

Abstract
Thirteen spontaneous multiple-antibiotic-resistant (Mar) mutants of Escherichia coli AG100 were isolated on Luria-Bertani (LB) agar in the presence of tetracycline (4 μg/ml). The phenotype was linked to insertion sequence (IS) insertions in marR or acrR or unstable large tandem genomic amplifications which included acrAB and which were bordered by IS3 or IS5 sequences. Five different lon mutations, not related to the Mar phenotype, were also found in 12 of the 13 mutants. Under specific selective conditions, most drug-resistant mutants appearing late on the selective plates evolved from a subpopulation of AG100 with lon mutations. That the lon locus was involved in the evolution to low levels of multidrug resistance was supported by the following findings: (i) AG100 grown in LB broth had an important spontaneous subpopulation (about 3.7 × 10−4) of lon::IS186 mutants, (ii) new lon mutants appeared during the selection on antibiotic-containing agar plates, (iii) lon mutants could slowly grow in the presence of low amounts (about 2× MIC of the wild type) of chloramphenicol or tetracycline, and (iv) a lon mutation conferred a mutator phenotype which increased IS transposition and genome rearrangements. The association between lon mutations and mutations causing the Mar phenotype was dependent on the medium (LB versus MacConkey medium) and the antibiotic used for the selection. A previously reported unstable amplifiable high-level resistance observed after the prolonged growth of Mar mutants in a low concentration of tetracycline or chloramphenicol can be explained by genomic amplification.
In natural environments, bacteria face low concentrations of antimicrobial drugs to which they may respond with various mechanisms of resistance. High levels of resistance mediated by spontaneous chromosomal mutations, plasmids, or transposons may be acquired; but low levels of resistance (also described as decreased drug susceptibilities) conferred by spontaneous chromosomal mutations are also selected. Although the latter resistance phenotypes may be too low to represent a clinical threat, they can serve as intermediate steps for the development of increased clinically relevant resistances (14). Among the chromosomally mediated mechanisms causing low levels of multidrug resistance, that dependent on MarA, which was initially discovered in Escherichia coli (14), is one of the best characterized (2).
The general transcriptional regulator MarA directly or indirectly regulates a set of 60 to 80 genes known as the mar regulon (6, 36). The MarA-dependent activation of transcription of acrAB, which codes for a multidrug efflux system, and the indirect inhibition of the expression of the OmpF porin lead to low-level resistance to multiple antibiotics and other antimicrobial agents (2, 29, 34, 45), defined as the multiple-antibiotic-resistance (Mar) phenotype. Transcription of the marA gene from the marRAB operon is activated by MarA (9, 27) and its two homologs, SoxS and Rob (5, 7, 17, 28), and is repressed by the transcriptional repressor MarR (4). In the presence of inducers such as sodium salicylate or 2,4-dinitrophenol (DNP), which inactivate MarR, transcription of marRAB is induced (1, 40). Once induction of marRAB transcription stops, active degradation of MarA by the Lon protease and another still unknown ATP-independent protease allows the reversion of the MarA overproduction and the associated multidrug resistance (16).
Mutations conferring a spontaneous low-level multidrug resistance phenotype have previously been found in the genes marR, soxR (which codes for the transcriptional activator of soxS), and acrR (which codes for the transcriptional repressor of acrAB) among laboratory (9, 18, 28) and clinical (25, 33, 43, 44) isolates of E. coli. We recently described an additional genetic mechanism of low-level multiple-antibiotic resistance in a spontaneous mutant of E. coli: increased amounts of AcrAB resulting from the amplification of the acrAB genes present on a large tandemly amplified unit of 149 kb bordered by IS186 elements (31).
In this paper, we describe a role for lon mutations in the selection of Mar mutants. We also further investigate the importance of tandem genetic amplifications that include acrAB as a mechanism for selection of high-level unstable multidrug resistance in E. coli.
MATERIALS AND METHODS
Media.
E. coli strains were grown on Luria-Bertani (LB) broth or agar media or on MacConkey agar medium at 37°C or 33°C, as specified. Ampicillin, chloramphenicol, nalidixic acid, tetracycline, and DNP were purchased from Sigma-Aldrich Co. (St. Louis, MO) and added to the media at concentrations dependent on the experiment.
Strains and isolation of spontaneous AG100 mutants.
The strains used during this work are presented in Table 1. The AG100 spontaneous mutants (mutant M013 and other mutants in Table 1) were isolated under conditions similar to those used for M113 (31). Specifically, a single colony of E. coli AG100 was grown at 37°C under vigorous shaking in LB broth up to a concentration of 108 cells/ml. This culture was diluted into fresh LB broth to 104 cells/ml, which were then distributed into 13 tubes which were grown until late logarithmic phase (A600, 1.0). One hundred microliters of each culture (representing about 108 bacteria) was spread onto LB agar plates containing 4 μg/ml of tetracycline. The first tetracycline-resistant mutants appeared after 2 to 3 days of incubation at 33°C. From each plate, one mutant that appeared after 3 days of incubation and one mutant that appeared after 4 days of incubation were picked. From those 26 mutants, 13 (representing 3 days or 4 days of emergence) were randomly chosen for this study. Each mutant was purified twice on LB agar plates supplemented with tetracycline (4 μg/ml), from which one colony was selected and grown in LB broth and stored at −80°C in 20% glycerol.
TABLE 1.
Strains used in the study
| Name | Genotypea | MarAb | Remark or construction | Reference or source |
|---|---|---|---|---|
| AG100 | argE3 thi-3 rpsL xyl mtl supE44 | − | Wild-type strain | 14 |
| AG100HN17 | AG100 lon-3::IS186 ppiD::Tn10 | + | Constructed by P1 transduction | 31 |
| AG100HN19 | AG100 ppiD::Tn10 | + | Constructed by P1 transduction | 31 |
| MG1655 | ilvG-rfb-50 rph-1 | − | Wild-type strain | 12 |
| AB1157 | thr-1 araC14 leuB6(Am)Δ (gpt-proA)62 lacY1 tsx-33 supE44(AS) galK2(Oc) hisG4(Oc) rfbD1 mgl-51 rpoS396(Am) rpsL31(StrR) kdgK51 xylA5 mtl-1 argE3(Oc) thi-1 | − | Wild-type strain | 12 |
| AG112 | AG100 marR (5-bp deletion near 5′ end) | ++ | Spontaneous AG100 mutant | 32 |
| AG112HN2-74 | AG100 marR dupIS186 | Not tested | Constructed by P1 transduction | 31 |
| M113 | AG100 lon3::IS186 dupIS186 dupIS3 | + | Spontaneous AG100 mutant | 31; this work |
| M113R | AG100 lon3::IS186 | + | Partial revertant of M113 | 31 |
| CAG12017 | MG1655 ppiD::Tn10 | Not tested | 41 | |
| CAG12154 | MG1655 gsk::Tn10 | Not tested | 41 | |
| M013 | AG100 Δ(lon-hupB-ppiD) dupIS5 | + | Plate 1, day 3f; 2,688-bp deletione | This work |
| M043 | AG100 lon4::IS186 marR::IS2 | +++ | Plate 4, day 3f | This work |
| M044 | AG100 lon4::IS186 marR::IS | +++ | Plate 4, day 4f | This work |
| M064 | AG100 lon1::IS186 acrR::IS | + | Plate 6, day 4f | This work |
| M073 | AG100 lon2::IS186 acrR::IS5 | + | Plate 7, day 3f | This work |
| M074 | AG100 lon2::IS186 acrR::IS | + | Plate 7, day 4f | This work |
| M083 | AG100 lon4::IS186 acrR::IS1 | + | Plate 8, day 3f | This work |
| M084 | AG100 lon4::IS186 unknownc | + | Plate 8, day 4f | This work |
| M093 | AG100 lon3::IS186 acrR::IS1 | + | Plate 9, day 3f | This work |
| M103 | AG100 lon3::IS186 acrR-acrABd | + | Plate 10, day 3f | This work |
| M104 | AG100 dupIS3 | − | Plate 10, day 4f | This work |
| M114 | AG100 lon4::IS186 marR::IS2 | +++ | Plate 11, day 4f | This work |
| M124 | AG100 lon4::IS186 acrR::IS | + | Plate 12, day 4f | This work |
marOR and acrR loci were amplified by PCR from all 13 mutants (mutants M013 to M124). The loci that did not present a large insertion were sequenced to look for other types of mutations. The nature of the inserted IS found was generally determined by sequencing or remains unknown (noted “IS”). The lon genes from all mutants were sequenced. For details about the lon::IS186 mutations found, see Table 3. unknown, the mutation that resulted in the growth of the mutant on the selective plate was not found.
