

Abstract
Cocaine, amphetamines and other psychostimulants inhibit synaptic dopamine uptake by interfering with dopamine transporter (DAT) function. The resultant potentiation of dopaminergic neurotransmission is associated with psychostimulant addiction. Fluctuations in dopamine uptake inhibition potency (DUIP) were observed for classical DAT blockers including cocaine, mazindol, methylphenidate (Ritalin™) and benztropine in CHO cells expressing wildtype DAT; cocaine potency also decreased in DAT-expressing non-neuronal COS-7 cells and neuronal N2A neuroblastoma cells. In contrast, the DAT substrate (+)-amphetamine did not display this DUIP fluctuation. In parallel experiments, no fluctuation was observed for the apparent binding affinities of these 5 drugs. The DUIP decrease appeared to correlate with an increase in cell surface DAT expression level, as measured by Bmax values and confocal microscopy. The fact that the DUIP profile of amphetamine diverged from that of the classical DAT blockers is consistent with the idea of fundamental differences between the mechanisms of abused psychostimulant DAT substrates and inhibitors. Identification of the cellular factors that underlie the DAT inhibitor DUIP fluctuation phenomenon may be relevant to anti-psychostimulant drug discovery efforts.
Keywords: Cocaine, amphetamine, dopamine, transporter, drug abuse, addiction
1. Introduction
The euphoric and addictive properties of cocaine and amphetamine are linked to binding of these abused drugs at the dopamine transporter (DAT) protein (Fischer and Cho, 1979; Ritz et al., 1987; Spealman et al., 1989; Giros et al., 1996). Cocaine and other classic DAT inhibitors such as mazindol, methylphenidate and benztropine block dopamine uptake from the synapse and thus potentiate dopamine receptor-mediated signal transduction in the nucleus accumbens and other areas of the brain associated with drug reward/reinforcement (Wise and Bozarth, 1985). The DAT substrate amphetamine also increases synaptic dopamine levels, but by mediating dopamine efflux from the presynaptic cell via the DAT (Fischer and Cho, 1979; Sitte et al., 1998). Mapping the DAT binding site(s) directly linked to the physiological actions of cocaine and amphetamine is a priority for advancing efforts in developing anti-psychostimulant medications. Identifying such sites, however, may be a complex undertaking, as DAT inhibitor binding affinity profiles containing two or more components have been frequently observed (Madras et al., 1989; Boja et al., 1991; Boja et al., 1992; Rothman et al., 1994; Reith and Coffey, 1994; Gracz and Madras, 1995). Indeed, whether a given psychostimulant’s DAT high affinity binding site and dopamine uptake inhibition site are identical is debatable.
The premise of a single site for both high affinity cocaine binding and physiologically relevant cocaine inhibition of dopamine uptake implies that assays measuring each should yield the same potency value if conducted identically. Instead, a lack of correlation between DAT inhibitor binding affinity and dopamine uptake inhibition potency (DUIP) at the wildtype (WT) DAT has been noted (Pristupa et al., 1994; Eshleman et al., 1999; Wang et al., 2003). Moreover, cocaine DUIP values may differ by almost a thousand fold (Reith and Coffey, 1994; Eshleman et al., 1999). In some cases, binding/uptake inhibition potency discrepancies and DUIP variation are likely due to differences in assay conditions (e.g., temperature, ionic strength, incubation period, cell intactness) or cell/tissue type (Rothman et al., 1993). In at least one case (Wang et al., 2003), however, the potency discrepancies at WT DAT were observed using identically conducted assays and intact cells from the same stable CHO cell line. Intriguingly, the same study demonstrated that a conservative point mutation (D79E) in transmembrane 1 of the DAT significantly decreased binding affinities for methylphenidate, mazindol and the cocaine analog WIN 35,428, yet the DUIPs for these blockers were unaffected. The decoupling of DAT high affinity binding and DUIP for most blockers tested again suggests that two or more DAT sites, conformations or populations may be responsible for the observed pharmacologic profiles (Wang et al. 2003; Ukairo et al., 2005).
