidence and/or delay in the onset of chronic diseases, including those affecting the nervous system (29). It has been proposed that AMPK, which senses alterations in cellular energy levels by monitoring of AMP:ATP ratios, may participate in CR responses through its effects on mitochondrial biogenesis, alterations in anabolic and catabolic pathways, and because it is activated in hypothalamic neurons and hepatocytes in response to a dietary restriction (19, 30). Many of these responses are also promoted by resveratrol, thus we investigated whether there was a molecular link among resveratrol, AMPK activation, and neuronal metabolism. Our results showed that resveratrol activates AMPK signaling in Neuro2a cells and primary cultured neurons. Using multiple modalities, including pharmacological activators and inhibitors as well as AMPK mutants, we show that AMPK activation, such as that stimulated by resveratrol, is necessary and sufficient for inhibition of cell proliferation, neurite outgrowth, and mitochondrial biogenesis in Neuro2a cells. Finally, using LKB1- and SIRT1-deficient DRG and cortical neurons, we showed that resveratrol-stimulated AMPK activation requires the upstream AMPK kinase LKB1; however, its activation is independent of SIRT1 within the time frames examined.
Resveratrol Activities in the Nervous System.
CR increases neuronal resistance to oxidative, metabolic, and excitotoxic insults (31–33) and has proved beneficial in animal models of neurodegenerative disease (34, 35). These beneficial effects of CR have stimulated the search for CR mimetics. Studies in yeast have indicated that resveratrol may promote longevity through pathways used by CR, pathways that are dependent on the NAD-dependent protein deacetylase SIRT1/Sir2 (7). In accord, Sir2 overexpression is associated with extended life span in a variety of species, whereas deletion of the Sir2 gene results in decreased longevity (36). Resveratrol was identified in a screen for Sir2 activators as the most potent agonist of Sir2 activity (37), thus it is thought that its effects are mediated by stimulating Sir2 deacetylase activity. However, other studies have concluded that resveratrol is not a direct activator of Sir2 proteins and that the increased longevity mediated by CR is independent of Sir2 (38, 39). Furthermore, inhibition of insulin signaling, a hallmark of CR and longevity, also appears to be promoted by resveratrol in a SIRT1-independent pathway (8). Interestingly, AMPK signaling also antagonizes insulin signaling (40), promotes mitochondrial biogenesis, and is necessary for longevity (41). These observations coupled with our demonstration that resveratrol can activate AMPK and lead to inhibition of ACC suggest that many, if not all, SIRT1-independent CR mimetic actions of resveratrol depend on the AMPK cascade. Indeed, the multiple beneficial effects of resveratrol may be due to its ability to alter the activity of multiple proteins involved in the cellular response to stress (i.e., SIRT1 and AMPK).
Numerous studies have shown that resveratrol can protect against brain damage after ischemia and hypoxia (1, 6). The neuroprotective activity of resveratrol has also been demonstrated in models of mechanical injury, Alzheimer's disease, and Huntington's disease (3–5). These protective actions may occur through hormesis, i.e., agents that produce low levels of stress (and therefore stimulate the organism to initiate a stress response) help the organism adapt to more intense stresses at later times. The stimulation of AMPK by resveratrol would be consistent with this idea because AMPK is activated at times when the energy levels of the cell are low. Indeed, the phenomenon of preconditioning lesions, which consist of a mild ischemic episode that promotes tolerance to subsequent severe ischemic insults, is well known (42). Pomegranate juice, which is rich in resveratrol, also provides neuroprotection against ischemic episodes (25). Both ischemic preconditioning and resveratrol result in AMPK activation in the brain, suggesting that perhaps they work through a common protective mechanism.
AMPK Activation and Neuronal Function.
AMPK function appears to play a crucial role in neuronal energy metabolism on two levels. First, at the organismal level, AMPK activation in hypothalamic neurons plays a central regulatory role in whole-body energy expenditure and peripheral glucose metabolism (43), and studies in AMPKα2 knockout mice revealed that AMPK in sympathetic neurons regulates whole-body insulin sensitivity (44). Moreover, AMPK activation in the rat brain protects neurons during energy deprivation (16), whereas mutation in the AMPK γ subunit in Drosophila leads to progressive neurodegeneration further supporting the role of AMPK in preserving neuronal integrity (45). Second, at the cellular level, AMPK functions as an important energy sensor. The principal regulator of AMPK activation is thought to be an increase in the AMP:ATP ratio. However, other mechanisms, including intracellular calcium mobilization (15, 46, 47), can also activate AMPK. Indeed, resveratrol must activate AMPK through one of these alternative mechanisms because we did not observe any increase in the AMP:ATP ratio in resveratrol-treated Neuro2a cells.