No MarA overproduction (−) or increased MarA overproduction (+ < ++ < +++), as observed by Western blot hybridization with anti-MarA antibodies.
The mutation was unstable.
The undefined mutation mapped to the acrRAB region.
Deletion starting at nucleotide 459285, according to the E. coli K-12 sequenced genome with GenBank accession number U00096.
Selective LB agar plate and day on which the mutant was isolated.
The general protocol for the isolation of other drug-resistant mutants was as follows: colonies isolated on LB agar plates were grown at 37°C in LB broth up to late logarithmic phase (A600, 1.0) unless specified otherwise. The cultures were then diluted 10−1 to 10−6 and 100 μl (LB medium) or 200 μl (MacConkey medium) was plated on LB or MacConkey agar with or without drugs and incubated for several days at 37°C. After each day of incubation, the new mutants that appeared on the selective plates were marked and their numbers were determined. Numeration of the bacteria initially plated was done by counting the CFU from 100 μl of the 10−6 dilution plated on LB or MacConkey agar.
Amplification of the resistance phenotype of AG112.
Fifteen microliters of an overnight culture of strain AG112 (mutant marR) in LB broth (growth 0) was added to 5 ml of LB broth supplemented with tetracycline (5 μg/ml). After an overnight growth at 37°C, 15 μl of the culture was used to start a new growth under similar conditions. A total of 30 such sequential growths were performed in the presence of tetracycline (5 μg/ml) (representing about 250 growth generations), followed by 15 growths in the absence of tetracycline (representing about 125 generations).
Fitness of wild-type and lon mutant bacteria in LB broth.
Colonies of strains AG100HN17 (lon3::IS186 ppiD::Tn10) and its wild-type equivalent, strain AG100HN19 (ppiD::Tn10), isolated on LB plates were grown in LB broth at 37°C up to mid-logarithmic phase (optical density at 600 nm, 0.4). The cultures were then mixed together, and 10 μl of this mix was used to inoculate two tubes containing 5 ml of LB broth prewarmed at 37°C. The cultures were then incubated at 37°C for 2 days (cultures 1), diluted 1:500, and incubated for 2 more days (allowing nine growth generations to occur; cultures 2). After each culture, the bacteria were spread on LB plates, and isolated colonies were tested for their resistance (strain AG100HN19) or sensitivity (strain AG100HN17) to 0.6 mM DNP (the latter phenotype is associated with a lon mutation in E. coli AG100) (33). After the first cultures, 71% and 73% of DNPr bacteria were present. This proportion increased to 86% and 90% after the second cultures.
Estimation of spontaneous lon::IS186 mutation rate.
The general formula for the calculation of the mutation rate (μ) used is:
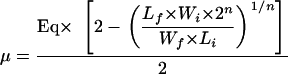 |
where Eq is the proportion of lon::IS186 mutants in a population at equilibrium; Wi and Wf are the proportions of wild-type bacteria at the beginning (Wi) and after n generations (Wf) in LB broth (experiment A; see below); and Li and Lf are the proportions of lon::IS186 at the beginning (Li) and after growth for n generations (Lf) in LB broth (experiment A; see below). The definitions of the other terms used to find the equation are as follows: n is the number of generations of the wild-type bacteria; NiW is the initial population of wild-type bacteria; NiL is the initial population of lon::IS186 mutants; NfW is the final population of wild-type bacteria and is ≈NiW × 2n; NgL is the final population of lon::IS186 mutants growing from an initial population of mutants and is equal to NiL × Yn, where Y is the factor of increase of the lon::IS186 population during one doubling of the wild type population; m is the number of new spontaneous lon::IS186 mutants; and NfL is the total final population of lon::IS186 mutants and is equal to NgL + m.
The rate of spontaneous lon::IS186 mutation (μ) is m/NfW. With NfL = NgL + m, we obtain μ = (NfL − NgL)/NFW (equation 1). We found that a culture of AG100 (wild type) grown in LB broth maintains an equilibrium proportion of lon::IS186 mutants (Eq) of 3.7 × 10−4. If we consider a population at equilibrium and growing for one doubling of the wild-type population, NfW = NiW × 21 and, to keep the subpopulation of lon::IS186 constant, NfL = NiL × 21. However, the lon::IS186 population grows at its own rate, Y, and NgL = NiL × Y1. By replacing NfL, NgL, and NfW in equation 1, we find μ = [(NiL × 2) − (NiL × Y)]/(NiW × 2) (equation 2). We can simplify NiL ≈ NiW × Eq in equation 2 to obtain equation 3: μ = 1/2 × Eq × (2 − Y). To find Y, we observed that, when AG100HN17 cells were mixed with AG100HN19 cells (wild type), the proportion of AG100HN17 cells (lon::IS186) decreased from 28% to 12% of the total population in n = 9 wild-type generations (experiment A). Therefore, NiW/NiL = Wi/Li = 0.72/0.28 (equation 4), and NfL/NfW = Lf/Wf = 0.12/0.88 (equation 5) after n = 9 generations. From equation 5, we obtain NfL = (Lf × NfW)/Wf and, finally, NfL = (Lf × NiW × 2n)/Wf (equation 6). By using equation 6 in equation 4, we find NfL = (Lf × Wi × NiL × 2n)/(Wf × Li) (equation 7), with NfL = m + NgL. However, because of the important proportion of lon::IS186 mutants present in the culture of the experiment A (12 to 28% of the total population) and the duration of the growth, the amount of new spontaneous lon::IS186 mutants (m) appearing during the growth is negligible and NfL ≈ NgL = NiL × Yn (equation 8). By using equation 8 in equation 7, we can solve for Y and find Y = [(Lf × Wi × 2n)/(Wf × Li)]1/n (equation 9). By using equation 9 in equation 3, we obtain the general formula for the calculation of the mutation rate (see above).
Drug susceptibility.
The drug susceptibility and the DNP sensitivity phenotypes were determined on LB agar plates by Etests (AB BIODISK, Solna, Sweden) or by the serial plate technique, as described previously (31).
Molecular biology techniques.
Western blot hybridizations with polyclonal antibodies to MarA or to AcrA (received from H. Zgurskaya) and P1 transductions were performed as described previously (31). The primers used for PCR amplifications (Table 2) were synthesized by the Tufts University Core Facility. DNAs extracted with the DNeasy tissue kit (QIAGEN, Inc.) were used for comparative PCR amplifications. For noncomparative PCRs, 100 μl of the bacterial cultures was pelleted and resuspended in 50 μl of water, heated at 98°C for 10 min, and centrifuged at 9,000 × g for 5 min. Three microliters of supernatant was routinely used per PCR mixture. PCR-amplified bands were quantified with a Gel doc 1000 camera system and Molecular Analyst software (Bio-Rad). The DNA fragments produced by PCR amplification were purified with a QIAquick PCR purification kit (QIAGEN), quantified by measurement of their A260, and sequenced at the Tufts University Core Facility.
TABLE 2.
