

Abstract
Beta defensins comprise a family of cationic, cysteine-rich antimicrobial peptides, predominantly expressed at epithelial surfaces. Previously we identified a unique five-cysteine defensin-related peptide (Defr1) that, when synthesized, is a mixture of dimeric isoforms and exhibits potent antimicrobial activity against Escherichia coli and Pseudomonas aeruginosa. Here we report that Defr1 displays antimicrobial activity against an extended panel of multidrug-resistant nosocomial pathogens for which antimicrobial treatment is limited or nonexistent. Defr1 fractions were collected by high-pressure liquid chromatography and analyzed by gel electrophoresis and mass spectrometry. Antimicrobial activity was initially investigated with the type strain Pseudomonas aeruginosa PAO1. All fractions tested displayed equivalent, potent antimicrobial activity levels comparable with that of the unfractionated Defr1. However, use of an oxidized, monomeric six-cysteine analogue (Defr1 Y5C), or of reduced Defr1, gave diminished antimicrobial activity. These results suggest that the covalent dimer structure of Defr1 is crucial to antimicrobial activity; this hypothesis was confirmed by investigation of a synthetic one-cysteine variant (Defr1-1cys). This gave an activity profile similar to that of synthetic Defr1 but only in an oxidized, dimeric form. Thus, we have shown that covalent, dimeric molecules based on the Defr1 β-defensin sequence demonstrate antimicrobial activity even in the absence of the canonical cysteine motif.
The emergence of multidrug-resistant bacterial pathogens continues to reduce the antibiotic options available to clinicians in the treatment of serious nosocomial infections. This threat particularly applies to pathogens responsible for infections in intensive care units; these include methicillin-resistant Staphylococcus aureus, Pseudomonas aeruginosa, Enterococcus faecalis, and Acinetobacter baumannii (15). The increased prevalence of resistant P. aeruginosa in cystic fibrosis lung infections and high-level innate resistance in other opportunistic pathogens, including the Burkholderia cepacia complex, Stenotrophomonas maltophilia, Candida albicans, and Ralstonia species, are also causes for concern (1). Clearly, there is a need for novel antimicrobial agents.
β-Defensins are cysteine-rich, cationic peptides displaying important roles within the mammalian innate immune system (6). In vitro and in vivo studies have confirmed that defensins have potent broad-spectrum bactericidal activity and suggest that bacterial resistance may have coevolved with, and influenced the diversity of, antimicrobial peptides (2, 8, 11). In addition, β-defensins have been shown to act upon T lymphocytes and immature dendritic cells playing key roles in the adaptive immune response (19). β-Defensins produced as propeptides are processed to a mature secreted peptide, which contains six canonical cysteine residues (cys1-cys5, cys2-cys4, cys3-cys6) with specific spacing and intramolecular disulfide connectivity (18). These are distinct from the disulfide connectivities displayed by the similar α-defensins (17). The mature β-defensins are 30 to 45 amino acids in length and are amphipathic, containing discrete cationic and hydrophobic areas.
A mechanism for defensin binding and disruption of bacterial membranes is the subject of intensive study (12). Active peptides have been shown to display an appropriate balance of hydrophobicity and net positive charge (5). The conserved disulfide bridges impose a structural core composed of β-sheets, and nonconserved residues on the surface are subject to selective pressure against rapidly evolving bacteria (10, 14). In direct contrast to the perceived structure-function relationship(s), however, it has been reported that antimicrobial activity is independent of intramolecular disulfide bridging (18).
Recently we described a murine β-defensin gene present in C57BL/6 mice that encoded a peptide with only five cysteine residues (Table 1) (9). This peptide (Defr1) displays a tyrosine in place of the first cysteine and yet still retains potent antimicrobial activity against gram-positive and gram-negative organisms. The gene is a variant allele of Defb8 which encodes six cysteines and is present in all the other inbred murine strains we have tested. A synthetic analogue of Defr1 (Defr1 Y5C) in which the tyrosine is restored to a cysteine exists as a noncovalent homodimer formed by monomers with β-defensin connectivity but displays poor antimicrobial activity (3). In contrast, we found that synthetic Defr1 was a mixture of isoforms with variable disulfide connectivities but that each isoform contained a single intermolecular S-S bond. We found that the antimicrobial activity was influenced by the covalent dimeric structure imposed by this novel defensin motif (3).
TABLE 1.