In the course of the above WT DAT CHO cell studies, it was noticed that the DUIP of cocaine appeared to fluctuate as a function of whether the cultured cells were “old” or “new” as defined by the number of twice weekly trypsin-mediated dilutions of the cell monolayer (i.e., "high" or "low" passage number). No such fluctuation in the binding affinity of cocaine, as measured by [3H]-WIN 35,428 displacement, was obvious for the same cells. In the present work, the cocaine DUIP fluctuation phenomenon was more thoroughly characterized and the investigation extended to other DAT blockers and amphetamine. Three possible influences on DAT function at the level of the cultured cell were initially addressed: the age of the cell line (measured by cell passage number), the density of the cell monolayer (i.e., percent confluence), and the effect of varying DAT expression level by manipulation of transfection conditions. To ensure that comparisons between DUIP and apparent binding affinity were legitimate for a given DAT inhibitor, [3H]-dopamine uptake assays, binding assays involving the cocaine analog [3H]-WIN 35,428, and versions of each assay that included competitor nonradioactive DAT blockers were conducted under identical conditions. To address in part the physiological relevance of such a phenomenon, neuronal as well as non-neuronal cell lines were similarly tested for DAT inhibitor DUIP fluctuations.
2. Results
Empirical observations suggested that the dopamine uptake inhibition potency (DUIP) of cocaine at wildtype (WT) DAT-bearing cultured cells was not constant. Specifically, the DUIP of a given DAT blocker seemed to fluctuate for a CHO cell line stably transfected with the WT DAT. This fluctuation appeared to be a property of the cell's age, measured by the number of cell "passages", or twice weekly trypsin-mediated dilutions of the cell monolayer. This hypothesis was more rigorously addressed by testing in parallel "low", "medium" and "high" passage WT DAT CHO cells, of arbitrarily set ranges of cell passages 9–20, 25–36, and 40–54, respectively. Indeed, a trend of decreasing cocaine DUIP with increasing cell passage number was observed, with a statistically significant difference detected between the cocaine DUIPs at low and high passage cells. In contrast, binding assays conducted in parallel indicated that the apparent binding affinity of cocaine at the WT DAT CHO cells did not vary with cell passage (Fig. 1 and Table 1). The [3H]-dopamine uptake inhibition and [3H]-WIN 35,428 displacement assays were conducted under identical conditions.
Fig. 1.
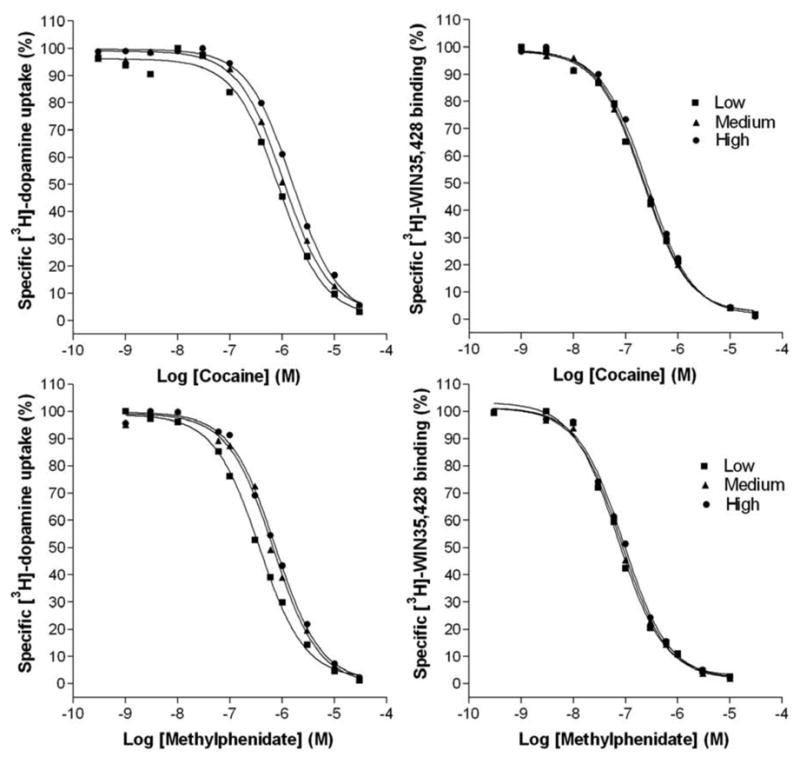
Cocaine (top) or methylphenidate (bottom) inhibition of [3H]-dopamine uptake (left) or [3H]-WIN 35,428 binding (right) under identical conditions at WT DAT CHO cells of “low” (squares), “medium” (triangles) or “high” (circles) passage number. The data are representative of at least 3 independent experiments.
