

Abstract
We present a novel approach for comparing 16S rRNA gene clone libraries that is independent of both DNA sequence alignment and definition of bacterial phylogroups. These steps are the major bottlenecks in current microbial comparative analyses. We used direct comparisons of taxon density distributions in an absolute evolutionary coordinate space. The coordinate space was generated by using alignment-independent bilinear multivariate modeling. Statistical analyses for clone library comparisons were based on multivariate analysis of variance, partial least-squares regression, and permutations. Clone libraries from both adult and infant gastrointestinal tract microbial communities were used as biological models. We reanalyzed a library consisting of 11,831 clones covering complete colons from three healthy adults in addition to a smaller 390-clone library from infant feces. We show that it is possible to extract detailed information about microbial community structures using our alignment-independent method. Our density distribution analysis is also very efficient with respect to computer operation time, meeting the future requirements of large-scale screenings to understand the diversity and dynamics of microbial communities.
Frequency analysis of phylogroups is the most widely used approach for studying structures in communities for higher organisms (14). Similar approaches have also been adapted for comparisons of microbiological clone libraries (25). The problem, however, is that there are no natural species barriers for asexual prokaryote species and thereby no rationale criteria for phylogroup definitions (9). Thus, a dependence on phylogroups severely limits the ability to describe and understand microbial communities (5). Furthermore, most of the microbial biodiversity is actually found within environmental clone libraries in which it is difficult to determine the boundaries of the phylogroups (8).
The challenge with phylogroup definitions has recently been addressed through direct microbial community comparison utilizing DNA sequence alignment-based phylogenetic reconstructions (12, 13, 21, 22, 24). The problem with DNA sequence alignments and phylogenetic reconstruction, however, is that large data sets cannot readily be analyzed. The reasons for this are both that there is no objective criterion for determining the correctness of an alignment and that the number of possible phylogenetic trees increases exponentially with the number of taxa analyzed. There are currently nearly 300,000 16S rRNA gene sequences in public databases, and that number is estimated to double every 7 months (8). Relying on alignments would therefore not suit the future needs for microbial community analyses.
We present a novel approach for comparing microbial communities that is phylogroup and DNA sequence alignment independent. Our concept is based on describing the evolutionary relatedness between bacteria in an absolute multidimensional coordinate space, using multimer transformation in combination with principal component analyses (20). The microbial community analyses are subsequently performed by direct comparisons of frequency distribution landscapes, representing densities of taxa within an absolute coordinate space. Density distribution comparisons are very efficient with respect to computer operation time (CPU), enabling analyses of very large clone libraries. A further benefit of our approach is that we can directly apply multivariate statistical tools, such as multivariate analyses of variance (MANOVA) and multivariate regression for the microbial community comparisons. We have also developed a permutation-based method for determining the significance of local differences in taxon distributions within the absolute coordinate space.
It is well known that gastrointestinal (GI) bacteria have a major impact on human health (1). Particularly important is the effect of the initial colonization on the maturation of the immune system of infants (2, 7, 10, 17). The aim of our work was to evaluate our density distribution analysis as a framework for determining structures in the human microbiota. This was carried out by reanalyzing the extensive human bacterial clone library provided by Eckburg et al. (6). We present a detailed description of significant differences in the microbiota with respect to different persons and to mucosal tissues from the six major subdivisions of the colon in addition to fecal samples. We also present initial results for an in-house-generated clone library for infant fecal microbiota, showing differences for age and mode of delivery (caesarean or vaginal). Finally, we compare our density distribution approach with the commonly used tools for microbial community analyses.
MATERIALS AND METHODS
Fecal samples.
From 14 children born at Østfold Hospital (Fredrikstad, Norway), fecal samples were collected at the ages of less than 1 month old and 4 months old. The sampling was performed during autumn 2002 and spring 2003. Nine of the children were delivered vaginally, and five were delivered by caesarian section. The mode of delivery, however, was confounded with prematurity (birth before week 38). All of the children delivered by caesarean section were premature, while only two of the children delivered by vaginal birth were premature. All the children were either fully or partly breast fed. There is no record, however, of the use of milk supplements.