Properties of Defr1 and related peptidesa
| Peptide | Sequence | Charge | ΔG (kcal mol−1) |
|---|---|---|---|
| Defr1 | DPVTYIRNGGICQYRCIGLRHKIGTCGSPFKCCK | +6 (+12) | −1.92 (−3.84) |
| Defr1 Y5C | DPVTCIRNGGICQYRCIGLRHKIGTCGSPFKCCK | +6 (+12) | −3.13 (−6.26) |
| Defr1-1cys | DPVTYIRNGGIAQYRAIGLRHKIGTAGSPFKCAK | +6 (+12) | −3.56 (−7.12) |
Amino acid sequence, charge, and hydrophobicity (ΔG) values as calculated by Wimley and White (16) are given for Defr1, Defr1Y5C, and Defr1-1cys. Values are for the monomeric forms of the peptides, with values for the dimeric forms in brackets. Boldface letters indicate the position of cysteine residues in a canonical six cysteine β-defensin motif.
In this study we evaluated the antimicrobial activity of Defr1 against a panel of bacteria and fungi selected to represent important multidrug-resistant nosocomial pathogens. Reverse-phase high-pressure liquid chromatography (HPLC) was performed using the synthetic preparation of Defr1, and 14 fractions were collected for examination of the activity of the specific isoforms of the peptide. Additionally, sodium dodecyl sulfate-polyacrylamide gel electrophoresis and mass spectrometry were used to examine the structures of the peptides within these fractions, all of which possess similar antimicrobial properties. Reduced Defr1 has poor antimicrobial activity, implying that its disulfide-linked, dimeric structure is important for activity (3); therefore, we synthesized a peptide with only one cysteine that formed a dimer of defined structure. This one-cysteine variant (Defr1-1cys) in an oxidized, dimeric form gave an activity profile similar to that of synthetic Defr1; however, this activity was greatly reduced after the single S-S bond was broken using dithiothreitol (DTT). Thus, this synthetic peptide based on the Defr1 β-defensin sequence shows potent antimicrobial activity in the absence of the characteristic β-defensin cysteine motif but only in an oxidized, disulfide-linked, covalent dimer form.
MATERIALS AND METHODS
Materials.
Defr1 Y5C, Defr1, and Defr1-1cys were chemically synthesized by the standard solid-phase methodology (CSS Albachem Ltd.) and were refolded and oxidized in air as described previously (3). Polymyxin B sulfate was purchased from Sigma.
Antimicrobial assays.
Test organisms were grown to midlogarithmic phase in Iso-Sensitest broth (Oxoid) growth media and then diluted to 1 × 106 to 5 × 106 CFU/ml in 10 mM potassium phosphate containing 1% (vol/vol) Iso-Sensitest broth, pH 7.4. Different concentrations of test peptide were incubated in 100 μl of cells (1 × 105 to 5 × 105 CFU) at 37°C for 3 h. Reduction of the peptides, where performed, was done by adding 10 mM DTT and incubating at room temperature overnight. The oxidation state of each peptide was determined by mass spectrometry. Serial dilutions (10-fold) of the incubation mixture were spread on Iso-Sensitest plates and incubated at 37°C, and the CFU levels were determined the following day. The minimum bactericidal concentration (MBC) is the concentration of peptide at which we observed >99.99% killing of the initial inoculum. All assays were performed in triplicate on three independent occasions. The MBC was obtained by taking the mean of all results, and experimental errors were within one doubling dilution.
The effect of salt on antimicrobial activity was tested by incubating 100 μl of 1 × 105 CFU of P. aeruginosa PAO1 in 10 mM potassium phosphate-1% (vol/vol) Iso-Sensitest (pH 7.4), which contained various concentrations of NaCl (0 to 200 mM). The bacteria were then challenged with peptide at various concentrations (doubling dilutions from 0 to 150 μg/ml), and the MBC for each concentration was determined.
Defr1 fractionation-HPLC.
Preparative reverse-phase HPLC for this study was performed by CSS Albachem Ltd. The peptide was fractionated with a gradient of 20% to 30% acetonitrile over 60 min on a Jupiter C5 Semi-Prep column (Phenomenex, Macclesfield, United Kingdom) (250 by 10 mm) at a flow rate of 5 ml/min. Fractions (5 ml) (i.e., 1 min) were collected throughout the region of interest in order to sample across the chromatographic peak (data not shown). The fractions were lyophilized, weighed, and resuspended in water to give solutions of 1 mg/ml.