Primers and PCR conditions
| Locus or mutation | Use | Temp (°C)a | Primer name | Sequence (5′→3′) | Coordinate and orientationb |
|---|---|---|---|---|---|
| acrR | PCR and sequencing | 55 | acrFR1 | CTAACGCCTGTAAATTCACG | 484960> |
| acrRV1 | CCAGGAAAAATCCTGGAGTCAG | 485662< | |||
| dupIS186 | Detection by PCR and sequencing | 60 | hokE1 | ACATGCTGACGAAATATGCCC | 607057> |
| lonR1 | ATGCGTTCAGAACGCTCAGG | 458137< | |||
| dupIS3 | Detection by PCR and sequencing | 61 | IS3-2 | TCCTGTTATGGGCGGTAGAC | 565814> |
| IS3-1 | TGGTACGGTATGTGAATATGC | 315767< | |||
| dupIS3-2 | Detection by PCR | 61 | IS3-4 | ATTCAGTGCGCTACGAGTAAC | 1092911> |
| IS3-3 | CTGTTGACGTCGTTTACGTAG | 392639< | |||
| dupIS5 | Detection by PCR and sequencing | 60 | IS5-8 | AGGTAAGCATTGCTGCTCTG | 573433> |
| IS5-7 | ATGCAAAGTCAACCCTGCAC | 274480< | |||
| Inversion between IS3 | Detection by PCR | 60 | IS3-1 | See above | 315767< |
| IS3-3 | See above | 392639< | |||
| lon promoter | PCR and sequencing | 60 | clpX1 | GGTTATGTGGGTGAAGACGT | 457103> |
| lonR2 | TTTTGACCTTGCTACGCGC | 459042< | |||
| Additional sequencing | clpX3 | TTTATGGCAAGCCGGAAGCG | 457884> | ||
| lonR1 | See above | 458137< | |||
| lon (end of the gene) | PCR and sequencing | 60 | lon1 | ACGTACATGTTAATAGATGGCG | 458033> |
| lonR2 | See above | 459042< | |||
| PCR and sequencing | 60 | lon2 | AAAATGATGTCTCCGATGTCG | 458946> | |
| lonR3 | CGCACGAACCACCGTTAA | 460052< | |||
| PCR and sequencing | 60 | lon3 | GACCATTGAAACCGCATGTG | 459944> | |
| lonR4 | GATCAATTGAGATTTATTCACTC | 460695< | |||
| Δlon-hup-ppiD | Detection by PCR and sequencing | 58 | lon2 | See above | 458946> |
| ppiDR2 | AGTGGAATCACCTTAACGGC | 463136< | |||
| lon::IS186 | Detection by PCR | 59 | IS186IR | ACCCTTAAGTTAGCGCTTATG | IS186<>c |
| lonR2 | See above | 459042< | |||
| marOR | PCR and sequencing | 54 | orab2 | GCTAGCCTTGCATCGCAT | 1617010> |
| or1R | GAATGGTAATAGCGTCAG | 1617631< | |||
| IS186 | Detection by PCR | 60 | 186-2 | CGGCCCCGGGGGGATGTC | 607437>d |
| 186-R1 | GCCACCTGTAAGCTCCAGATG | 608448<d |
Hybridizing temperature of the PCR.
Coordinate of the 5′ nucleotides are given according to the E. coli K-12 genome with GenBank accession number U00096; >, forward primer; <, reverse primer.
Primer hybridizes in the left and right inverted sequences of IS186, facing outward from IS186 (example of sites in E. coli K-12 genome, positions 607233 < and 608551 >).
Primers hybridize in IS186 and can hybridize with all the IS186 present in E. coli K-12.
Computer simulation.
Computer simulation of the growth over n generations of a bacterial population with a given initial ratio of wild-type to lon mutants was done with Excel software from the Microsoft Office suite. The definitions of the terms used (see also above) are as follows: Eq(n) is the proportion of lon::IS186 mutants in a population grown for n generations; NfW(n) and NfW(n − 1) are the final populations of wild-type bacteria after n and n − 1 growth generations, respectively; NfL(n) and NfL(n − 1) are the final populations of lon::IS186 mutants after n and n − 1 growth generations, respectively; NgL(n) is the growth of the lon::IS186 population during generation n; NgW(n) is the growth of the wild-type population during generation n; and m(n) is the number of new spontaneous lon::IS186 mutants appearing during generation n.
For each growth generation, the Excel software calculated the following values: NgL(n) = NfL(n − 1) × Y; NfW(n) = [NfW(n − 1) × 2] − m(n), which we can simplify as NfW(n) ≈ NfW(n − 1) × 2 [because m(n) ≪ NfW(n − 1)]; m(n) = NfW(n) × μ; NFL(n) = NgL(n) + m(n); and Eq(n) = NfL(n)/[NfL(n) + NfW(n)]. Any arbitrary initial values NfL(0) and NfW(0) (the amounts of lon::IS186 and wild-type bacteria at the beginning of growth, respectively) can be used to calculate Eq(n).
RESULTS
Isolation of spontaneous Mar mutants of E. coli.
Thirteen mutants of E. coli AG100 were isolated on LB plates supplemented with tetracycline (4 μg/ml) after 3 or 4 days of incubation at 33°C (see Materials and Methods). Like mutant M113, which was isolated under similar conditions (31), they all formed visible colonies in 1 day when they were replated on similar medium, implying that the mutational event that allowed the growth probably occurred during incubation on the selective plate. All mutants had a Mar phenotype, with observed decreased susceptibilities to chloramphenicol, ampicillin, nalidixic acid, norfloxacin, and tetracycline (Table 3). Western blot hybridization with anti-MarA antibodies revealed the overproduction of MarA in all mutants except M104 (data not shown). Compared to the control marR mutant AG112, there was stronger overproduction of MarA in mutants M043, M044, and M114 and weaker overproduction in mutants M013, M064, M073, M074, M083, M084, M093, M103, and M124 (Table 1).
TABLE 3.
Antibiotic susceptibilities of spontaneous AG100 mutants selected on LB agar supplemented with tetracycline (4 μg/ml)
| Strain | MIC (μg/ml)a
|
|||||
|---|---|---|---|---|---|---|
| Tet | Nal | Chl | Rif | Nor | Amp | |
| AG100 | 2 | 6 | 4 | 16 | 0.02 | 2 |
| M013 | 6 | ND | 16 | 16 | 0.13 | 3 |
| M043 | 16 | 8 | 64 | 24 | 0.38 | 6 |
| M044 | 4 | 12 | 32 | 24 | 0.25 | 6 |
| M064 | 8 | 12 | 32 | 24 | 0.19 | 6 |
| M073 | 12 | 8 | 32 | 24 | 0.19 | 6 |
| M074 | 4 | 8 | 48 | 24 | 0.19 | 6 |
| M083 | 4 | 8 | 32 | 16 | 0.25 | 4 |
| M084 | 4 | 6 | 16 | 16 | 0.13 | 3 |
| M093 | 4 | 8 | 32 | 24 | 0.25 | 6 |
| M103 | 6 | 6 | 24 | 24 | 0.25 | 4 |
| M104 | 6 | 8 | 32 | 16 | 0.25 | 3 |
| M114 | 12 | 8 | 64 | 24 | 0.38 | 8 |
| M124 | 6 | 8 | 24 | 16 | 0.25 | 3 |
The phenotypes were determined by Etests. Tet, tetracycline; Nal, nalidixic acid; Chl, chloramphenicol; Rif, rifampin; Nor, norfloxacin; Amp, ampicillin; ND, not determined.
Presence of lon mutations in Mar mutants.
Twelve of 13 mutants were DNP sensitive, a phenotype associated with a lon mutation in strain AG100 (31). PCR amplification of the lon promoter of the 13 mutants revealed an additional 1.3-kb-long sequence consisting of four different lon::IS186 mutations in 11 mutants (Table 4). The remaining DNP-sensitive mutant (mutant M013) had a 2.688-kb-long deletion ranging from lon to ppiD and starting at position 459,285, according to the E. coli K-12 genome sequence (GenBank accession number U00096). Only M104 showed wild-type susceptibility to DNP, and sequencing verified the absence of a mutation in lon.
