

Abstract
The broad spectrum of antimicrobial activity, oral bioavailability, extensive tissue distribution, and once-daily intravenous or oral dosing of gatifloxacin, an expanded-spectrum 8-methoxy fluoroquinolone, make it a potentially useful agent for the treatment of pediatric infections. A population pharmacokinetic model was developed to describe the pharmacokinetics of gatifloxacin in children. Data for analysis were obtained from a single-dose safety/pharmacokinetic study utilizing intensive blood sampling in patients aged 6 months to 16 years. Each subject received a single oral dose of gatifloxacin as a suspension, at doses of 5, 10, or 15 mg/kg of body weight. A total of 845 samples were obtained from 82 patients. A one-compartment model with first-order absorption and elimination was the most appropriate to describe the gatifloxacin concentrations. Covariate analysis using forward selection and backward elimination found that apparent clearance was related to body surface area, and apparent volume of distribution was related to body weight. No effect of age on drug clearance could be identified once clearance was corrected for body surface area. Based on pharmacokinetic simulations, the 10-mg/kg (maximum, 400 mg) once-daily dose of gatifloxacin is expected to provide drug exposure similar to that in healthy adults. The population pharmacokinetic model described herein will be used for Bayesian analyses of sparse pharmacokinetic sampling in phase II/III clinical trials and for Monte Carlo simulation experiments. The success of this strategy provides a model for future pediatric drug development programs.
Gatifloxacin is an expanded-spectrum 8-methoxy fluoroquinolone effective against a broad spectrum of pathogens (18, 20, 21). Its spectrum of activity, high oral bioavailability, extensive tissue distribution, and once-daily, interchangeable intravenous or oral dosing make it an attractive potential addition to the therapeutic armamentarium for pediatric infectious disease (26, 28, 34). Although gatifloxacin is not FDA approved for the treatment of otitis media in children, studies support its effectiveness in patients with recurrent otitis media and/or acute treatment failure (3, 15, 16, 24) and provide supporting evidence for its activity in children against key respiratory tract pathogens such as Streptococcus pneumoniae.
In adults, orally administered gatifloxacin is well absorbed in a dose-independent fashion, with an absolute bioavailability of approximately 96% (23). The rate and extent of absorption are not significantly affected by food (22). Gatifloxacin undergoes limited biotransformation and is eliminated primarily unchanged in the urine (70 to 80%), with an elimination half-life of approximately 8 to 10 h (14, 28). Gatifloxacin exhibits linear pharmacokinetics (PK), with clearance being independent of dose (14). The mean area under the concentration-time curve from 0 to 24 h (AUC0-24) ± standard deviation for adults given multiple daily doses of 400 mg is 34.4 ± 4.1 mg·h/liter (28).
The pharmacokinetics of gatifloxacin in pediatric patients were investigated in a single-dose safety/pharmacokinetic study utilizing intensive blood sampling and noncompartmental pharmacokinetic analysis methods (9). While that study did examine the potential impact of gatifloxacin disposition on pediatric-dose selection, it did not fully consider all of the potentially relevant factors that can contribute to variability in pharmacokinetics, which are afforded by population-based techniques. In general, population pharmacokinetic models have the additional advantage of providing precise estimates of the variance-covariance between pharmacokinetic parameters, useful for subsequent simulation experiments, and are applicable to sparse sampling strategies often employed in phase II/III pediatric studies (5, 31, 32). Thus, the data collected in the above-described study were used to develop a population pharmacokinetic model describing the patient factors responsible for the intersubject variability in gatifloxacin pharmacokinetics. This model is suitable for use, in combination with sparse-concentration sampling strategies often employed in pediatric studies, to estimate exposure in future studies, potentially leading to the identification of PK and pharmacodynamic (PD) targets for efficacy against pediatric respiratory tract infections. Furthermore, this model should also be suitable for the conduct of robust simulation experiments critical to defining appropriate dosing regimens as well as to evaluating the comparative utility of anti-infective agents (2, 7).
MATERIALS AND METHODS
Patients and data collection.