Gel electrophoresis.
Electrophoresis of Defr1, Defr1 fractions, and Defr1-1cys was performed under reducing and native, nonreducing conditions. Peptide (2.5 μg) was dissolved in 2.5 μl of 0.01% acetic acid-5 μl of 2× sample buffer (Novex Tricine-sodium dodecyl sulfate sample buffer LC1676). Reduction of samples was performed by adding 10 mM of NuPAGE sample-reducing agent (Invitrogen NP0004) and incubating at room temperature for 1 h. The entire sample was loaded on a 16% Tricine gel (Invitrogen). The gel was fixed and stained with Coomassie blue stain.
Mass spectrometry.
All data were taken on a Q-ToF mass spectrometer (Micromass) fitted with a nanoelectrospray source. Nanospray tips were pulled using a Sutter P97 tip puller with a voltage applied through a short length of platinum wire. Samples were prepared at 10 μM in 1:1 MeOH/H2O-0.1% formic acid.
Analytical HPLC data.
Peptides were separated on a Phenomenex Jupiter C5 column (250 by 2 mm) with a 20% to 30% acetonitrile gradient over 60 min at 0.2 ml/min by use of a Waters 2795 LC system attached to a Micromass Platform II mass spectrometer. Synthetic Defr1 was prepared at 20 μM in 1:1 MeOH/H2O, with a total injection of 40 μl.
RESULTS
Antimicrobial activity of Defr1 and Defr1 Y5C against a panel of pathogens.
We have extended our previous study where we investigated the antimicrobial activity and specificity of Defr1 (3), and here we used sensitive reference strains of Escherichia coli, P. aeruginosa, and Bordetella bronchiseptica and an extended panel which represented multidrug-resistant isolates of major nosocomial pathogens (Table 2). These included four nonclonal multidrug-resistant isolates of P. aeruginosa responsible for epidemic outbreaks of respiratory infection in individuals with cystic fibrosis. Polymyxin B was also included as a reference cationic peptide presently in clinical use.
TABLE 2.
Antimicrobial activity of Defr1 against a spectrum of gram-positive and gram-negative organisms and a fungus
| Organism and straina | Description or origin | MBC (μg/ml)b
|
||||
|---|---|---|---|---|---|---|
| Pol | Defr1 | Defr1 Y5C | Defr1-1cys
|
|||
| Oxidized | Reduced | |||||
| Gram-negative bacteria | ||||||
| P. aeruginosa | ||||||
| PAO1 | Reference strain | 0.5 | 6 | 50 | 6 | >50 |
| C3425 | Manchester | 1 | 6 | 50 | ||
| H183 | Liverpool | 1 | 6 | 100 | ||
| C4269 | Brisbane | 1 | 6 | >100 | ||
| C3781 | Melbourne | 0.5 | 6 | 100 | ||
| E. coli | ||||||
| ATCC 25922 | Reference strain | 0.25 | 8 | >100 | 12 | >50 |
| B. cepacia | ||||||
| ATCC 25416 | Type strain | >512 | >100 | >100 | ||
| B. cenocepacia | ||||||
| ATCC BAA-245 | Type strain | >512 | >100 | >100 | >100 | >100 |
| S. maltophilia | ||||||
| C1980 | Clinical isolate | 32 | 12.5 | >100 | ||
| C3625 | Clinical isolate | 32 | 6 | >100 | ||
| C3626 | Clinical isolate | 32 | 3 | >100 | ||
| C3627 | Clinical isolate | 64 | 6 | >100 | ||
| B. bronchiseptica | ||||||
| J3083 | Reference strain | 0.12 | 6 | >100 | ||
| R. pickettii | ||||||
| R4050/C3079 | Reference strain | >512 | 3 | >100 | ||
| R. eutropha | ||||||
| ATCC 17697 | Reference strain | 128 | 3 | >100 | ||
| A. baumannii | ||||||
| ATCC 19606 | Type strain | 1 | 3 | 12.5 | 6 | >50 |
| Gram-positive bacteria | ||||||
| E. faecalis ATCC 700802 | Reference strain | 256 | 6 | 100 | 12 | >50 |
| S. aureus ATCC 25923 | Reference strain | 256 | 10 | >100 | 12 | >50 |
| S. aureus J2918-MRSAc | Clinical isolate | 64 | 6 | >100 | ||
| Fungus | ||||||
| C. albicans J2922 | Clinical isolate | >512 | 3 | 25 | ||
Resistance phenotypes of clinical isolates for ciprofloxacin (Cip), meropenem (Mer), tobramycin (Tob) colistin (Col), and methicillin (Met): strain C3425, Mer resistant [Mer-R], Col-R; H183, Tob-R; C4269, Tob-R; C3781, Mer-R, Tob-R; C1980, Cip-R, Mer-R, Tob-R; C3625, Cip-R, Mer-R, Col-R; C3626, Cip-R, Mer-R; C3627, Mer-R, Tob-R; J2918, Cip-R, Met-R.