TABLE 4.
lon mutations found in spontaneous AG100 mutants selected in the presence of tetracycline
| Mutant(s) | lon mutation | Orientationa | IS186 insertion site and sequences duplicatedb |
|---|---|---|---|
| M013 | Δ(lon-hupB-ppiD) | ||
| M064 | lon1::IS186 | I | 5′-GGGGGAAACAT-≫IS186≫-AAACATCCCCATATACTG-3′ |
| M073, M074 | lon2::IS186 | II | 5′-GGGGGAAACAT-≪IS186≪-AAACATCCCCATATACTG-3′ |
| M093, M103, M113c | lon3::IS186 | I | 5′-GGGGGAAACAT-≫IS186≫-GAAACATCCCCATATACTG-3′ |
| M043, M044, M083, M084, M114, M124 | lon4::IS186 | II | 5′-GGGGGAAACAT-≪IS186≪-GAAACATCCCCATATACTG-3′ |
The orientation of the inserted IS186 is given as described previously (39).
IS186 insertion sites and duplications of the insertion site (underlined and in boldface) are indicated in the portion of the E. coli K-12 chromosome from nucleotides 458010 to 458032 (GenBank accession number U00096); the orientation of IS186 is indicated with arrowheads (≫ or ≪); the −10 motif of the σ32 lon promoter is double underlined.
As described previously (31).
IS insertions in acrR and marR caused the Mar phenotype in nine mutants.
A lon mutation alone does not confer a Mar phenotype, but a Mar phenotype can appear in lon mutants when a second mutation increases the amount of AcrAB (31). We searched for possible mutations in acrR, marR, and its operator, marO, in the 12 lon mutants and in M104. Six mutants had an insertion sequence (IS) in acrR (mutants M064, M073, M074, M093, M083, and M124; Table 1). Sequencing showed that IS1 and IS5 were involved. P1 transductions with donor strains carrying a wild-type acrR gene (strains CAG12017 and CAG12154 with a ppiD::Tn10 or gsk::Tn10 marker 18 and 20 kb from acrAB, respectively) were performed in recipient strain M083 (lon4::IS186 acrR::IS1) and confirmed that the acrR::IS1 mutation caused the Mar phenotype. Western blot analysis with anti-AcrA antibodies showed that AcrA overproduction in lon3::IS186 acrR::IS1 mutant M093 was four times higher than the level of production in AG100 and similar to the amount found in double mutant lon3::IS186 marR (mutant AG112HN48) (31), where it caused the Mar phenotype.
Three mutants (mutants M043, M044, and M114; Table 1) had an IS insertion in marR. Insertion of IS2 sequences in marR (confirmed in M043 and M114) in orientation II relative to the orientation of the marRAB promoter both inactivates marR and allows the transcription from the original promoter to proceed through the IS2 element and into the downstream genes, marA and marB (9), causing overexpression of MarA. As observed previously with the constructed mutant AG112HN48 (31), the additional marR mutation found in lon mutants M043, M044, and M114 led to the overproduction of larger amounts of MarA (Table 1), which further increased the amount of AcrA and caused the Mar phenotype. After sequencing of the mutants, no mutations in acrR or marOR were found in the four remaining mutants (mutants M013, M084, M103, and M104). However, the uncharacterized mutation in M103 was mapped by P1 transduction to the acrAB region by using the donor strain CAG12017 (ppiD::Tn10) (data not shown).
Two large unstable tandem amplifications including acrAB caused the Mar phenotype in two mutants.
We found that M013, M084, and M104, in which no mutations in acrR, marO, or marR were found, had an unstable Mar phenotype (data not shown). In fact, the high degree of instability of the Mar phenotype of M084 did not allow us to isolate on LB plates colonies of M084 harboring the original Mar phenotype of the mutant.
The instability of the Mar phenotype in these three mutants was reminiscent of that of M113 (31). However, no unstable large tandem duplications similar to the dupIS186 found in M113 (31) were detected by PCR in any of the 13 mutants by use of primers lonR1 and hokE1 (Table 2; Fig. 1A and B). To look for different tandem amplifications that include acrAB, two IS5 sequences found in the same orientation and on each side of acrAB (IS5 sequences starting at positions 273,179 and 573,814 on the E. coli K-12 sequence with GenBank accession number U00096) were tested as possible ends of 300-kb-long amplified units carrying acrAB. PCR primers IS5-7 and IS5-8 were designed to detect the tandem amplifications that we named dupIS5 (Table 2; Fig. 1A). Of the 13 mutants and mutant M113 tested by PCR, only mutant M013 had a dupIS5 (Fig. 1B). Sequencing of the amplified PCR fragment confirmed the ybcQ-IS5-mmuP junction between two amplified units. Two pairs of IS3 sequences present in the same orientation and positioned on each side of acrAB (positions 314,453 and 566,000 on the E. coli chromosome) were also tested. PCR primers IS3-1 and IS3-2 were designed to detect the dupIS3 tandem amplification consisting of 252-kb-long units bordered by the two IS3 sequences (Table 2; Fig. 2A). Both M104 and previously described mutant M113 (31) were found to have dupIS3 when they were tested by PCR (Fig. 2B). Sequencing of the amplified PCR fragment confirmed the presence of the intD-IS3-ykgA junction between two amplified units. Similarly, primers IS3-3 and IS3-4 were designed to detect by PCR a 703-kb-long amplification bordered by the IS3 sequences located at positions 390,933 and 1,093,468 on the E. coli K-12 chromosome (Table 2; Fig. 2A). No such tandem amplification could be detected by PCR in any of the 13 mutants or mutant M113 (data not shown).
FIG. 1.

Detection of large tandem duplications. (A) Representation of the tandem genetic amplifications detected. The acrAB locus is shown as a black dot on the chromosome of E. coli. ISs are shown as black (IS3), dark gray (IS5), and white (IS186) arrows. The PCR primers used to detect the dupIS3, dupIS5, and dupIS186 events are shown at their hybridization sites along the chromosome. (B) PCR detection of the dupIS3, dupIS5, and dupIS186 mutations in AG100, M113, and the 13 mutants studied. Template DNA corresponded to suspensions of strains cultivated in LB broth, as described in Materials and Methods. For detection of dupIS3, the template DNA was diluted 1:5 to compensate for the high background of spontaneous dupIS3. (C) Detection of dupIS5 mutations in AG100 and MG1655. DNA from three independent growths of AG100 or MG1655 in LB medium with or without nalidixic acid (5 μg/ml) was extracted and used in comparative PCR of 35 cycles. A total of 150 ng (dilution 1), 30 ng (dilution 2), 0.24 ng (dilution 5), or 0.048 ng (dilution 6) of template DNA was used. Amplified bands were quantified under UV light (see Materials and Methods).
FIG. 2.

Isolation of drug-resistant mutants from wild-type AG100 compared with that from its lon mutant on two different media. Drug-resistant mutants of AG100 (wild-type) or M113R (lon3::IS186) were selected at 37°C on LB (A) or MacConkey (B) agar plates containing antibiotics for 6 days (see Materials and Methods). After each day of incubation, the new colonies that appeared were counted. Experiments were done in duplicate, except for the selection on LB medium with tetracycline at 4 μg/ml (four experiments) and on MacConkey medium with chloramphenicol at 10 μg/ml (single experiment). The curves (dots and thick line for AG100, rectangles and thin line for M113R) represent the number of drug-resistant mutants which appeared per bacterium initially plated on the selective medium since day 1 of the selection. The dotted line (triangles) represents the number of colonies with a lon::IS186 insertion isolated per AG100 bacterium initially plated since the beginning of the experiment. a, percentage of the new colonies of AG100 appearing on the specific day which carried a lon::IS186 insertion; b, percentage of AG100 colonies with a lon::IS186 insertion among all the colonies which appeared since the beginning of the selection; c, number of colonies isolated from AG100 after each day of incubation that were chosen randomly and analyzed by PCR for the presence of a lon::IS186 mutation (see Materials and Methods).