Fresh stool samples were immediately frozen at −20°C. The samples were then transported to the microbial community test laboratory. Upon arrival, the samples were dissolved in 5 ml of 50 mM glucose, 25 mM Tris-HCl, and 10 mM EDTA. The samples were stored at −40°C and processed further within 1 month.
DNA purification, PCR amplification, cloning, and DNA sequencing.
Fecal suspensions were thawed on ice and slightly vortexed, and the bacteria in 500 μl of the suspension were disrupted mechanically for 40 s at maximum speed using 0.5 g 106-μm glass beads (Sigma-Aldrich, Steinheim, Germany) in a Fast-Prep bead beater (Bio 101, La Jolla, CA). DNA was then purified with the DNeasy tissue kit (QIAGEN, Hilden, Germany) following the manufacturer's recommendations, eluting the DNA in a 100-μl volume. Three independent DNA purifications were carried out for each fecal sample.
We used the primers 5′ TCCTACGGGAGGCAGCAGT 3′ (forward) and 5′ GGACTACCAGGGTATCTAATCCTGTT 3′ (reverse), targeting generally conserved 16S rRNA gene regions (16). These primers generate an amplicon of 466 bp, corresponding to the region between residues 331 and 797 when applied to the Escherichia coli 16S rRNA gene. We chose this amplicon because the quantitative properties are well documented. The relatively short amplicon is a trade-off between the robustness of the PCR and the phylogenetic information gained (19). Short sequences are also relatively resistant to the generation of chimerical amplicons. PCR amplification, cloning, and DNA sequencing were performed as previously described (19).
Clone libraries.
The human adult clone library consists of 11,831 clones. A detailed description of this library has previously been published (6). The infant clone library consists of 390 clones, and details about this library are described here. The composition of the library was of 108 sequences from children aged less than 1 month and 282 sequences from 4-month-old children. There were 218 sequences from children delivered by vaginal birth and 172 from children delivered by caesarean section for the mode of delivery category. This library has been deposited in the GenBank database (accession no. EF063741 to EF064130).
AIBIMM.
The sequences were transformed into multimer frequencies (n = 5) by the in-house-developed computer program PhyloMode (www.matforsk.no/web/sampro.nsf/downloadE/Microbial_community). The transformation was based on sliding a window of 5 nucleotides along a DNA sequence and counting the frequencies of the different multimers encountered. The sizes of the multimer windows were chosen as tradeoffs between detecting phylogenetic signals (homologous multimer equalities), avoiding base composition biases due to nonhomologous multimer equalities, and the requirements for computer operation time (20). The multimer frequency data were compressed using principal component analysis (PCA) as previously described for the alignment-independent bilinear multivariate modeling (AIBIMM) approach (20). The phylogenetic content in the PCA was evaluated by cross-validation, while the potential presence of chimerical sequences was determined empirically by conflicting multimer loadings.
AIBIMM is related to the Tetra approach previously published by Teeling et al. (23). The main difference, however, is that Tetra is designed for the detection of skewed tetranucleotide distribution in whole genomes, while AIBIMM is designed to detect phylogenetic signals in single genes (20).
A special consideration when using AIBIMM is that the sequences should have approximately the same starting and ending points. If the sequences have different lengths, then this should be corrected for by weighting using the “normalize data” option in PhyloMode, so that the weighted numbers of multimers are equal for all taxa. Clustering in the PCA plot indicates close relatedness between the taxa if the residual variance is low, while if the residual variance is high, clustering could be because the taxa are not separated within the model. Deep branches can also be difficult to resolve since perfect matches for the entire multimers are required for a phylogenetic signal. A detailed description for the phylogenetic interpretation of the AIBIMM data is given by Rudi et al. (20).
MANOVA.