Table 1.
Dopamine uptake inhibition potencies and binding affinities of DAT ligands at WT DAT CHO cells as a function of cell passage number
| Cell Passage | |||
|---|---|---|---|
| Low | Medium | High | |
| [3H]-DA uptake inhibition | IC8050 (nM) | ||
| Cocaine | 719 ± 94 | 1055 ± 52 | 1503 ± 127a |
| Mazindol | 44 ± 4 | 74 ± 5 | 93 ± 17a |
| Methylphenidate | 352 ± 17 | 640 ± 22 | 662 ± 45a |
| Benztropine | 236 ± 40 | 395 ± 45 | 413 ± 27a |
| (+)-Amphetamine | 671 ± 88 | 756 ± 86 | 906 ± 39 |
| [3H]-WIN 35,428 inhibition | Ki (nM) | ||
| Cocaine | 179 ± 22 | 197 ± 22 | 251 ± 28 |
| Mazindol | 15 ± 2 | 19 ± 2 | 20 ± 2 |
| Methylphenidate | 74 ± 7 | 72 ± 5 | 85 ± 6 |
| Benztropine | 78 ± 6 | 85 ± 9 | 92 ± 10 |
| (+)-Amphetamine | 524 ± 31 | 478 ± 52 | 557 ± 78 |
| [3H]-WIN 35,428 binding | |||
| Kd (nM) | 17 ± 1 | 17 ± 2 | 18 ± 1 |
| Bmax (pmol/mg) | 9 ± 2 | 11 ± 2 | 19 ± 3a |
P < 0.05 versus low passage cells for that assay (one-way ANOVA, Newman-Keuls post hoc test)
Inhibition of [3H]-dopamine uptake or [3H]-WIN35,428 binding by classical DAT blockers under identical conditions at WT DAT CHO cells of different passage number. IC50 and Ki values were derived from experiments incubating the stably-transfected cells with nonradioactive DAT inhibitors in the presence of [3H]-dopamine or [3H]-WIN 35,428 at 22°C in KRH buffer. Passages of 9–20, 25–36 and 40–54 were classified as “low”, “medium” and “high”, respectively. (The ranges for an individual drug were narrower, as testing with a given inhibitor was completed before the next inhibitor was addressed). Mean ± SEM for at least 3 independent experiments.
The classical DAT blockers methylphenidate (Fig. 1), mazindol and benztropine were similarly tested, as was (+)-amphetamine, a dopamine uptake inhibitor that is itself a DAT substrate. All three classic blockers mirrored the cocaine DUIP fluctuation pattern. Curiously, no significant DUIP fluctuation was observed for amphetamine, although a trend toward decreasing DUIP may be present. Binding affinities were unvarying for all of the aforementioned DAT inhibitors, as well as for WIN 35,428 (Table 1). As noted previously (Wang et al., 2003; Ukairo et al., 2005), the classic DAT blockers were more potent in inhibiting [3H]-WIN 35,428 binding than in inhibiting [3H]-dopamine uptake at the WT DAT; amphetamine was approximately equipotent under these conditions. The dichotomy between these DAT blockers and amphetamine was especially apparent from the ratio of IC50 and Ki values for uptake inhibition and apparent binding affinity, respectively (Table 2; Ki values for [3H]-dopamine uptake inhibition (not shown) were virtually identical to the Table 1 IC50 values, allowing direct comparison of uptake and binding inhibition constants). A statistically significant increase in the Vmax of dopamine uptake by high passage WT DAT CHO cells was also observed relative to low passage cells. The increase in net dopamine uptake by high passage cells appeared to be attributable to an increase in transporter number, evidenced by a concomitant increase in the Bmax value for WIN 35,428 binding at high passage cells. The DAT transport capacity did not vary appreciably between low and high passage cells (Fig. 2 and Table 3).