Occurrence of spontaneous dupIS3 and dupIS5 genetic amplifications.
A comparative PCR amplification of marA and ybaO, present outside and inside the amplified units, respectively, was carried out with template DNA of M104 (carrying dupIS3) and M013 (carrying dupIS5) grown in LB medium supplemented with 5 μg/ml of nalidixic acid (a condition that we found could maintain dupIS186 amplification [31]). An average of 2.9 and 2.5 copies of the amplified unit were present in mutants M104 and M013, respectively. Using the same template DNAs and DNA extracted from three independent growths of wild-type strain AG100 in LB broth, we performed comparative PCR amplifications with primers IS3-1 and IS3-2 and primers IS5-7 and IS5-8 (for the detection of dupIS3 and dupIS5, respectively). We hypothesized that in the AG100 cultures, most bacteria which carried a spontaneous duplication had only two copies of the amplified unit because the growth was performed without positive selection for the duplications. With this hypothesis, the comparative PCR showed that 3.4% ± 1.2% and 0.21% ± 0.06% of AG100 bacteria had a spontaneous dupIS3 or dupIS5 (with two copies of the amplified units), respectively, after overnight growth in LB broth (data not shown). Comparative PCR amplifications were done to detect whether dupIS5 was present in AG100 and MG1655 grown overnight in LB supplemented or not supplemented with nalidixic acid at 5 μg/ml (Fig. 1C). We found that similar amounts of template DNA had 147 ± 46 (AG100) and 195 ± 81 (MG1655) times more dupIS5 after growth in nalidixic acid (5 μg/ml) than after growth without nalidixic acid. Using the findings that in the absence of nalidixic acid, 0.21% of AG100 had a dupIS5 with two copies of the amplified unit (see above) and that an average of 2.6 copies of the amplified unit were present after growth in 5 μg/ml of nalidixic acid (see above and reference 31), we could estimate that 24% of the AG100 isolates had an unstable dupIS5 (with 2.6 copies of the amplified unit) after overnight growth in LB supplemented with nalidixic acid (5 μg/ml).
This revealed that spontaneous large tandem duplications in the region of acrAB and including acrAB were (i) detected in different E. coli strains, (ii) frequent in the absence of selective pressure, and (iii) selected and present in an important proportion of the bacteria after overnight growth in a selective medium.
Large tandem duplications involved in stepwise amplification of multiple-antibiotic resistance.
The resistance of AG112 (mutant marR) was amplified in a similar way, as described by George and Levy (14) (see Materials and Methods; Table 5). After 250 generations, AG112 grown in the presence of tetracycline (always kept at 5 μg/ml), acquired a very high level of resistance to multiple antibiotics (e.g., 125 μg/ml for tetracycline; Table 5). This amplified high-level multidrug resistance was mostly lost after growth for 125 additional generations in the absence of tetracycline (Table 5). Such instability had been observed previously (14), without elucidation of the mechanism.
TABLE 5.
Amplification of drug resistance of AG112
| Continuous growtha | MIC (μg/ml)b
|
dupIS3 detectedc | ||||
|---|---|---|---|---|---|---|
| Tet | Chl | Nal | Nor | Amp | ||
| None | 8 | 24 | 12 | 0.38 | 6 | 1 |
| 250 generations in LB + tetracycline | 128 | 256 | 192 | 2 | 32 | 1,590 |
| 125 additional generations in LB | 24 | 24 | 24 | 0.75 | 12 | 1.5 |
AG112 (marR) was sequentially grown in LB broth with tetracycline (5 μg/ml) for about 250 generations and then sequentially grown in the absence of tetracycline for 125 generations (see Materials and Methods).
The susceptibilities to the antibiotics (MICs) were determined by Etests. Tet, tetracycline; Chl, chloramphenicol; Nal, nalidixic acid; Nor, norfloxacin; Amp, ampicillin.
The amplification of acrAB in the whole population was assessed by comparative PCR with primers IS3-1 and IS3-2, which detect the dupIS3 events. The amplification obtained with the template DNA from AG112 before sequential growth in the presence of drug was arbitrarily given the value “1”, and the results after 250 and 125 generations are the number of dupIS3 amplifications detected in similar amounts of template DNA compared to the amounts found in AG112 before the sequential growths.
Using a comparative PCR approach, we quantified the dupIS3 amplifications and found that similar amounts of template DNA of AG112 had 1,575 ± 20 times more dupIS3 after the 250 generations in tetracycline than before growth of the bacteria in the presence of the drug (data not shown). After the additional 125 generations in the absence of antibiotic, the number of dupIS3 amplifications detected dropped and was almost identical to that found initially (Table 5). So, amplification of the antibiotic resistance correlated with genetic amplifications containing acrAB, and loss of the amplified resistance related to loss of the acrAB amplifications.
Occurrence of lon mutations among drug-resistant mutants selected on LB or MacConkey agar in the presence of different selective drugs.
To further investigate the link between lon mutations and selection of drug-resistant mutants, spontaneous mutants of AG100 were selected on LB or MacConkey agar in the presence of tetracycline, chloramphenicol, or nalidixic acid (Fig. 2). The concentrations of antibiotics used represented about 2× MIC (tetracycline), 2 to 2.5× MIC (chloramphenicol), and 1.2× MIC (nalidixic acid) for AG100 (MICs on LB medium). At any given antibiotic concentration tested, we found that fewer spontaneous mutants were selected on MacConkey agar than on LB agar plates. Therefore, in order to get enough spontaneous mutants for analysis, selections at lower concentrations of antibiotics were also performed on MacConkey agar plates. Selection of mutants with M113R (lon3::IS186) was done in parallel to determine the frequency of isolation of drug-resistant mutants by a lon::IS186 strain under similar selective conditions (Fig. 2). Because of the high frequency of lon::IS186 mutations among the 13 mutants isolated on LB agar in the presence of tetracycline (see above), we determined by PCR amplification and for each selective condition the proportion of drug-resistant AG100 mutants which carried a lon::IS186 mutation (Fig. 2).
When M113R was plated on LB and MacConkey agars, it produced 10 to >100 times more tetracycline- or chloramphenicol-resistant mutants than AG100 did (Fig. 2; compare the results for days 1 and 2 of the selection). Even when selection was done in the presence of nalidixic acid (7 μg/ml), M113R still gave more drug-resistant mutants than AG100 did (although <10 times more), despite the increased sensitivity of lon mutants to this drug (31). When selections with similar amounts of drugs were compared, selection on MacConkey medium affected the lon mutant more than it affected the wild-type strain and reduced the proportion of drug-resistant mutants carrying an additional lon mutation (Fig. 2). For example, selection of drug-resistant mutants on MacConkey agar supplemented with 4 μg/ml of tetracycline was ∼2.5 log less efficient than selection on LB agar for AG100 but ∼4.5 log less efficient for M113R (lon-3::IS186) (Fig. 3). When selection was on MacConkey agar supplemented with chloramphenicol (8 μg/ml), the frequency of appearance of colonies was reduced by a factor of ∼0.5 log for M113R and was augmented by a factor of ∼1 log for AG100 (compared to the frequency of appearance of colonies by selection on LB with chloramphenicol 10 μg/ml). When M113R and AG100 were plated on LB medium supplemented with 8 μg/ml of chloramphenicol, M113R, but not AG100, grew as microcolonies visible after several days of incubation (growths were performed for a maximum of 6 days; data not shown). This finding probably reflects the previously observed small decreased susceptibilities of lon mutants to chloramphenicol (31). A similar observation was made on LB medium supplemented with tetracycline (4 μg/ml), revealing a small decreased susceptibility of the lon mutants to tetracycline previously undetected when the MIC was tested by Etests and the results were read after 24 h of growth (31). In Fig. 2, the M113R drug-resistant mutants selected after 5 and 6 days corresponded to the large colonies appearing on these days rather than the numerous small colonies present. The subpopulation of lon::IS186 mutants originally plated on the selective plates was ∼3.7 × 10−4 (see below). However, after 3 days of incubation on LB medium with tetracycline (4 μg/ml) or chloramphenicol (10 μg/ml) and after 5 days on MacConkey agar with chloramphenicol (8 μg/ml), the number of drug-resistant mutants carrying an additional lon::IS186 mutation selected from AG100 was equivalent to the number of mutants expected if subpopulations of ∼3 × 10−3 (selections on LB medium) and ∼1 × 10−2 (selection on MacConkey agar) lon::IS186 mutants had initially been present on the plates (Fig. 2). This finding suggests that new spontaneous lon mutants appeared during incubation on the selective media.