Variance analyses were performed directly on the multimer frequency data by using the 50-50 MANOVA software ([11] www.matforsk.no/ola). Classical MANOVA tests perform poorly in cases with several highly correlated responses, and the tests collapse when the number of responses exceeds the number of observations (which is the case for the multimer data). In 50-50 MANOVA, the dimensionality of the data is reduced by using principal component decompositions and the final tests are still based on the classical test statistics and their distributions (11).
Multivariate regression.
The covariance between the Y and X matrices (sample information and multimer frequencies, respectively) was determined by using partial least-squares (PLS) regression. Briefly, PLS regression models both the X and the Y matrices simultaneously to find the latent variables in X that will best predict the latent variables in Y. The PLS regression analyses were performed using the multivariate statistics software package Unscrambler (CAMO Technologies, Inc., Woodbridge, NJ). The calibrated model was validated using random cross-validation, a process in which 5% of randomly chosen samples were kept out during validation, and the process is repeated 20 times (for details, see the Unscrambler user manual; CAMO Technologies, Inc., Woodbridge, NJ).
fLAND.
Our calculations with respect to frequency landscape distribution (fLAND) analyses were performed using MATLAB (MathWorks, Natick, MA). We have also developed a computer program for making fLAND analyses more easily accessible for microbiologists. The program can be downloaded from www.matforsk.no/fLAND. It includes a user manual and a version of the program that is dependent on MATLAB and a stand-alone version that does not require MATLAB.
In the work presented here, fLANDs were obtained by counting the number of taxa within each (0.5 by 0.5) interval for the two first PCs in a global AIBIMM model (20). The distributions were then transformed to represent relative frequencies. The relative frequency distribution for each interval fLANDij is given by the following equation:
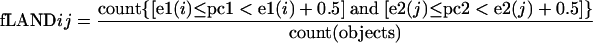 |
where [e1(i),e2(j)] are the coordinates of the lower left edge of the fLANDij interval in the scatter plot formed by pc1 and pc2, “count” represents the sum of the objects (in this case taxa) satisfying the criteria given in the parenthesis, and “objects” represents all taxa.
The relative frequency of samples belonging to different categories within each interval was obtained by counting the taxa belonging to each category and dividing this number by the total number of taxa assigned to the same category using the formula described above. To find the difference fLANDdiffij among all categories from k = 1 to K in one specific interval ij, we summarized the squared difference between the relative frequencies for each category and the average frequencies for all categories. We used the following formula:
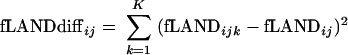 |
In order to calculate the probability of observing a difference at least this large between categories “just by chance,” we performed significance testing by permutations (15). We generated 999 random data sets. The taxon count for each category and each interval remained the same, but the taxon assignment to categories was permuted. For each interval ij, the following hypotheses were tested:
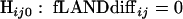 |
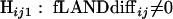 |
The null hypothesis Hij0 for a specific fLANDij stated that there was no significant difference between the categories in interval ij. The distribution of fLANDdiffij,random represented the sampling distribution under the condition that Hij0 was true. The P value was calculated as the probability that fLANDdiffij,random had a value at least as extreme as fLANDdiffij,observed:
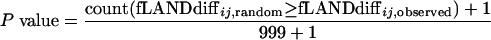 |
This is the probability of obtaining differences at least as large as the observed difference given Hij0. The one extra occurrence added to both the numerator and the denominator is representing the fLANDdiffij,observed itself. In our significance determinations, we excluded all segments with only one taxon in the original data in order to lower the risk of false discovery with respect to type I errors given the small data sets. We did not, however, use advanced corrections for multiple testing due to the risk of increasing the probability of type II errors and because correction for multiple testing is a controversial issue (18).
RESULTS AND DISCUSSION
Overall compositions of the human GI microbiota.