Values represent the MBCs for oxidized Defr1, Defr1 Y5C, and polymyxin (Pol) as a control. MBC data for Defr1 and Y5C against PAO1, E. coli ATCC 25922, S. aureus ATCC 25923, E. faecalis ATCC 292126, and C. albicans J2922 have been previously reported (3) and are repeated here as comparators.
MRSA, methicillin-resistant Staphylococcus aureus.
Synthetic Defr1 showed bactericidal activity against the majority of bacterial pathogens investigated and also antifungal activity against C. albicans (Table 2). MBCs were generally less than 10 μg/ml for both gram-positive and gram-negative bacteria. In separate experiments, bactericidal activity of Defr1 was shown to occur rapidly, within 30 min of exposure (data not shown). Polymyxin B showed good activity against P. aeruginosa, E. coli, and B. bronchiseptica but poor activity against other members of the strain panel. Only the B. cepacia and B. cenocepacia isolates showed high-level resistance to Defr1 (MBC > 100 μg/ml), a result which is in agreement with the characteristic resistance of these bacteria to conventional antibiotics and AMPs (13). We previously reported the bactericidal activity of Defr1 against B. cenocepacia J2315, with which, even at 50 μg/ml of peptide, only 40% killing of the bacterial inoculum was achieved (9). Importantly, in this new study the antimicrobial activities of Defr1 against both multiresistant and susceptible pathogens were similar, and in all cases, the activity of 5-cysteine Defr1 was greater than that of the 6-cysteine analogue (Defr1 Y5C). Indeed, when tested against P. aeruginosa, E. coli, S. aureus, methicillin-resistant Staphylococcus aureus, and S. maltophilia, the Defr1 disulfide dimer exhibited 10-fold-greater bactericidal activity.
In order to determine whether any particular fraction of the Defr1 preparation contained a more active isoform, we resynthesized Defr1 and attempted to resolve the isoforms by separation using reverse-phase HPLC and a shallow elution gradient. We collected 14 5-ml fractions, and each was lyophilized, analyzed by native gel electrophoresis on a 16% Tricine gel (Fig. 1 upper panel), and subsequently tested for antimicrobial activity against the P. aeruginosa type strain PAO1.
FIG. 1.

Analysis of fractions from reverse-phase HPLC separation of Defr1. Upper panel: native gel electrophoresis of Defr1 fractions in 16% Tricine gels. The 5-ml fractions taken from the HPLC preparation of Defr1 were subjected to electrophoresis in nondenaturing gels. Lower panel: electrospray ionization-mass-spectrum ion envelopes of fractions 2, 9, and 13 from the Defr1 HPLC. Spectrum a corresponds to fraction 2, spectrum b corresponds to fraction 9, and spectrum c corresponds to fraction 13. The mass of the dominant species (indicated above each x axis by an “A” followed by a numeral) is given for each spectrum; the mass values determined agree with the mass expected for the fully disulfide bridged form of the ion in each case, with an accuracy of ±1 Da. Peaks are labeled “An,” with the indicated numbers corresponding to the individual charge states given by the molecular ions of A in the form (M + nH)n+. Some small impurity peaks are noted. Similar spectra were obtained for all fractions despite their having somewhat dissimilar gel mobilities.