The data used in this analysis were collected in a nonrandomized, open-label, parallel-group, single-dose, dose escalation study of the safety and pharmacokinetics of orally administered gatifloxacin in 84 hospitalized or outpatient pediatric patients (9). The study consisted of the following four age strata: 6 months to ≤2.0 years, >2.0 to ≤6.0 years, >6.0 to ≤12.0 years, and >12.0 to ≤16.0 years. Within each age stratum, patients were administered one dose of 5, 10, or 15 mg/kg of body weight of gatifloxacin as an oral suspension, up to a maximum dose of 600 mg. All patients meeting the eligibility criteria as defined in the protocol (82 of the 84 patients) were included in the analysis. Eleven blood samples were collected from each patient at the following times after dosing: 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h.
Additional information was collected regarding patient demographics and covariate factors. This information included weight, height, age, gender, race, and serum creatinine concentration. Creatinine clearance was calculated from the measured serum creatinine concentration using the Schwartz formula (30). Body surface area (BSA) was calculated using the method of Gehan and George (12). Based on the distribution of patient race, each patient was categorized as white, black, or other.
Drug analysis.
Plasma specimens were analyzed for gatifloxacin using a previously described, validated high-performance liquid chromatography fluorescence method (23). The lower limit of quantitation of gatifloxacin in plasma was 0.01 μg/ml; standard curves were linear (r2 ≥ 0.983) over the concentration range of 0.01 to 5 μg/ml. The between-run and within-run levels of precision for quality control samples were no greater than coefficients of variation of 9.5 and 10.1% respectively, with deviations from nominal concentrations being within ±5.6%.
Population pharmacokinetic model development.
The gatifloxacin concentration-time data were fit to a one-compartment model using the NONMEM computer program (version 5) (6). Due to the large number of samples per patient, the first-order conditional estimation method with interaction was used (4). When a one-compartment model with first-order absorption and elimination was evaluated (subroutines ADVAN2 and TRANS2), the apparent oral clearance (CL/F, where F is oral bioavailability), apparent volume of distribution (V/F), and first-order absorption rate constant (ka) parameters were estimated. Interindividual variability in CL/F, V/F, and ka values was modeled using an exponential-error model. The two error models evaluated to estimate residual variability were a proportional-error model and a combined model of additive errors plus proportional errors.
Patient covariates were added to the basic model using a forward-selection procedure. The covariates evaluated included age, weight, body surface area, serum creatinine, creatinine clearance, gender, and race. Initially, Bayesian estimates of clearance generated from the basic model were plotted against each covariate to determine potential functional forms for the relationship (i.e., linear, piece-wise linear, and nonlinear). Each covariate of interest was then added to the model for clearance (or elimination rate) and volume of distribution separately, and the results of these runs were compared. All covariate effects whose addition resulted in a significant reduction in the value of the objective function (a decrease of at least 3.84 [α = 0.05, 1 degree of freedom]) were included in the full multivariable model. The resulting multivariable model was used to examine the appropriateness of the interindividual-variability and residual-error models. A recursive backward-elimination procedure was then performed to further refine the model and eliminate correlated predictors. During each step of backward elimination, the covariate whose removal resulted in the smallest nonsignificant increase in the objective function (a change of no more than 10.83 [α = 0.001, 1 degree of freedom]) was removed from the model. This process was repeated until no further covariate effects could be removed from the model without a statistically significant difference in the minimum value of the objective function. The final model was then assessed for the appropriateness of the interindividual-variability and residual-error models.
Statistical analysis.
Statistical significance was assessed by the change in the log likelihood value obtained for various models. For each analysis, NONMEM computed the minimum value of the objective function, a statistic that is proportional to minus twice the log likelihood of the data. In the case of hierarchical models, the change in the minimum value of the objective function produced by the inclusion of a parameter is asymptotically distributed as χ2, with the number of degrees of freedom equal to the number of parameters added to or deleted from the model. When alternative models could not be cast as hierarchical, the change in the objective function was used only as a qualitative measure of statistical significance.
The goodness-of-fit of each NONMEM analysis was assessed by the examination of the following: scatter plots of predicted concentrations versus measured concentrations and predicted concentrations versus weighted residuals, the precision of the parameter estimates as measured by the percent standard error of the mean (standard error/parameter estimate × 100%), and changes in the estimates of interindividual variability and residual variability for the specified model.
Model validation.