The fLAND analyses showed a relatively structured description of the diversity of the dominating bacterial groups in the human adult feces and colon (Fig. 1A). In particular, there was a very clear differentiation of the Bacteroidetes from the rest of the bacteria. Bacteria belonging to the Firmicutes, on the other hand, were separated into three groups. The Clostridia were separated into two distinct groups (1 and 2), while the rare Firmicutes formed a relatively loose association (designated Firmicutes/misc) with the other rare bacteria identified in the samples (6). The bacteria in this group were not separated by the principal components used for the fLAND analyses due to their low abundance. A total of 579 out of 6,400 segments in the fLAND density plot contained one or more taxa, while 141 segments contained only single taxa. Surprisingly, only 22 segments contained 46% of the taxa in the library. Hierarchical clustering for the first three principal components further supported the group structure of the density distribution data (Fig. 1B). The reason for not using more components in the dendrogram construction is that our main interest is to describe the microbial community structure for the abundant bacterial groups and not the phylogenetic reconstruction of the complete bacterial assemblage.
FIG. 1.

Global fLAND distribution (A) and the phylogenetic position of the major bacterial groups (B) in human adult microbiota. (A) A 0.5-by-0.5-interval density distribution based on AIBIMM analyses for the 11,831 taxa from the human adult clone library is shown. The major bacterial groups identified are marked. The color coding represents the natural logarithm of the densities within each segment. (B) Complete linkage dendrogram based on Euclidean distances for the first three principal components (explaining 52% of the variance). The dendrogram is based on a single taxon from each of the 579 segments. The dendrogram is collapsed to represent the same group structure as for the density distribution plot. Groups with low explained variance are red, while groups with high explained variance are black.
The infant fecal microbiota was dominated by two segments within Bifidobacterium, constituting 34% of all the taxa analyzed. A total of 104 out of 2,500 segments in the fLAND density plot contained one or more taxa, while 60 segments contained single taxa (Fig. 2A). Due to the relatively low abundance, the Bacteroidetes showed low separation levels for the first two PCs. The Bacteroidetes, however, were well separated in the third PC (results not shown).
FIG. 2.

Global fLAND distribution (A) and differences with respect to age (B) and mode of delivery (C) for human infant microbiota. (A) A 0.5-by-0.5-interval density distribution based on AIBIMM analyses for the 390 taxa from the human infant clone library is shown. The major bacterial groups identified are marked. The color coding represents the natural logarithm of the densities within each segment. Shown is a natural logarithm for the ratio between the libraries with respect to age (B) and mode of delivery (C). Segments with significant differences (P < 0.05) are marked. The following color coding was used: for age (B), green indicates an age of less than 1 month and red indicates an age of 4 months; for mode of delivery (C), green indicates delivery by caesarean section and red indicates vaginal delivery.
Structures in the human GI microbiota.
Using fLAND, we found very high individual variations in the human adult microbiota (Fig. 3). The structure in these data was that person A was dominated by Clostridia (groups 1 and 2) and Bacteroidetes III. Person B had a relatively wide level of diversity of bacteria within the Firmicutes/misc group, in addition to Bacteroidetes I. Person C had a relatively low diversity level, with a dominance of Bacteroidetes. The PLS regression confirmed these observations. The PLS model required three PCs to explain approximately 30% of the total variance with respect to persons (Y variance). The validated correlation coefficient was 0.54.
FIG. 3.

fLAND intervals with significant (P < 0.01) differences between persons A, B, and C. The relative distribution (corrected for the differences in library size) is shown. The significance threshold was determined by using permutation testing as described in Materials and Methods. The coordinates for the interval's lower left edge are shown in parentheses as (PC1, PC2). The total number of taxa for each interval is also shown.
The spatial distribution of taxa within the colon showed that person B had the highest number of 27 significant fLAND intervals, while person C showed an intermediate number of 24 intervals and person A had the lowest number with 17 intervals (Fig. 4). However, when excluding the segments with overrepresentation from the fecal libraries, the numbers are more equal, with 13 intervals for person A, 14 for person B, and 11 for person C.
FIG. 4.