In a study published while this paper was being prepared, the activation of AMPK in liver by resveratrol was demonstrated (48). Here, we show that resveratrol also produces dramatic effects on neuronal function and that these effects are mediated by AMPK activation. Although the exact mechanism of resveratrol-mediated AMPK activation is unclear, it is under the influence of LKB1 and, in CNS neurons, also of CaMKKβ. Whether resveratrol directly activates these kinases or alters other cellular pathways is unknown; however, it is unlikely to involve their rapid deacetylation by SIRT1 because resveratrol activation of AMPK at 2 h occurs normally in neurons lacking SIRT1. Because both SIRT proteins and AMPK are involved in the cellular response to metabolic stress, it will be interesting to determine whether there are additional interactions between these two protein families in neurons and other cells.
Materials and Methods
Mice.
Lkb1-floxed mice were the gift of Ronald A. Depinho (Dana–Farber Cancer Institute, Boston, MA). SIRT1 heterozygous mice were kindly provided by Frederick W. Alt (Harvard University Medical School, Boston, MA).
Cell Culture and Reagents.
Neuro2a cells obtained from American Type Culture Collection (Manassas, VA) were grown in MEM with 1.5 g/liter sodium bicarbonate/0.1 mM nonessential amino acids/0.1 mM sodium pyruvate/2 mM l-glutamine/10% FCS. For differentiation, cells were switched to serum-starvation medium (containing 0.2% FCS). For neurite outgrowth measurements, experiments were done in quadruplicate, and 100 neurites per well were randomly measured with MetaMorph software (Molecular Devices, Sunnyvale, CA). DRG sensory and cortical neurons were established from E13.5 mouse embryos, maintained in neurobasal medium supplemented with B27 (and nerve growth factor for DRG neurons), and infected with lentiviruses as described previously (4, 16). Resveratrol was a gift from Sirtris Pharmaceuticals (Cambridge MA), and AICAR was obtained from Toronto Research (North York, ON, Canada). Splitomycin and CC were purchased from Biomol (Plymouth Meeting, PA) and Calbiochem (San Diego, CA). Sirtinol and nicotinamide were purchased from Sigma (St. Louis, MO).
Plasmids and Viruses.
The dnAMPK and caAMPK plasmids were gifts from Russell Jones (University of Pennsylvania, Philadelphia PA), and pCXN-Cre was a gift from Inder Verma (Salk Institute, San Diego, CA). All constructs were subcloned into the lentiviral shuttle vector FCIV and verified by nucleotide sequence analysis. Viruses were prepared as described previously (4).
Protein and mRNA Analysis.
Immunoprecipitations and Western blot analysis were performed by standard methods with antibodies directed against total AMPK, phosphorylated AMPK, total ACC, or phosphorylated ACC that were obtained from Cell Signaling Technology (Beverley, MA). The phospho-AMPK antibody detects endogenous AMPKα1 and α2 when phosphorylated at Thr172. The LKB1 antibody was purchased from Upstate (Lake Placid, NY). Quantitative RT-PCR analysis was performed by using Sybr-Green methodology on a model 7700 instrument (Applied Biosystems, Foster City, CA) as described previously (4). Primer sequences were those used in previous studies (22).
Measurement of Intracellular ATP and AMP Levels.
Neuro2a cells grown in the presence or absence of 10 μM resveratrol were washed twice in PBS and lysed with 500 μl of 0.2 M HCLO4 and centrifuged at 5,000 × g for 10 min. The supernatant was neutralized with 50 μl of 2 N KOH on ice, centrifuged, and filtered through a 10-kDa followed by a 3-kDa cutoff filter (Millipore, Billerica, MA). The resulting filtrate was diluted 1:1 in 100 mM phosphate buffer (pH 7.0) and analyzed by HPLC (Waters, Milford, MA) with an LC-18T reverse-phase column (Supelco, Bellefonte, PA) at a flow rate of 1 ml/min, and the absorbance at 254 nm was recorded. Each elution peak was compared with AMP and ATP standards (Sigma) to confirm its identity.
Supplementary Material
Acknowledgments
We thank Eugene Johnson, Craig Press, Sanjay Jain, Robert Baloh, and Yo Sasaki for fruitful discussions and reading of the manuscript and Yo Sasaki for ATP and AMP measurements by HPLC. This work was supported by National Institutes of Health (NIH) Neuroscience Blueprint Core Grant NS057105 (to Washington University), the HOPE Center for Neurological Disorders, and NIH Grants AG13730 and NS39358 (to J.M.).