FIG. 3.

Genome instability of wild type versus that of lon mutant. (A) Schematic representation of the genomic inversion and tandem amplification detected. ISs are presented as black (IS3) or dark gray (IS5) arrows along the E. coli chromosome. The PCR primers used to detect the inversion and the duplication are shown on their hybridization sites along the chromosome. (B) Detection of genetic inversions and duplications in a wild-type strain and a lon mutant. DNA from three independent growths of AG100 (wild type) or M113R (lon3::IS186) in LB broth was extracted and quantified by determination of the A260. Comparative PCR of 35 cycles was done with 150 ng (dilution 1), 50 ng (dilution 2), 5.5 ng (dilution 4), or 1.9 ng (dilution 5) of template DNA. The amplified bands were quantified (see Materials and Methods).
From six spontaneous AG100 Mar mutants previously selected in our laboratory on LB or nutrient agar (NP3.5GP) in the presence of pine oil (29; M. C. Moken and L. M. McMurry, unpublished data), one mutant selected on nutrient agar was DNP sensitive and had the additional 1.3-kb-long sequence in the lon promoter, characteristic of an IS186 insertion.
lon mutations selected among other E. coli strains.
To determine if the observations made above were strain specific, two different E. coli strains (strains MG1655 and AB1157) and two different stocks of strain AG100 (the AG100 strain used for this work and an older culture of AG100 stored at −80°C since ∼1987) were used to select drug-resistant mutants on LB medium with tetracycline (3 or 4 μg/ml). The two frozen stocks of AG100 both allowed the selection of similar proportions of DNP-sensitive tetracycline-resistant mutants. As observed with AG100, most of the tetracycline-resistant mutants isolated with AB1157 were also DNP sensitive and thus probably had an additional lon mutation (data not shown). The DNP sensitivity associated with the lon mutation in AG100 and AB1157 was not seen with a MG1655 lon mutant (31). Therefore, PCR amplifications were used to detect the presence of an IS inserted in the promoter of lon in MG1655 tetracycline-resistant mutants selected after 2 to 5 days of incubation. Two of 20 mutants presented an additional sequence inserted in the lon promoter.
We conclude that the very high association between lon and mar mutations occurred in all three E. coli K-12 strains tested. Although the concentration of tetracycline used for selection of MG1655 mutants might not have been optimal for the selection of mutants carrying an additional lon mutation, the very high association between lon and mar mutations seen with AG100 may be lower in MG1655.
High rate of spontaneous lon::IS186 mutations in strain AG100 grown in the absence of selective pressure.
Most of the lon mutations detected were IS186 insertions in the lon promoter. To determine the proportion of spontaneous lon::IS186 mutants present in AG100 grown in the absence of selective pressure (LB broth), we used a comparative PCR approach with primers IS186IR and lonR2, designed to detect IS186 inserted in the lon promoter in any orientation (Table 2). Six colonies of AG100 isolated on LB agar and two isolated colonies of M113R were grown overnight in LB broth, followed by a new growth in LB broth up to an A600 of 1.0, from which DNA was extracted. Amplifications with M113R (presence of one lon::IS186 per bacterium) were used as a reference. We found that an average subpopulation of ∼3.7 × 10−4 (±2.6 × 10−4) lon::IS186 mutants were present when AG100 was grown in LB broth.
To estimate the rate of spontaneous lon::IS186 mutations in AG100 grown in LB medium, we compared the fitness of a wild type and a lon mutant in this medium (see Materials and Methods). We found that after nine generations, the proportion of lon mutants dropped from 28% to 12%, revealing that the growth of the strain with a wild-type lon gene was favored over that of the lon mutant. Using the formula for the determination of the mutation rate (see Materials and Methods), we calculated the frequency of spontaneous lon::IS186 mutations and found that μ was equal to ∼4 × 10−5 lon::IS186 mutation per bacterium and generation. The proportion of lon::IS186 mutants had been determined after >30 generations of growth. Using the values Eq of 3.7 × 10−4 and μ of 4 × 10−5, we calculated Y to equal 1.78 (see Materials and Methods). Computer simulation (see Materials and Methods) confirmed that the same equilibrium (Eq = 3.7 × 10−4) was reached no matter what the initial ratio of wild type to mutant was and that an isolated AG100 bacterium grown in LB broth would almost attain its equilibrium proportion of lon::IS186 mutants in only 30 generations (Eq = 3.5 × 10−4 after 30 generations).
Increased genome instability in lon mutants.
We compared the stability of the genome of a lon mutant with that of its wild-type parental strain. Three colonies of AG100 and M113R (lon3::IS186) isolated on LB plates were independently grown overnight in LB broth. Primers IS5-7 and IS5-8 (Table 2) were chosen to detect and quantify by PCR spontaneous large tandem amplifications events (mutations dupIS5; Fig. 3A), and primers IS3-3 and IS3-1 (Table 2) were chosen to detect and quantify genomic inversions bordered by IS3 sequences located at positions 314453 and 390933, according to the E. coli K-12 genome sequence with GenBank accession number U00096 (Fig. 3A). Comparative PCR amplifications (Fig. 3B) revealed that about 10 times more spontaneous tandem amplifications and genomic inversions were detected in DNA from M113R than in DNA from AG100. Thus, a lon mutation increased the genome instability and favored spontaneous large tandem duplications, causing a multidrug resistance.
DISCUSSION
The majority of the Mar mutants selected on tetracycline had two mutations.
Twelve of 13 spontaneous Mar mutants of E. coli selected on LB plates in the presence of tetracycline (4 μg/ml) and analyzed in detail had at least two mutations (Table 1). Twelve had a mutation in lon, most with an IS186 insertion in the promoter of the gene (Table 4), which is a hot spot for IS186 insertion (39). The presence of five different lon mutations among the 13 mutants selected revealed that they were independent mutational events and were not siblings of a single spontaneous lon mutation which occurred at the beginning of the bacterial growth. Despite the overproduction of AcrAB-TolC pump linked to MarA overproduction in lon mutants (31), a lon mutation alone does not produce a significant increase in multidrug resistance (31). Although increased polysaccharide biosynthesis that led to a mucoid phenotype of lon mutants (15, 26) and that was observed after several days of growth on LB agar was thought to cause increased antibiotic resistance in biofilms via decreased permeability (22), a recent study showed that antibiotic resistance in biofilms of Pseudomonas aeruginosa was dependent on periplasmic glucans biosynthesis (23). The absence of increased antibiotic resistance in E. coli lon mutants agrees with the findings of that study; a second locus or a subsequent mutation, as shown in this work, is the likely basis for increased drug resistance in mucoid lon mutants. Among the second mutations that were found in the lon mutants and that further increased the amount of AcrAB and caused the Mar phenotypes were marR and acrR mutations (Table 1). Two Mar mutants selected in the presence of tetracycline had a large genetic amplification, including acrAB (dupIS3 in mutants M104 and dupIS5 in M013; Fig. 1A). As previously described for M113, those duplications increased the amount of AcrAB and conferred an unstable Mar phenotype both in the presence (as in M104) and in the absence (as in M013) of a wild-type lon gene (31).
lon is a mutator gene which increases IS transpositions and genome rearrangements.