fLAND intervals with significant (P < 0.01) differences for each person (A, B, or C) with respect to the seven sample types analyzed. The relative distribution (corrected for the differences in library size) is shown with respect to each person (A, B, or C). The significance threshold was determined by using permutation testing as described in Materials and Methods. The coordinates for the interval are shown in parentheses as (PC1, PC2). The total number of taxa for each interval is also shown.
The overall pattern was that there was an overrepresentation of bacteria belonging to the Firmicutes/misc for the fecal bacteria, while the Clostridia group 1 was underrepresented. The Bacteroidetes group, on the other hand, contained segments that were both over- and underrepresented in the fecal library. Using PLS regression, we found a correlation between fecal and mucosal libraries for all three persons, except for person B with the transverse colon, with the fecal libraries having the highest correlations (Fig. 5) .
FIG. 5.

Cross-validated PLS regression coefficients for the respective libraries for each subject. The PLS regression analysis was carried out as described in Materials and Methods. Abbreviations: AC, ascending colon; C, cecum; DC, descending colon; R, rectum; SC, sigmoid colon; TC, transverse colon.
For data from the human adult library, we were not able to run the 50-50 MANOVA using a computer with a 3-GHz Pentium 4 processor and 1 GB memory due to the size of the library. The 50-50 MANOVA program has been developed for purposes other than microbial community comparisons (11), so the limitation could be that the program has not been optimized for the analysis of such large data sets. The infant library, however, is much smaller, enabling MANOVA with our desktop computers. By using MANOVA, we found significant variance in the data related to all of the categories of samples analyzed (age and mode of delivery) (Table 1).The ages of the children explained 3.30% of the variance (P < 0.01), while the mode of delivery explained 0.75% (P = 0.05) and the interaction between age and mode of delivery explained 0.54% (P < 0.01). Most of the variance (95.4%) in the data, however, could not be explained by the categories evaluated. This is probably due to high stochastic variation in the microbiota among the children analyzed. We obtained a high linear correlation between age and the microbiota using PLS regression (validated correlation coefficient of 0.87). There were also relatively strong correlations between the total microbiota and the mode of delivery category with a validated correlation coefficient of 0.30. fLAND analyses showed that there were nine segments in the fLAND density plots that were significantly different between the two age categories analyzed (Fig. 2B). The segments at coordinates [given as (PC1, PC2)] (1, 1), (2, 1), and (2, 2) were classified as Escherichia using an RDP II hierarchical classifier, while the segment (13, 31) was classified as Bacteroides and the segments (45, 20), (46, 20), (48, 20), (49, 21), and (50, 20) were classified as Bifidobacterium. We also found significant differences for five segments with respect to mode of delivery (Fig. 2C). According to the RDP II hierarchical classifier, the segment (13, 30) belongs to Bacteroides, segment (13, 39) belongs to Staphylococcaceae, and segments (47, 19), (47, 20), and (50, 20) belong to Bifidobacterium.
TABLE 1.
Explained variance and significance by 50-50 MANOVA for the infant clone library
| Category | Explained variance (%) | df | P value |
|---|---|---|---|
| Age | 3.3 | 1 | <0.01 |
| Mode of delivery | 0.75 | 1 | 0.05 |
| Mode of delivery and age | 0.54 | 1 | <0.01 |
| Error | 95.4 | 383 |
Statistical testing of microbial community comparisons in an absolute coordinate space.
The statistical tests applied for our multimer frequency data highlight different aspects of microbial community structures. MANOVA determines the total explained variance in the data with respect to the categories analyzed (11). The regression analyses, on the other hand, show how well the categories can be predicted from the community data (3), and local differences in taxon distributions within an absolute coordinate space can be determined by fLAND analyses.
A lack of rigid statistical testing has been a major obstacle in identifying biological phenomena in microbial communities. Two recent reports have addressed the issue of statistical testing within large 16S rRNA gene clone libraries from intestinal samples using alignment-based microbial community comparisons (6, 12). These reports illustrate the complexity of the statistical analyses for the phylogenetically reconstructed microbial community data that are based on relative pair-wise comparisons. Obviously, absolute distances are much easier to compare than relative distances are. Basing the microbial comparisons on an absolute coordinate space would therefore simplify the comparative analyses.