Abbreviations
- ACC
acetyl-CoA carboxylase
- AICAR
5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- AMPK
AMP-activated kinase
- ca
constitutively active
- CaMKKβ
calcium/calmodulin-dependent protein kinase β
- CC
Compound C
- CR
caloric restriction
- dn
dominant-negative
- DRG
dorsal root ganglia
- En
embryonic day n
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator 1α
- Sir
silent information regulator
- Tfam
mitochondrial transcription factor A.
Footnotes
Conflict of interest: J.M. and Washington University have a financial interest in Sirtris Pharmaceuticals. Sirtris Pharmaceuticals did not support this work.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610068104/DC1.
References
- 1.Baur JA, Sinclair DA. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 2.Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Néri C. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 3.Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Curr Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 4.Araki T, Sasaki Y, Milbrandt J. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 5.Han YS, Zheng WH, Bastianetto S, Chabot JG, Quirion R. Br J Pharmacol. 2004;141:997–1005. doi: 10.1038/sj.bjp.0705688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. Brain Res. 2002;958:439–447. doi: 10.1016/s0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- 7.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J. Biochem J. 2006;397:519–527. doi: 10.1042/BJ20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang CZ, Wang Y, Di A, Maqnuson MA, Ye H, Roe MW, Nelson DJ, Bell GI, Philipson LH. Biochem Biophys Res Commun. 2005;330:1073–1079. doi: 10.1016/j.bbrc.2005.03.093. [DOI] [PubMed] [Google Scholar]
- 10.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Am J Physiol. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 11.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 12.Hardie DG, Scott JW, Pan DA, Hudson ER. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Salt IP, Hawley SA, Davies SP. Biochem J. 1999;338:717–722. [PMC free article] [PubMed] [Google Scholar]
- 14.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Proc Natl Acad Sci USA. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. J Biol. 2003;2:28.1–28.16. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenquelli BG, Hardie DG. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Woods A, Dickerson K, Heath R, Hong S, Momcilovic M, Johnstone SR, Carlson M, Carling D. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Culmsee C, Monnig J, Kemp BE, Mattson MP. J Mol Neurosci. 2001;17:45–48. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 19.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Noriyuki O, Walsh K. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minokoshi Y, Alquier T, Furukawa N, Kim Y, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, et al. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 22.Motoshima H, Goldstein BJ, Igata M, Araki E. J Physiol (London) 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, et al. Proc Natl Acad Sci USA. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, et al. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 25.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. Proc Natl Acad Sci USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly DP, Scarpulla RC. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 27.Hartman RE, Shah A, Fagan AM, Schwetye KE, Parsadanian M, Schulman RN, Finn MB, Holtzman DM. Neurobiol Dis. 2006;24:506–515. doi: 10.1016/j.nbd.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Virgili M, Contestabile A. Neurosci Lett. 2000;281:123–126. doi: 10.1016/s0304-3940(00)00820-x. [DOI] [PubMed] [Google Scholar]
- 29.Prolla TA, Mattson MP. Trends Neurosci. 2001;24(Suppl 11):S21–S31. doi: 10.1016/s0166-2236(00)01957-3. [DOI] [PubMed] [Google Scholar]
- 30.Pallottini V, Montanari L, Cavallini G, Bergamini E, Gori Z, Trentalance A. Mech Age Dev. 2004;125:633–639. doi: 10.1016/j.mad.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, et al. Proc Natl Acad Sci. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, Hagepanos A, Ingram DK, Lane MA, Mattson MP. Proc Natl Acad Sci. 2003;100:6216–6220. doi: 10.1073/pnas.1035720100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Proc Natl Acad Sci. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartke A. Endocrinology. 2005;146:3718–3723. doi: 10.1210/en.2005-0411. [DOI] [PubMed] [Google Scholar]
- 35.Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE. Neurobiol Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Kaeberlein M, McVey M, Guarente L. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 38.Kaeberlein M, Steffen KK, Hu D, Dang N, Kerr EO, Tsuchiya M, Fields S, Kennedy BK. Science. 2006;312:1312. doi: 10.1126/science.1124608. [DOI] [PubMed] [Google Scholar]
- 39.Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. PLoS Biol. 2004;2:1381–1387. doi: 10.1371/journal.pbio.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Yoon M, Choi S, Kang I, Kim S, Kim Y, Choi Y, Ha J. J Biol Chem. 2001;276:19102–19110. doi: 10.1074/jbc.M011579200. [DOI] [PubMed] [Google Scholar]
- 41.Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gidday JM. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 43.Long Y, Zierath JR. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viollet B, Andrelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Mennoun M, et al. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tschäpe JA, Hammerschmied C, Mühlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. EMBO J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahmann N, Woods A, Carling D, Heller R. Mol Cell Biol. 2006;26:5933–5945. doi: 10.1128/MCB.00383-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}