The Mar phenotype in 9 of the 12 mutants harboring a lon mutation was caused by IS insertions (Table 1). This finding suggests that a lon mutation increases IS transposition events. Other studies have implicated Lon in the regulation of transposition of several IS elements of E. coli. The stability of the transposase of IS903 (11), possibly IS1 (38) and Tn5 (24, 30) elements, is dependent on the Lon protease, which seems to regulate their transposition activity. Transposition is also regulated by numerous host factors, such as the E. coli Dam methyltransferase (24), a substrate of the Lon protease (8). In mitochondria, the stability of the genome depends on the activity of the Pim1/Lon protease (10) and the E. coli Lon protease restored the mitochondrial DNA stability in a PIM1/LON mutant (42). Similarly, we found a 10-fold increased instability of the genome of lon mutants, revealing that lon mutations favor genome rearrangements. This effect of Lon might be related to its DNA binding activity (13) or to its role in the control of the cell cycle methylation (8). Therefore, lon mutations confer a mutator phenotype which affects genome stability and the frequency of IS transposition. Although no other regions of the chromosome were analyzed, the increased genome instability linked to a lon mutation probably affects the entire genome of E. coli.
lon mutations favor the appearance and selection of drug-resistant mutants.
A lon mutant allowed the selection of spontaneous drug-resistant mutants at a higher frequency (10 to >100 times) than a wild-type strain did. We found that under certain selective conditions, most drug-resistant mutants selected from a wild-type E. coli strain arose from a subpopulation of spontaneous lon mutants. In the presence of nalidixic acid (but not tetracycline or chloramphenicol) or on MacConkey medium (but not LB medium), lon mutants were disadvantaged compared to a wild-type strain and fewer or no spontaneous drug-resistant mutants selected from a wild-type strain arose from the lon subpopulation. Although we did not test this hypothesis, the effect of MacConkey medium on lon mutants might be linked to the presence of bile salts, which, like nalidixic acid, induce the SOS system (37), a stress condition known to be harmful to lon mutants (21). This study suggests that spontaneous lon-mutated subpopulations of E. coli play an important role in the appearance and selection of spontaneous low-level drug-resistant mutants under selective conditions that are not detrimental to the growth of lon mutants.
From our findings, we propose the following mechanism for the evolution of E. coli to multidrug resistance. A high frequency of spontaneous lon::IS186 mutations (estimated to be 4 × 10−5) caused an important subpopulation of lon mutants to be initially present on the selective media. The size of this subpopulation depends on the fitness of lon mutants and wild-type bacteria in the medium (in LB broth, the proportion of lon mutants was ∼3.7 × 10−4). We found that new spontaneous lon mutants also appeared among the wild-type population during the time of the selection. Furthermore, when selection occurred on certain media (i.e., LB medium with tetracycline at 4 μg/ml or LB medium with chloramphenicol at 10 μg/ml), the lon mutants had a fitness advantage over wild-type bacteria and could slowly grow. This slow growth increased the probability that new mutations causing a higher-level drug resistance occurred in the lon subpopulation rather than in a wild-type bacterium. This was further favored by the mutator phenotype caused by the lon mutation.
Previous studies of Mar mutants revealed that IS insertions were rare and that other types of mutations (i.e., point mutations) were usually found (9, 19, 25, 33, 43, 44). This finding suggests that the connection between lon and mar mutations that we observed might be weak in clinically isolated E. coli strains. Although IS186 sequences, which caused most of the lon mutations characterized so far, were detected in about 50% of the E. coli natural isolates that we tested (data not shown) and some lon::IS186 mutations have been characterized in natural isolates (39), we showed that, via their negative effect on the fitness of the mutants, growth conditions disadvantageous to lon mutants reduce the role played by the lon subpopulation in the evolution to drug resistance.
A role for genetic amplifications for evolution of drug-resistance?
In Salmonella enterica serovar Typhimurium cultures, any genetic locus appears to be spontaneously duplicated in a subpopulation of up to 3% of the bacteria (3). Similarly, we estimated that ∼3.4% and ∼0.21% of AG100 bacteria grown in LB broth in the absence of selective pressure had a dupIS3 and a dupIS5, respectively. However, AG100 drug-resistant mutants are selected at a low frequency (Fig. 2), revealing that in most bacteria dupIS3 and dupIS5 do not allow efficient growth on the selective media. It is possible that amplification of acrAB to more than two copies is required for efficient growth in the presence of the drugs and that this would occur in only a fraction of the bacteria originally carrying a duplication of acrAB. The persistence of only two copies of acrAB in most bacteria would mediate only a small reduced susceptibility to drugs and a small fitness advantage. Although we did not test this hypothesis, the consequence would be that mutants acquiring an increased level of drug resistance would then preferentially evolve from this slowly growing subpopulation rather than from the wild-type population, as observed with the lon subpopulation (this work) or during selection of Lac+ adaptive revertant mutants (20). In agreement with this hypothesis, we observed that an important population (∼24%) of bacteria carried a dupIS5 after an overnight growth of AG100 in the presence of nalidixic acid (5 μg/ml).
Our findings reveal that the genetic amplification of acrAB is an important mechanism for multidrug resistance. For example, such amplifications might explain a multidrug-resistant mutant of Salmonella enterica (mutant BN18/21) with an increased level of production of AcrAB but no mutations in the acrR, acrAB promoter, soxRS, and marRAB loci (35).
Amplification to clinically significant multidrug resistance is linked to acrAB amplification.
We found that the unstable amplification of antibiotic resistance following the serial growth of a mar mutant in the presence of a constant sublethal low concentration of tetracycline (5 μg/ml), as originally described by George and Levy (14), was linked to unstable genetic amplifications carrying acrAB. A high level of mutidrug resistance had also previously been observed with a constructed double marR dupIS186 mutant (31). Although the original mar mutant used was already resistant to 5 μg/ml of tetracycline (Table 5), selection for the genetic amplifications must have somehow increased the fitness of the bacteria under the growth conditions used. The cause of this increased fitness remains unknown and could be linked to any locus present on the amplified units. This mechanism, independent of the presence of a lon mutation, allows natural E. coli mar mutants facing low antibiotic concentrations for a prolonged period of time to develop transient clinically significant high antibiotic resistances (Table 5).
This work demonstrates the unexpected role of spontaneous lon mutants in the evolution of E. coli to low levels of multidrug resistance under selective conditions that are not detrimental to lon mutants. We also easily found new large genetic amplifications that carried acrAB and that caused unstable low levels of multidrug resistance, revealing that this newly uncovered mechanism of transient resistance might be frequent. Interestingly, those genetic amplifications could also be linked to unstable high levels of multidrug resistance that could represent a clinical threat. In addition to the role played by lon mutations, our observations also suggest a role for spontaneous large genetic duplications carrying acrAB in the evolution mechanism to low levels of multiple-drug resistance.
Acknowledgments
We thank L. M. McMurry for helpful discussions during this work, C. A. Gross for strains CAG12017 and CAG12154, H. Zgurskaya for the anti-AcrA antibodies, and AB BIODISK for the generous gift of the Etest strips used for this study.
This work was supported by U.S. National Institutes of Health PHS grant AI56021.
Footnotes
Published ahead of print on 12 January 2007.