Comparison of tools for microbial community analyses.
The human adult clone library has already been extensively investigated using the DOTOUR program for phylotype determinations and ∫ LIBSHUFF for microbial community comparisons (6). We used these data in comparison with our alignment-independent method. Due to the relatively good coverage in the RDP II database for the bacteria found in the infant intestine, we used this library for RDP II comparison.
The DOTUR program identified 395 bacterial phylotypes from aligned sequence data, while we identified 579 intervals with one or more taxa in our fLAND analysis. This illustrates that our density distribution analyses gives a separation that is slightly higher than the phylotype determinations. The ∫ LIBSHUFF analyses showed no significant mucosal library differences for the same subject with two exceptions. The library from the ascending colon from subject A was a subset of the other libraries, and the descending colon was a subset of the ascending colon for subject B (6). A notable difference between the fLAND and the ∫ LIBSHUFF analyses was that bacteria within the fLAND segment (73, 31) showed a significant overrepresentation (P < 0.01) in the ascending colon for subject A compared to those for the rest of the mucosal sites (Fig. 4). This is in contrast to the conclusion that the microbiota in the ascending colon is a subset of the other libraries.
The RDP II classifier cannot be used for libraries with a relatively large portion of bacteria that are not well characterized (see the user recommendation at the RDP II homepage, rdp.cme.msu.edu). This is certainly the case for the human adult library. For the infant library, however, the RDP II classifier gave only 1.3% unassigned strains in the bacterial domain. We therefore used the infant library for evaluating RDP II Library Compare. The major difference between fLAND analyses and RDP II Library Compare is that Library Compare did not separate the two dominating segments of Bifidobacterium; consequently, it did not detect the major structures in our data related to these groups.
A summary of commonly used approaches for microbial community comparisons is presented in Table 2. Most of the microbial community comparison tools available are based on comparing phylogenetically reconstructed data based on DNA sequence alignments using a relative distance measure (13, 21, 22). The available alignment-independent tools, on the other hand, are generally based on predefined models for known categorical groups (4). Our fLAND analysis is different from the other approaches with respect to the combination of alignment independence, phylogenetic description, and the use of absolute distances.
TABLE 2.
Properties of microbial community comparison tools
Conclusion.
Using the fLAND approach, we are able to give a detailed description of the human GI microbiota for both infants and adults. Coordinate-based classification systems are compatible with relation databases (20). Since the coordinate space is absolute, direct comparisons for any microbial communities are possible using, e.g., a universal fLAND coordinate system (covering the known bacterial biodiversity). The fLAND distribution analyses are also very efficient with respect to CPU time and have high resolution, enabling analyses of very large sets of data. Thus, future databases based on coordinate classification may enable global comparisons of microbial communities. In this way, we can start to understand global distribution patterns for bacteria in the human gut and in other microbial ecosystems.
Acknowledgments
This work was supported by Helse Øst, Hedmark Sparebank, and a research levy on certain agricultural products.
We thank P. B. Eckburg and D. A. Relman for providing the human microbiota data set.
Footnotes
Published ahead of print on 2 March 2007.