Due to the relatively small sample size, a cross-validation approach was taken. In this approach, the data were split into 10 groups of similar demographic characteristics in order to provide 10 distinct validation data sets (“test data sets”) of eight patients each. For the first test set, the final population model was fit to data from the remaining 74 patients (the “training set”) to obtain mean population pharmacokinetic parameter estimates. The model, with parameter values fixed to those determined from the training data set, was then applied to the test data set in order to predict the measured concentrations. Because the total sample size was 82 subjects, 2 subjects were never included in a test set but were included in the “training data sets.” This process was then repeated for the remaining nine test data sets. The bias and precision of the predicted concentrations were assessed by means of the percent prediction error (PEP) and the absolute PEP (APEP) calculated using equations 1 and 2 below:
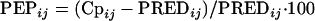 |
(1) |
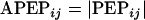 |
(2) |
where PEPij is the percent prediction error between the measured value of the ith plasma concentration in the jth subject and the predicted value of the ith plasma concentration in the jth subject, APEPij is the absolute value of PEPij, Cpij is the measured value of the ith plasma concentration in the jth subject, and PREDij is the predicted value of the ith plasma concentration in the jth subject.
Simulation of exposure using alternative dosing regimens.
The pediatric dosing regimen for gatifloxacin oral suspension previously under consideration was as follows: 10 mg/kg/dose given once daily up to a maximum dose of 400 mg once daily. As the maximum dose allowed in this study was 600 mg once daily, it was necessary to conduct simulations in order to evaluate the suitability of the regimen under consideration. These simulations were conducted by creating a data set of 1,134 hypothetical pediatric subjects with age-appropriate weight and body surface area characteristics. The age appropriateness of the patient characteristics was achieved by first simulating age using a uniform distribution and then simulating the patient's height and weight using the CDC reference charts.
Using the fixed- and random-effect models from the final population PK model, two AUC0-24 values were generated for each simulated patient, one using a 10-mg/kg regimen with no upper dose limit and one using the proposed regimen of 10 mg/kg up to 400 mg. An estimate for the free-drug AUC-to-MIC ratio (fraction unbound AUC [fuAUC]/MIC90) was obtained by dividing the free-drug AUC (0.8·AUC0-24) estimated for the proposed regimen (10 mg/kg up to 400 mg) by the MIC90 of gatifloxacin to Streptococcus pneumoniae (0.5 μg/ml) (19).
RESULTS
Data.
The initial pharmacokinetic database consisted of information from 84 patients and 860 gatifloxacin plasma concentrations. Data from two patients (13 concentrations) were excluded due to concomitant administration of medications that were prohibited by the protocol and resulted in outlier or undetectable plasma concentrations. Two additional samples collected 24 h postdose were undetectable and were also excluded. Thus, data from a total of 82 patients and 845 plasma concentrations were utilized for the final pharmacokinetic analysis. Table 1 summarizes the demographic characteristics of the patients in the pharmacokinetic analysis. The majority of patients were male (63.4%) and white (54.9%). The mean age was approximately 7 (7) years. Subjects were relatively equally distributed among the age strata, with 22 subjects being ≤2.0, 18 being between 2.0 and 6.0, 22 being between 6.1 and 12.0, and 20 being >12.0 years of age. Scatter plots of gatifloxacin plasma concentrations versus time since the last dose are illustrated in Fig. 1, stratified by dose group.
TABLE 1.
Demographic summary of the patients included in the population pharmacokinetic model development
| Dose (mg/kg) of gatifloxacin (no. of children) | Mean value (% CV) by patient characteristica
|
No. of subjects (%)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| By gender
|
By race
|
||||||||
| Wt (kg) | Age (yr) | CLCR (ml/min/1.73 m2) | BSA (m2) | Male | Female | White | Black | Other | |
| 5 (22) | 28.2 (63.9) | 7.37 (70.2) | 165 (20.0) | 0.97 (45.3) | 16 (72.7) | 6 (27.3) | 12 (54.5) | 7 (31.8) | 3 (13.6) |
| 10 (33) | 28.3 (69.6) | 7.17 (75.2) | 154 (28.9) | 0.96 (49.6) | 18 (54.5) | 15 (45.5) | 17 (51.5) | 11 (33.3) | 5 (15.2) |
| 15 (27) | 26.8 (68.5) | 6.36 (74.1) | 141 (26.8) | 0.92 (45.5) | 18 (66.7) | 9 (33.3) | 16 (59.3) | 9 (33.3) | 2 (7.41) |
| All doses combined (82) | 27.8 (67.0) | 6.96 (72.9) | 153 (26.3) | 0.95 (46.7) | 52 (63.4) | 30 (36.6) | 45 (54.9) | 27 (32.9) | 10 (12.2) |
CV, coefficient of variation; CLCR, creatinine clearance.