Each fraction apparently displayed a different band intensity of between ∼5 to 13 kDa on the 16% Tricine gel, perhaps due to the presence of different isoforms within the sample, a phenomenon we had observed previously (3). Fractions 2, 9, and 13 were selected due to their noticeably different mobilities and were subjected to electrospray ionization mass spectrometry analysis (Fig. 1, lower panel) and in the reduced form after treatment with DTT (data not shown). The ion series results were essentially identical for all fractions, with each peptide producing a series of ions of similar charges and intensities (between [M + 7H]7+ and [M + 10H]10+). The deconvoluted mass measured for each fraction is consistent with the theoretical mass (7,567.0 Da), suggesting that each contained a fully oxidized Defr1. This mass analysis revealed that despite apparent mobility differences on the native, Tricine gel, the three fractions chosen were all dimeric forms of the peptide, with masses which corresponded to a state in which all of their cysteines are oxidized, i.e., representing a fully disulfide-bridged, dimeric peptide, where the dimer is formed via an intermolecular disulfide bridge. All of the fractions displayed LC profiles similar to that of the unfractionated parental Defr1 (data not shown); combined with the heterogeneity observed in the gel electrophoresis profile across the elution gradient, this suggests that the Defr1 mixture is complex and difficult to resolve.
Each of the 14 fractions in Fig. 1 was tested for antimicrobial activity, and each had an MBC of 6 μg/ml, which is the same as that of the parental preparation of Defr1. Treatment of the peptide fractions with excess DTT resulted in the loss of the dimer structure (confirmed by mass spectrometry; data not shown) and a corresponding marked decrease in antimicrobial activity to an MBC greater than 50 μg/ml (Fig. 2 and 3).
FIG. 2.
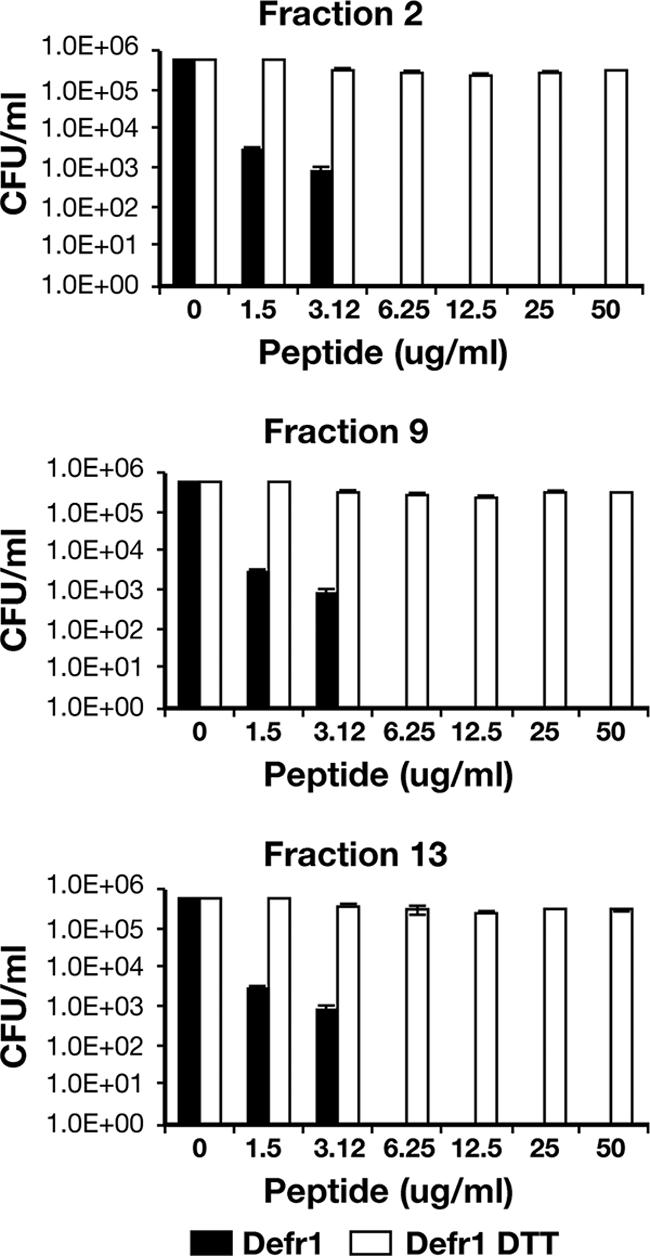
Antimicrobial activity before and after DTT treatment of fractions 2, 9, and 13. The data represent the numbers of CFU of P. aeruginosa PAO1 surviving after incubation with each of the three reduced and unreduced Defr1 fractions at the concentrations shown. All assays were performed in triplicate and were repeated on two independent occasions.
FIG. 3.