REFERENCES
- 1.Bäckhed, F., R. E. Ley, J. L. Sonnenburg, D. A. Peterson, and J. I. Gordon. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915-1920. [DOI] [PubMed] [Google Scholar]
- 2.Björkstén, B., E. Sepp, K. Julge, T. Voor, and M. Mikelsaar. 2001. Allergy development and the intestinal microflora during the first year of life. J. Allergy Clin. Immunol. 108:516-520. [DOI] [PubMed] [Google Scholar]
- 3.Bjørnstad, A., F. Westad, and H. Martens. 2004. Analysis of genetic marker-phenotype relationships by jack-knifed partial least squares regression (PLSR). Hereditas 141:149-165. [DOI] [PubMed] [Google Scholar]
- 4.Cole, J. R., B. Chai, R. J. Farris, Q. Wang, S. A. Kulam, D. M. McGarrell, G. M. Garrity, and J. M. Tiedje. 2005. The ribosomal database project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294-D296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curtis, T. P., and W. T. Sloan. 2004. Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr. Opin. Microbiol. 7:221-226. [DOI] [PubMed] [Google Scholar]
- 6.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 308:1635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favier, C. F., E. E. Vaughan, W. M. De Vos, and A. D. Akkermans. 2002. Molecular monitoring of succession of bacterial communities in human neonates. Appl. Environ. Microbiol. 68:219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank, D. N., and N. R. Pace. 2005. Another ribosomal RNA sequence milestone—and a call for better annotation. ASM News 71:501-502. [Google Scholar]
- 9.Gevers, D., F. M. Cohan, J. G. Lawrence, B. G. Spratt, T. Coenye, E. J. Feil, E. Stackebrandt, Y. V. de Peer, P. Vandamme, F. L. Thompson, and J. Swings. 2005. Re-evaluating prokaryote species. Nat. Rev. Microbiol. 3:733-739. [DOI] [PubMed] [Google Scholar]
- 10.Kirjavainen, P. V., T. Arvola, S. J. Salminen, and E. Isolauri. 2002. Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning? Gut 51:51-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langsrud, O. 2002. 50-50 multivariate analysis of variance for collinear responses. J. Royal Stat. Soc. Ser. D 51:305-317. [Google Scholar]
- 12.Ley, R. E., F. Backhed, P. Turnbaugh, C. A. Lozupone, R. D. Knight, and J. I. Gordon. 2005. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102:11070-11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozupone, C., and R. Knight. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228-8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magurran, A. E. 2005. Ecology: linking species diversity and genetic diversity. Curr. Biol. 15:R597-R599. [DOI] [PubMed] [Google Scholar]
- 15.Moore, D. S., and G. P. McCabe. 2005. Introduction to the practice of statistics, 5th ed. W. H. Freeman, New York, NY.
- 16.Nadkarni, M. A., F. E. Martin, N. A. Jacques, and N. Hunter. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257-266. [DOI] [PubMed] [Google Scholar]
- 17.Park, H. K., S. S. Shim, S. Y. Kim, J. H. Park, S. E. Park, H. J. Kim, B. C. Kang, and C. M. Kim. 2005. Molecular analysis of colonized bacteria in a human newborn infant gut. J. Microbiol. 43:345-353. [PubMed] [Google Scholar]
- 18.Perneger, T. V. 1998. What's wrong with Bonferroni adjustments. BMJ 316:1236-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudi, K., T. Maugesten, S. E. Hannevik, and H. Nissen. 2004. Explorative multivariate analyses of 16S rRNA gene data from microbial communities in modified-atmosphere-packed salmon and coalfish. Appl. Environ. Microbiol. 70:5010-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudi, K., M. Zimonja, and T. Næs. 2006. Alignment independent bi-linear multivariate modeling (AIBIMM) for global analyses of 16S rRNA phylogeny. Int. J. Syst. Evol. Microbiol. 56:1565-1575. [DOI] [PubMed] [Google Scholar]
- 21.Schloss, P. D., and J. Handelsman. 2006. Introducing TreeClimber, a test to compare microbial community structures. Appl. Environ. Microbiol. 72:2379-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schloss, P. D., B. R. Larget, and J. Handelsman. 2004. Integration of microbial ecology and statistics: a test to compare gene libraries. Appl. Environ. Microbiol. 70:5485-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teeling, H., J. Waldmann, T. Lombardot, M. Bauer, and F. O. Glockner. 2004. TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics 5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Templeton, A. 1983. Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the evolution of humans and apes. Evolution 37:221-244. [DOI] [PubMed] [Google Scholar]
- 25.Wang, X., S. P. Heazlewood, D. O. Krause, and T. H. Florin. 2003. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J. Appl. Microbiol. 95:508-520. [DOI] [PubMed] [Google Scholar]