FIG. 1.

Scatter plots (by dose) of observed gatifloxacin concentrations versus time since the last dose.
Structural pharmacokinetic model.
Initially, a one-compartment model with first-order absorption and linear elimination was tested in which the parameters of clearance and volume of distribution were estimated. During the structural-model development, while NONMEM data converged, the goodness-of-fit plots indicated slight model misspecification. In addition, the pharmacokinetic parameter estimates from this preliminary model were inconsistent with expected values based on prior adult and pediatric noncompartmental analyses. It was felt that this might be due to identifiability issues secondary to the importance of subject size in relation to CL/F and V/F. Thus, the impact of subject size on both CL/F and V/F was assessed simultaneously prior to the addition of the remaining covariates (such as age and creatinine clearance, etc.). This approach incorporates physiologic development (using body size as a surrogate) in the base model, with additional effects of age being examined during covariate model building.
Covariate and interindividual-variability models.
Table 2 details the results of covariate model building. After a screening of various size functions on CL/F and V/F, a linear relationship between body surface area and CL/F combined with a linear relationship between weight and V/F best described the data. Forward-selection and backward-elimination procedures failed to yield any covariates that significantly improved the model and confirmed that both of these relationships improved the model.
TABLE 2.
Summary of covariate-model building for the clearance-based modela
| Function and model | Step | Parameter | Covariate added | Covariate removed | MVOF | MVOF change | df | P value |
|---|---|---|---|---|---|---|---|---|
| Forward selection (basic structural model) | 0 | 320 | ||||||
| 1 | V | Wt | 197 | −123 | 1 | <0.0001 | ||
| 2 | CL | BSA | 91.2 | −106 | 1 | <0.0001 | ||
| 3 | CL | Age | 87.1 | −4.10 | 1 | 0.04 | ||
| 4 | NS | NS | ||||||
| Backward elimination (full multivariable model) | 0 | 87.1 | ||||||
| 1 | CL | Age | 91.2 | 4.10 | 1 | 0.04 | ||
| 2 | SS | SS |
NS, none of the remaining covariates were statistically significant (P < 0.05); SS, all of the remaining covariates were statistically significant (P < 0.001); MVOF, minimum value of the objective function; MVOF change, change in the MVOF relative to that of the comparator model.
Final pharmacokinetic model.
The details of the final population model are provided in Table 3. In brief, the final population pharmacokinetic model was a one-compartment model with first-order absorption and elimination. The covariate relationships in the final model were between BSA and CL/F and between weight and V/F. Interindividual variabilities in CL/F, V/F, and ka values were estimated to be 30%, 25%, and 84%, respectively. The correlation between the interindividual variability in CL/F values and the interindividual variability in V/F values was estimated to be 24%. Residual variability, estimated using a proportional-error model, was 25%. The typical values of CL/F, V/F, and ka can be calculated using equations 3 through 5 below:
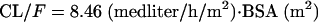 |
(3) |
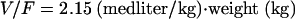 |
(4) |
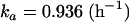 |
(5) |
The goodness-of-fit plots for this model are provided in Fig. 2. The model fit the measured concentrations with minimal bias and moderate precision (the residual error was 25%).
TABLE 3.
Parameter and variance estimates for final population pharmacokinetic modela
| Parameter | Population mean
|
Magnitude of IIV
|
||
|---|---|---|---|---|
| Final estimate | % SEM | Final estimate (% CV) | % SEM | |
| CL/F (liter/h/m2) | 8.46 | 3.50 | 29.7 | 18.9 |
| V/F (liter/kg) | 2.15 | 3.30 | 24.5 | 27.0 |
| ka (h−1) | 0.936 | 10.3 | 84.0 | 16.2 |
| Covariance of the IIV CL/F and IIV V/F | 24.4 | 24.5 | ||
| Residual variability (% CV) | 25.1 | 10.1 | ||
The minimum value of objective function is −254. IIV, interindividual variability; CV, coefficient of variation.