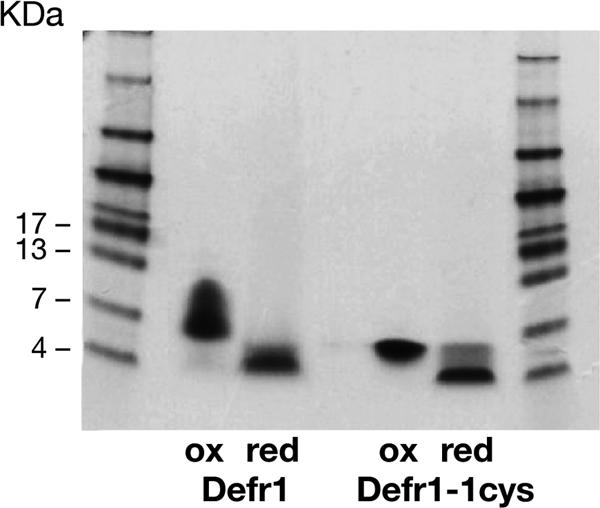
Gel electrophoresis of Defr1-1cys showing dimerization and reduction (red) to a monomeric form with DTT. Unfractionated, oxidized (ox) Defr1 and Defr1-1cys were analyzed on a Tricine gel before and after reduction with 10 mM DTT.
Antimicrobial activity of a single-cysteine dimer.
In an attempt to confirm the relationship between covalent dimerization and potent antimicrobial activity, a one-cysteine variant (Defr1-1cys) was synthesized (Table 1). The penultimate cysteine was retained, as this was considered to be the cysteine without a partner in the five-cysteine peptide Defr1 when canonical β-defensin disulfide connectivities (C1-C5, C2-C4, C3-C6) were assumed. The other cysteines in the peptide were replaced with an alanine. Following exposure to air, Defr1-1cys spontaneously forms a disulfide-bridged dimer with a mass of 7,318.4 Da (theoretical mass, 7,318.54 Da) as opposed to the theoretical value of 7,320.5 Da for a non-cysteine-bridged noncovalent dimer (data not shown). Defr1-1cys was tested for antimicrobial activity against a selection of gram-positive and gram-negative organisms (Table 2).
Defr1-1cys displays antimicrobial activity identical to that of Defr1 against the panel of organisms tested. Only the gram-negative strain B. cenocepacia J2315 was not killed at a low MBC. Again upon reduction in the presence of DTT, the dimer structure of Defr1-1cys was lost (Fig. 3) and the antimicrobial activity was diminished (Table 2).
The salt sensitivity of Defr1 is unusual, as this cationic peptide's antimicrobial action against P. aeruginosa is largely insensitive to salt concentration levels. The salt sensitivity of Defr1-1cys was also tested and was found to be equivalent to that previously reported for Defr1 (3, 9). In NaCl-free conditions, the MBC was 6 μg/ml. At 50 and 100 mM NaCl, 25 μg/ml of Defr1-1cys peptide effectively killed the 105 bacteria in the MBC assay. At 150 mM and 200 mM NaCl, 50 μg/ml of the single cysteine peptide was required to kill the bacteria.
DISCUSSION
Defr1 is a murine beta defensin-like peptide expressed in the airways, testis, and heart (9). This peptide lacks the first cysteine of the characteristic six-cysteine canonical motif. Analyses of functional characteristics show this peptide to possess antimicrobial activity against both gram-positive and gram-negative organisms. The precise mechanism of action of antimicrobial peptides is unclear but is thought to include electrostatic binding and disruption of bacterial cell membranes (12). Defr1 forms a covalent dimeric structure using an intermolecular disulfide bond and to date has been considered a unique example within the defensin family (3, 9).
Defr1 is shown here to express potent antimicrobial activity against a range of bacterial pathogens, including multidrug-resistant nosocomial isolates; the exceptions are B. cepacia and B. cenocepacia, which are characteristically resistant to antimicrobial peptides. In comparison, Defr1 Y5C, the peptide analogue which retains the canonical β-defensin connectivity (cys1-cys5, cys2-cys4, cys3-cys6) and has been shown previously by mass spectrometry analysis not to be a covalent dimer (3), displays poor antimicrobial activity against the same panel of organisms.