REFERENCES
- 1.Alekshun, M. N., and S. B. Levy. 1999. Alteration of the repressor activity of MarR, the negative regulator of the Escherichia coli marRAB locus, by multiple chemicals in vitro. J. Bacteriol. 181:4669-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alekshun, M. N., and S. B. Levy. 1997. Regulation of chromosomally mediated multiple antibiotic resistance: the mar regulon. Antimicrob. Agents Chemother. 41:2067-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, P., and J. Roth. 1981. Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc. Natl. Acad. Sci. USA 78:3113-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ariza, R. R., S. P. Cohen, N. Bachhawat, S. B. Levy, and B. Demple. 1994. Repressor mutations in the marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 176:143-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ariza, R. R., Z. Li, N. Ringstad, and B. Demple. 1995. Activation of multiple antibiotic resistance and binding of stress-inducible promoters by Escherichia coli Rob protein. J. Bacteriol. 177:1655-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbosa, T. M., and S. B. Levy. 2000. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J. Bacteriol. 182:3467-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennik, M. H., P. J. Pomposiello, D. F. Thorne, and B. Demple. 2000. Defining a rob regulon in Escherichia coli by using transposon mutagenesis. J. Bacteriol. 182:3794-3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calmann, M. A., and M. G. Marinus. 2003. Regulated expression of the Escherichia coli dam gene. J. Bacteriol. 185:5012-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, S. P., H. Hachler, and S. B. Levy. 1993. Genetic and functional analysis of the multiple antibiotic resistance (mar) locus in Escherichia coli. J. Bacteriol. 175:1484-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Contamine, V., and M. Picard. 2000. Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiol. Mol. Biol. Rev. 64:281-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derbyshire, K. M., M. Kramer, and N. D. Grindley. 1990. Role of instability in the cis action of the insertion sequence IS903 transposase. Proc. Natl. Acad. Sci. USA 87:4048-4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dewitt, S. K., and E. A. Adelberg. 1962. Transduction of the attached sex factor of Escherichia coli. J. Bacteriol. 83:673-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu, G. K., M. J. Smith, and D. M. Markovitz. 1997. Bacterial protease Lon is a site-specific DNA-binding protein. J. Biol. Chem. 272:534-538. [PubMed] [Google Scholar]
- 14.George, A. M., and S. B. Levy. 1983. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 155:531-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottesman, S., and V. Stout. 1991. Regulation of capsular polysaccharide synthesis in Escherichia coli K12. Mol. Microbiol. 5:1599-1606. [DOI] [PubMed] [Google Scholar]
- 16.Griffith, K. L., I. M. Shah, and R. E. Wolf, Jr. 2004. Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 51:1801-1816. [DOI] [PubMed] [Google Scholar]
- 17.Jair, K. W., X. Yu, K. Skarstad, B. Thony, N. Fujita, A. Ishihama, and R. E. Wolf, Jr. 1996. Transcriptional activation of promoters of the superoxide and multiple antibiotic resistance regulons by Rob, a binding protein of the Escherichia coli origin of chromosomal replication. J. Bacteriol. 178:2507-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jellen-Ritter, A. S., and W. V. Kern. 2001. Enhanced expression of the multidrug efflux pumps AcrAB and AcrEF associated with insertion element transposition in Escherichia coli mutants selected with a fluoroquinolone. Antimicrob. Agents Chemother. 45:1467-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komp Lindgren, P., A. Karlsson, and D. Hughes. 2003. Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob. Agents Chemother. 47:3222-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kugelberg, E., E. Kofoid, A. B. Reams, D. I. Andersson, and J. R. Roth. 2006. Multiple pathways of selected gene amplification during adaptive mutation. Proc. Natl. Acad. Sci. USA 103:17319-17324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis, K. 2000. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 64:503-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mah, T. F., and G. A. O'Toole. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 9:34-39. [DOI] [PubMed] [Google Scholar]
- 23.Mah, T. F., B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart, and G. A. O'Toole. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306-310. [DOI] [PubMed] [Google Scholar]
- 24.Mahillon, J., and M. Chandler. 1998. Insertion sequences. Microbiol. Mol. Biol. Rev. 62:725-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maneewannakul, K., and S. B. Levy. 1996. Identification of mar mutants among quinolone-resistant clinical isolates of Escherichia coli. Antimicrob. Agents Chemother. 40:1695-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markovitz, A. 1964. Regulatory mechanisms for synthesis of capsular polysaccharide in mucoid mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 51:239-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin, R. G., K. W. Jair, R. E. Wolf, Jr., and J. L. Rosner. 1996. Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 178:2216-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, P. F., L. F. Gambino, M. C. Sulavik, and S. J. Gracheck. 1994. Genetic relationship between soxRS and mar loci in promoting multiple antibiotic resistance in Escherichia coli. Antimicrob. Agents Chemother. 38:1773-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moken, M. C., L. M. McMurry, and S. B. Levy. 1997. Selection of multiple-antibiotic-resistant (mar) mutants of Escherichia coli by using the disinfectant pine oil: roles of the mar and acrAB loci. Antimicrob. Agents Chemother. 41:2770-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy, Z., and M. Chandler. 2004. Regulation of transposition in bacteria. Res. Microbiol. 155:387-398. [DOI] [PubMed] [Google Scholar]
- 31.Nicoloff, H., V. Perreten, L. M. McMurry, and S. B. Levy. 2006. Role for tandem duplication and Lon protease in the AcrAB-TolC-dependent multiple antibiotic resistance (Mar) in an Escherichia coli mutant without mutations in marRAB or acrRAB. J. Bacteriol. 188:4413-4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oethinger, M., W. V. Kern, A. S. Jellen-Ritter, L. M. McMurry, and S. B. Levy. 2000. Ineffectiveness of topoisomerase mutations in mediating clinically significant fluoroquinolone resistance in Escherichia coli in the absence of the AcrAB efflux pump. Antimicrob. Agents Chemother. 44:10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oethinger, M., I. Podglajen, W. V. Kern, and S. B. Levy. 1998. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob. Agents Chemother. 42:2089-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okusu, H., D. Ma, and H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178:306-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olliver, A., M. Valle, E. Chaslus-Dancla, and A. Cloeckaert. 2004. Role of an acrR mutation in multidrug resistance of in vitro-selected fluoroquinolone-resistant mutants of Salmonella enterica serovar Typhimurium. FEMS Microbiol. Lett. 238:267-272. [DOI] [PubMed] [Google Scholar]
- 36.Pomposiello, P. J., M. H. Bennik, and B. Demple. 2001. Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J. Bacteriol. 183:3890-3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prieto, A. I., F. Ramos-Morales, and J. Casadesus. 2004. Bile-induced DNA damage in Salmonella enterica. Genetics 168:1787-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouquette, C., M. C. Serre, and D. Lane. 2004. Protective role for H-NS protein in IS1 transposition. J. Bacteriol. 186:2091-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SaiSree, L., M. Reddy, and J. Gowrishankar. 2001. IS186 insertion at a hot spot in the lon promoter as a basis for Lon protease deficiency of Escherichia coli B: identification of a consensus target sequence for IS186 transposition. J. Bacteriol. 183:6943-6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seoane, A. S., and S. B. Levy. 1995. Characterization of MarR, the repressor of the multiple antibiotic resistance (mar) operon in Escherichia coli. J. Bacteriol. 177:3414-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singer, M., T. A. Baker, G. Schnitzler, S. M. Deischel, M. Goel, W. Dove, K. J. Jaacks, A. D. Grossman, J. W. Erickson, and C. A. Gross. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teichmann, U., L. van Dyck, B. Guiard, H. Fischer, R. Glockshuber, W. Neupert, and T. Langer. 1996. Substitution of PIM1 protease in mitochondria by Escherichia coli Lon protease. J. Biol. Chem. 271:10137-10142. [DOI] [PubMed] [Google Scholar]
- 43.Wang, H., J. L. Dzink-Fox, M. Chen, and S. B. Levy. 2001. Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: role of acrR mutations. Antimicrob. Agents Chemother. 45:1515-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webber, M. A., and L. J. Piddock. 2001. Absence of mutations in marRAB or soxRS in acrB-overexpressing fluoroquinolone-resistant clinical and veterinary isolates of Escherichia coli. Antimicrob. Agents Chemother. 45:1550-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White, D. G., J. D. Goldman, B. Demple, and S. B. Levy. 1997. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J. Bacteriol. 179:6122-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]