FIG. 2.

Goodness-of-fit plots for the final population pharmacokinetic model. The solid line represents the line of unity for the two upper panels and the 0 reference line for the bottom two panels.
Model validation.
In general, the model performed well, with an overall median (25th- to 75th-percentile) percent prediction error (bias) of 0.316 (−30.0, 31.3); median bias estimates for individual data sets ranged from −12.6% to +16.7. The overall median (25th- to 75th-percentile) absolute percent prediction error (precision) was 30.7 (13.4, 52.3); median precision estimates for individual data sets ranged from 23.7% to 38.2%. The estimates were similar with each test population, the confidence interval from the 25th to 75th percentiles universally included 0 (for bias estimates), and the magnitude of values fell within the range of the residual error.
Simulated exposure using an alternative dosing regimen.
Results of the simulation suggest that, if the dose is not truncated at 400 mg, older (i.e., heavier) pediatric patients are likely to experience gatifloxacin exposure well above the range experienced by younger (i.e., lighter) patients. For those patients with a body weight above 40 kg, the median (minimum to maximum) predicted AUC0-24 using a uniform dose of 10 mg/kg would be 39.5 mg·h/liter (20.3 to 94.2) compared to the median (minimum to maximum) of 29.8 mg·h/liter (12.4 to 84.3) for patients weighing less than 40 kg. The median (minimum to maximum) predicted AUC0-24 using a hypothetical fixed dose of 400 mg for the patients weighing more than 40 kg was 29.8 mg·h/liter (13.9 to 63.2) (see Fig. 3).
FIG. 3.

Scatter plot of the individual AUC estimates (AUC0-24, μg·h/ml) versus patient age (years) for the simulated population. The dose used for this simulation was 10 mg/kg, with an upper limit of 400 mg. The solid line represents the best-fit line of AUC versus age.
For the entire simulated patient population, which ranged in age from 6 months to 16 years, the median (minimum to maximum) predicted AUC0-24 using a hypothetical fixed maximum dose of 400 mg was 29.8 mg·h/liter (12.4 to 84.3). The predicted median (minimum to maximum) fuAUC/MIC90 was 47.7 (19.9 to 135); 93.2% of patients (1,058 of 1,134 patients) had a simulated fuAUC/MIC90 above the target value of 30.
DISCUSSION
In order to facilitate analysis of future pediatric studies involving gatifloxacin, a population pharmacokinetic model describing the relevant patient factors contributing to variability in gatifloxacin pharmacokinetics in this pediatric population was undertaken. A prior noncompartmental analysis had been performed (9) but was limited, as only subjects with complete pharmacokinetic profiles could be included. Furthermore, steady-state concentrations at specific times during a dose interval cannot be predicted with noncompartmental-analysis methods. Population pharmacokinetic methods offer us the ability (i) to estimate gatifloxacin exposure (in plasma and other tissues and fluids) in pediatric patients by using a limited number of pharmacokinetic samples and (ii) to evaluate the appropriateness of the chosen dose in comparison with other anti-infective agents for similar indications, using simulation methods. Last, the population-based, mixed-effect approach enabled pharmacokinetic evaluation of incomplete and, therefore, unbalanced data sets (19 patients with one or more missing postdose samples) that alone would have been insufficient to yield accurate individual parameter estimates using the noncompartmental approach. To that end, we were able to obtain estimates of the individual pharmacokinetic parameters that were more accurate than the noncompartmental estimates (9) for these 82 pediatric patients. In a comparison of our 77 patients, for whom parameters were derived by both noncompartmental and population methods, the agreement between estimates of apparent oral clearance, AUC0-24, and half-life were excellent. In addition, parameter variance estimates from the population analysis are relatively unbiased, as they account for both the residual variability and the covariance among parameters. Although one report found that a linear, two-compartment model best described the plasma pharmacokinetics in adults (28), the most robust model that could be fit to this pediatric data set was a one-compartment model with first-order absorption and elimination; the data did not support a two-compartment model.