Synthetic Defr1 has been shown previously to exhibit various disulfide connectivities (3). In an attempt to identify active fractions, samples from a reverse-phase HPLC column were collected, analyzed by native gel electrophoresis, and investigated for antimicrobial activity. All fractions displayed visibly different but similar band intensities on a Tricine gel (Fig. 1, upper panel) and yet retained antimicrobial activities equivalent to that seen with the unfractionated Defr1. Further analysis of fractions 2, 9, and 13 was conducted due to their noticeable mobility differences on gel electrophoresis. Analysis by mass spectrometry showed that all fractions possessed a dominant molecular species which had a mass corresponding to a covalently bound dimeric form of the Defr1 peptide (Fig. 1, lower panel), although some small additional peaks in each fraction were present due to synthetic impurities and nonspecific adducts formed in the electrospray process. LC analyses of the synthetic Defr1 and of resolved fractions from it resulted in chromatograms which exhibit very close correlation (data not shown). Their chromatographic profiles also resemble that observed with our first batch of synthetic Defr1 (data not shown).
Upon reduction of both the fractions and the parental preparation of Defr1, antimicrobial activity was greatly reduced (Fig. 2 and reference 3), indicating that a covalent dimeric structure is necessary for the potent antimicrobial function associated with the Defr1 sequence. This is supported by the fact that the noncovalent dimer Defr1 Y5C with β-defensin connectivity displays poor antimicrobial activity even in the oxidized form (3).
Studies of the relationship between the structure and function of defensins have begun to appear. For example, Pazgier et al. (12) have shown that the antimicrobial activity of fully oxidized β-defensin HBD1 against E. coli is dominated by cationic residues at the C terminus of the peptide. In an earlier study, Wu et al. had demonstrated that a linear isomer of HBD3 (where all the cysteines had been replaced with α-aminobutyric acid, so these residues of the isomer were unable to form disulfide bonds) still retained its antimicrobial activity against a panel of pathogens (18). Interestingly, de Leeuw et al. reported that killing of E. coli by α-defensin HD5 is independent of the peptide structure whereas the activity against S. aureus is structure dependent (4). It appears that it is difficult to explain the bactericidal properties of individual defensins against specific pathogens solely based on their primary structures.
In the work presented here, the hypothesis that the antimicrobial activity of Defr1 appears to depend on covalent dimerization of the molecule was confirmed by the activity of Defr1-1cys. In this peptide, every cysteine except the penultimate one is replaced with an alanine residue (Table 1). Importantly, this molecule also forms a disulfide-linked dimer and displays very similar antimicrobial activity, which is diminished in the reduced, monomeric from. A similar phenomenon was also observed in the naturally-occurring Sushi 3 peptide derived from the Sushi 3 domain of Factor C, the lipopolysaccharide-sensitive serine protease of the horseshow crab coagulation cascade (7). The antimicrobial peptides of the Sushi peptide (34 amino acids, with a single cysteine) were shown to be due to its interaction with lipopolysaccharide and correlated directly with its ability to form a single S-S dimer.
Charge and hydrophobicity have been shown previously to be important in the antimicrobial function of HBD3 derivatives (5). However, the different antimicrobial activities of Derf1 and Defr1-1cys versus Defr1 Y5C are not easily explained by simple differences in their physical properties, since all three peptides have similar charge and hydrophobicity characteristics (Table 1). The three peptides Defr1, Defr1 Y5C, and Defr1-1cys have a cationic charge of +12 in the dimeric form, and they share similar hydrophobic scores (−1.92, −3.13, and −3.56 kcal mol−1, respectively) on a scale which measures the partitioning of peptides at a membrane to aqueous-phase interface. Common to Defr1 and Defr1-1cys and absent in Defr1 Y5C is a covalent intermolecular S-S bond which appears to be the governing factor in imparting potent bactericidal activity for these peptides.
Synthetic peptides based on the Defr1 β-defensin sequence are antimicrobially potent even in the absence of the canonical cysteine motif in a covalent, dimeric form. The mode of action, toxicity, and in vivo efficacy of this naturally occurring murine AMP remain to be determined. However, our results suggest that further studies of these novel peptide antibiotics containing this simple S-S structural motif may provide a source of optimized dimers capable of killing multiresistant pathogens.
Acknowledgments
This research was supported by the EPSRC, the Royal Society, the Cystic Fibrosis Trust UK, the MRC, and the University of Edinburgh.
We thank Bob Bateman and Waters Micromass Technologies and the British Mass Spectrometry Society. We thank both Nick Hastie for his enthusiasm for this project and Pat Langridge-Smith.
We have no financial or other relationships that present a conflict of interest with respect to this study.
Footnotes
Published ahead of print on 12 March 2007.