A primary benefit of population analysis methods over traditional methods is the ability to quantify the impact of patient factors on the pharmacokinetic parameters of the drug (5, 31, 32, 33). In this analysis, relationships between body size and gatifloxacin pharmacokinetic parameters were of the most importance. Body surface area best explained the intersubject variability in gatifloxacin clearance, while weight best described the variability in volume. The linear relationship between BSA and apparent clearance is consistent with the predominant renal elimination of gatifloxacin and the flat linear relationship between BSA and the glomerular filtration rate in older infants and children (30). Beyond these size-related relationships, no other covariate, including age, serum creatinine, and estimated creatinine clearance, improved the model. In this case, body size is a likely surrogate for developmental variability in the clearance organ, and no further influence of age could then be identified. While one might expect estimated creatinine clearance to be a significant independent covariate in the model, the patients in this study all had normal renal function, reducing our ability to evaluate the impact of reduced renal function. In addition, all patients had very low serum creatinine values (the maximum value was 1.0 g/dl) reported to one decimal place, reducing the precision of creatinine clearance estimates. Finally, both BSA and creatinine clearance are estimated from subject height; thus, the high degree of correlation seen between these two covariates was expected.
Although BSA was found to be a better predictor of clearance than body weight, the two measures are highly correlated, and body weight is simpler to use for dosing pediatric patients. As the nonlinear relationship between weight and BSA is most pronounced in the youngest patients, there is a potential for underdosing of the smallest children or overdosing of the largest children when using a weight-based dosing scheme in situations where clearance is best correlated with BSA. However, given the inherent interindividual variability in clearance and the relatively wide therapeutic index of gatifloxacin, any added precision gained from BSA-based dosing is unlikely to be clinically significant. A more clinically relevant issue is that of the maximum dose to be used in pediatric patients. In children weighing less than 40 kg, a dose of 10 mg/kg provides exposures similar to those seen in adults from studies utilizing 400 mg. However, children weighing more than 40 kg have higher gatifloxacin exposure (in terms of AUC0-24) than the younger age groups. As can be seen in Fig. 3, limiting the maximum dose to 400 mg once daily brings the gatifloxacin exposure in the older (i.e., heavier) children in line with the exposure seen in the younger (i.e., lighter) children. As the established PK-PD target for efficacy in Streptococcus pneumoniae infections is a fuAUC/MIC of 30 (1, 10, 11, 27), the dose of 10 mg/kg appears to be appropriate from an effectiveness standpoint as well, given the present MIC90 for gatifloxacin in large cohorts of Streptococcus pneumoniae isolates (19).
The results of this analysis demonstrate the applicability of a population-based pharmacokinetic analysis to gatifloxacin data obtained from a pediatric population. Validation of this approach is supported by both internal assessment (i.e., cross-validation) and external comparison to later studies (13, 25). The value of the population-based approach for gatifloxacin in pediatrics, as well as for pediatric drug development in general, resides with the potential to make use of sparse data sets obtainable from future studies to evaluate exposure-response relationships for safety and/or efficacy in pediatric subpopulations defined by disease state and/or development (e.g., patients with renal compromise, neonates, and infants). Such analyses can be extremely useful in providing dose justification and may help to streamline the pediatric drug development process, allowing children earlier access to potentially useful therapeutic agents.
Acknowledgments
This work was supported in part by a grant from Bristol-Myers Squibb and the following grants from the National Institute of Child Health and Human Development (Network of Pediatric Pharmacology Research Units), Bethesda, MD: 1U10 HD045937-01 (San Diego, CA), 5U01 HD031323-10 (Cleveland, OH), 2U10 HD031313-12 (Kansas City, MO), and 5U10 HD031324-11 (Little Rock, AR). G.L.K. is the Marion Merrell Dow/Missouri Chair in Pediatric Pharmacology, and R.F.J. is the Horace C. Cabe Professor of Pediatrics.
Footnotes
Published ahead of print on 12 January 2007.