REFERENCES
- 1.Al-Aloul, M., J. Crawley, C. Winstanley, C. A. Hart, M. J. Ledson, and M. J. Walshaw. 2004. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax 59334-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bals, R., M. J. Goldman, and J. M. Wilson. 1998. Mouse beta-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect. Immun. 661225-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campopiano, D. J., D. J. Clarke, N. C. Polfer, P. E. Barran, R. J. Langley, J. R. Govan, A. Maxwell, and J. R. Dorin. 2004. Structure-activity relationships in defensin dimers: a novel link between beta-defensin tertiary structure and antimicrobial activity. J. Biol. Chem. 27948671-48679. [DOI] [PubMed] [Google Scholar]
- 4.de Leeuw, E., S. R. Burks, X. Li, J. P. Kao, and W. Lu. 2007. Structure-dependent functional properties of human defensin 5. FEBS Lett. 581515-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klüver, E., S. Schulz-Maronde, S. Scheid, B. Meyer, W. G. Forssmann, and K. Adermann. 2005. Structure-activity relation of human beta-defensin 3: influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry 449804-9816. [DOI] [PubMed] [Google Scholar]
- 6.Lehrer, R. I. 2004. Primate defensins. Nat. Rev. Microbiol. 2727-738. [DOI] [PubMed] [Google Scholar]
- 7.Li, P., T. Wohland, B. Ho, and J. L. Ding. 2004. Perturbation of lipopolysaccharide (LPS) micelles by Sushi 3 (S3) antimicrobial peptide. The importance of an intermolecular disulfide bond in S3 dimer for binding, disruption, and neutralization of LPS. J. Biol. Chem. 27950150-50156. [DOI] [PubMed] [Google Scholar]
- 8.Morrison, G., F. Kilanowski, D. Davidson, and J. Dorin. 2002. Characterization of the mouse Beta defensin 1, Defb1, mutant mouse model. Infect. Immun. 703053-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison, G. M., M. Rolfe, F. M. Kilanowski, S. H. Cross, and J. R. Dorin. 2002. Identification and characterization of a novel murine beta-defensin-related gene. Mamm. Genome 13445-451. [DOI] [PubMed] [Google Scholar]
- 10.Morrison, G. M., C. A. Semple, F. M. Kilanowski, R. E. Hill, and J. R. Dorin. 2003. Signal sequence conservation and mature peptide divergence within subgroups of the murine beta-defensin gene family. Mol. Biol. Evol. 20460-470. [DOI] [PubMed] [Google Scholar]
- 11.Moser, C., D. J. Weiner, E. Lysenko, R. Bals, J. N. Weiser, and J. M. Wilson. 2002. β-Defensin 1 contributes to pulmonary innate immunity in mice. Infect. Immun. 703068-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pazgier, M., D. M. Hoover, D. Yang, W. Lu, and J. Lubkowski. 2006. Human beta-defensins. Cell. Mol. Life Sci. 631294-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahly, H., S. Schubert, J. Harder, P. Rautenberg, U. Ullmann, J. Schroder, and R. Podschun. 2003. Burkholderia is highly resistant to human beta-defensin 3. Antimicrob. Agents Chemother. 471739-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semple, C. A., A. Maxwell, P. Gautier, F. M. Kilanowski, H. Eastwood, P. E. Barran, and J. R. Dorin. 2005. The complexity of selection at the major primate beta-defensin locus. BMC Evol. Biol. 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent, J. L. 2003. Nosocomial infections in adult intensive-care units. Lancet 3612068-2077. [DOI] [PubMed] [Google Scholar]
- 16.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3842-848. [DOI] [PubMed] [Google Scholar]
- 17.Wu, Z., B. Ericksen, K. Tucker, J. Lubkowski, and W. Lu. 2004. Synthesis and characterization of human alpha-defensins 4-6. J. Pept. Res. 64118-125. [DOI] [PubMed] [Google Scholar]
- 18.Wu, Z., D. M. Hoover, D. Yang, C. Boulegue, F. Santamaria, J. J. Oppenheim, J. Lubkowski, and W. Lu. 2003. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human beta-defensin 3. Proc. Natl. Acad. Sci. USA 1008880-8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, D., O. Chertov, S. N. Bykovskaia, Q. Chen, M. J. Buffo, J. Shogan, M. Anderson, J. M. Schroder, J. M. Wang, O. M. Howard, and J. J. Oppenheim. 1999. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286525-528. [DOI] [PubMed] [Google Scholar]