REFERENCES
- 1.Ambrose, P. G., D. M. Grasela, T. H. Grasela, J. Passarell, H. B. Mayer, and P. F. Pierce. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 45:2793-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrose, P. G., S. M. Bhavnani, and R. C. Owens, Jr. 2003. Clinical pharmacodynamics of quinolones. Infect. Dis. Clin. N. Am. 17:529-543. [DOI] [PubMed] [Google Scholar]
- 3.Arguedas, A., L. Sher, E. Lopez, X. Saez-Llorens, K. Hamed, K. Skuba, and P. F. Pierce. 2003. Open label, multicenter study of gatifloxacin treatment of recurrent otitis media and acute otitis media treatment failure. Pediatr. Infect. Dis. J. 22:949-955. [DOI] [PubMed] [Google Scholar]
- 4.Beal, S. L., and L. B. Sheiner. 1998. NONMEM users guides—part VII: conditional estimation methods. NONMEM Project Group, University of California, San Francisco.
- 5.Bellissant, E., V. Sebille, and G. Paintaud. 1998. Methodological issues in pharmacokinetic-pharmacodynamic modelling. Clin. Pharmacokinet. 35:151-166. [DOI] [PubMed] [Google Scholar]
- 6.Boeckman, A. J., S. L. Beal, and L. B. Sheiner. 1994. NONMEM users guides—part V: introductory guide. NONMEM Project Group, University of California, San Francisco.
- 7.Bradley, J. S., M. N. Dudley, and G. L. Drusano. 2003. Predicting efficacy of antiinfectives with pharmacodynamics and Monte Carlo simulation. Pediatr. Infect. Dis. J. 22:982-992. [DOI] [PubMed] [Google Scholar]
- 8.Reference deleted.
- 9.Capparelli, E. V., M. D. Reed, J. S. Bradley, G. L. Kearns, R. F. Jacobs, B. D. Damle, J. L. Blumer, and D. M. Grasela. 2005. Pharmacokinetics of gatifloxacin in infants and children. Antimicrob. Agents Chemother. 49:1106-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drusano, G. L., D. E. Johnson, M. Rosen, and H. C. Standiford. 1993. Pharmacodynamics of a fluoroquinolone antimicrobial agent in a neutropenic rat model of Pseudomonas sepsis. Antimicrob. Agents Chemother. 37:483-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fantin, B., J. Leggett, S. Ebert, and W. A. Craig. 1991. Correlation between in vitro and in vivo activity of antimicrobial agents against gram-negative bacilli in a murine infection model. Antimicrob. Agents Chemother. 35:1413-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gehan, E. A., and S. L. George. 1970. Estimation of human body surface area from height and weight. Cancer Chemother. Rep. 54:225-235. [PubMed] [Google Scholar]
- 13.Grasela, D., C. Rubino, F. Lacreta, A. Arguedas, L. Sher, E. Lopez, X. Saez-Lorenz, M. Swingle, B. Cirincione, M. Sokolowski, and P. Pierce. 2001. Abstr. 41st Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-37.
- 14.Grasela, D. M. 2000. Clinical pharmacology of gatifloxacin, a new fluoroquinolone. Clin. Infect. Dis. 31:S51-S58. [DOI] [PubMed] [Google Scholar]
- 15.Hamed, K. A., A. Arguedas, X. Saez-Llorens, A. Rodriguez, J. Y. Yang, P. Pierce, and R. Echols. 2003. Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., abstr. G-1848.
- 16.Hamed, K. A., L. Sher, A. Arguedas, M. Husseman, D. Biswas, P. Pierce, and R. Echols. 2003. Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., abstr. G-1850.
- 17.Reference deleted.
- 18.Huczko, E., B. Conetta, D. Bonner, L. Valera, T. Stickle, A. Macko, and J. Fung-Tomc. 2000. Susceptibility of bacterial isolates to gatifloxacin and ciprofloxacin from clinical trials 1997-1998. Int. J. Antimicrob. Agents 16:401-405. [DOI] [PubMed] [Google Scholar]
- 19.Jones, R. N., C. M. Rubino, S. M. Bhavnani, and P. G. Ambrose. 2003. Worldwide antimicrobial susceptibility patterns and pharmacodynamic comparisons of gatifloxacin and levofloxacin against Streptococcus pneumoniae: report from the Antimicrobial Resistance Rate Epidemiology Study Team. Antimicrob. Agents Chemother. 47:292-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones, R. N., D. J. Biedenbach, M. E. Erwin, M. L. Beach, and M. A. Pfaller. 1999. Activity of gatifloxacin against Haemophilus influenzae and Moraxella catarrhalis, including susceptibility test development, E-test comparisons, and quality control guidelines for H. influenzae. J. Clin. Microbiol. 37:1999-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones, R. N., D. M. Johnson, M. E. Erwin, M. L. Beach, D. J. Biedenbach, M. A. Pfaller, et al. 1999. Comparative antimicrobial activity of gatifloxacin tested against Streptococcus spp. including quality control guidelines and etest method validation. Diagn. Microbiol. Infect. Dis. 34:91-98. [DOI] [PubMed] [Google Scholar]
- 22.Lacreta, F. P., G. D. Kollia, G. Duncan, and D. M. Grasela. 2001. Lack of effect of food on the bioavailability of gatifloxacin. J. Infect. Dis. Pharmacother. 4:43-54. [Google Scholar]
- 23.LaCreta, F. P., S. Kaul, G. D. Kollia, G. Duncan, D. M. Randall, and D. M. Grasela. 2000. Interchangeability of 400-mg intravenous and oral gatifloxacin in healthy adults. Pharmacotherapy 20:59S-66S. [DOI] [PubMed] [Google Scholar]
- 24.Leibovitz, E., L. Piglansky, S. Raiz, D. Greenberg, K. A. Hamed, J. M. Ledeine, J. Press, A. Leiberman, R. M. Echols, P. F. Pierce, M. R. Jacobs, and R. Dagan. 2003. Bacteriologic and clinical efficacy of oral gatifloxacin for the treatment of recurrent/nonresponsive acute otitis media: an open label, noncomparative, double tympanocentesis study. Pediatr. Infect. Dis. J. 22:943-949. [DOI] [PubMed] [Google Scholar]
- 25.Leibovitz, E., C. Rubino, B. Damle, B. Cirincione, G. Duncan, D. Grasela, K. Hamed, R. Echols, and R. Dagan. 2003. Abstr. 43rd Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-14.
- 26.Lober, S., S. Ziege, M. Rau, G. Schreiber, A. Mignot, P. Koeppe, and H. Lode. 1999. Pharmacokinetics of gatifloxacin and interaction with an antacid containing aluminum and magnesium. Antimicrob. Agents Chemother. 43:1067-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mouton, J. W., M. L. van Ogtrop, D. Andes, and W. A. Craig. 1999. Use of pharmacodynamic indices to predict efficacy of combination therapy in vivo. Antimicrob. Agents Chemother. 43:2473-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakashima, M., T. Uematsu, K. Kosuge, H. Kusajima, T. Ooie, Y. Masuda, R. Ishida, and H. Uchida. 1995. Single- and multiple-dose pharmacokinetics of AM-1155, a new 6-fluoro-8-methoxy quinolone, in humans. Antimicrob. Agents Chemother. 39:2635-2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reference deleted.
- 30.Schwartz, G. J., L. P. Brion, and A. Spitzer. 1987. The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr. Clin. N. Am. 34:571-590. [DOI] [PubMed] [Google Scholar]
- 31.Sheiner, L. B. 1984. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab. Rev. 15:153-171. [DOI] [PubMed] [Google Scholar]
- 32.Tett, S. E., N. H. G. Holford, and A. J. McLachlan. 1998. Population pharmacokinetics and pharmacodynamics: an underutilized resource. Drug Inf. J. 32:693-710. [Google Scholar]
- 33.Vozeh, S., J. L. Steimer, M. Rowland, P. Morselli, F. Mentre, L. P. Balant, and L. Aarons. 1996. The use of population pharmacokinetics in drug development. Clin. Pharmacokinet. 30:81-93. [DOI] [PubMed] [Google Scholar]
- 34.Wise, R., J. M. Andrews, J. P. Ashby, and J. Marshall. 1999. A study to determine the pharmacokinetics and inflammatory fluid penetration of gatifloxacin following a single oral dose. J. Antimicrob. Chemother. 44:701-704. [DOI] [PubMed] [Google Scholar]