Table 2.
Ratios of dopamine uptake inhibition potency-to-binding affinity constants for DAT ligands at WT DAT CHO cells as a function of cell passage number
| DUIP:Affinity Ratio Cell Passage | |||
|---|---|---|---|
| Low | Medium | High | |
| Cocaine | 4.0 | 5.3 | 5.9 |
| Mazindol | 2.9 | 3.8 | 4.7 |
| Methylphenidate | 4.8 | 8.8 | 7.8 |
| Benztropine | 3.0 | 4.6 | 4.5 |
| (+)-Amphetamine | 1.3 | 1.6 | 1.6 |
Fig. 2.

Kinetics of WT DAT CHO cell dopamine uptake as a function of “low” (squares), “medium” (triangles) or “high” (circles) passage number. The data are representative of 4 independent experiments.
Table 3.
Kinetics of WT DAT CHO cell dopamine uptake as a function of cell passage number
| Cell Passage | |||
|---|---|---|---|
| Low | Medium | High | |
| Vmax (pmol/min/mg) | 681 ± 18 | 1136 ± 85 | 1645 ± 202a |
| Km (μM) | 1.1 ± 0.2 | 1.4 ± 0.2 | 2.0 ± 0.4 |
| Bmax (pmol/mg) | 9 ± 2 | 11 ± 2 | 19 ± 3a |
| Vmax/Bmax | 76/min | 103/min | 87/min |
P < 0.05 versus low passage cells for that assay (one-way ANOVA, Newman-Keuls post hoc test)
Km and Vmax values were derived from uptake experiments incubating WT DAT cells with dopamine at 0.1 Ci/mmol for 5 min at 22°C. Bmax values were derived from [3H]WIN35,428 saturation binding experiments. Mean ± SEM of at least 3 independent experiments.
To address whether the decrease in cocaine DUIP for high passage WT DAT CHO cells was simply due to higher DAT levels per cell monolayer, uptake and binding assays employed cell monolayers of different densities (% confluence). The monolayer was defined as 100% confluent when all cells appeared to contact neighboring cells so as not to leave open spaces on the culture dish. (A confluence level of 150% reflects overcrowding of the cell monolayer.) A comparison of WT DAT CHO cell monolayers at 20%, 100% and 150% confluence suggested that cell density had no bearing on either cocaine DUIP or cocaine binding affinity (Table 4). DAT levels were next manipulated with respect to individual cells.
Table 4.
Dopamine uptake inhibition potencies and binding affinities of cocaine at WT DAT CHO cells as a function of cell density (% confluence)
| Cell Density (Confluence) | |||
|---|---|---|---|
| 20% | 100% | 150% | |
| Cocaine | |||
| IC50 (nM) | |||
| [3H]-Dopamine uptake inhibition | 505 ± 108 | 534 ± 109 | 540 ± 62 |
| Ki (nM) | |||
| [3H]-WIN 35,428 inhibition | 260 ± 25 | 240 ± 12 | 201 ± 16 |
Cocaine inhibition of [3H]-dopamine uptake or [3H]-WIN 35,428 binding was measured under identical conditions at intact WT DAT CHO cell monolayers at 20%, 100%, and 150% confluence. Mean ± SEM for at least 3 experiments.
Naïve COS-7 cells were transiently transfected with a cDNA plasmid encoding the WT DAT; the amounts of plasmid employed in the transfections were 25, 50 or 100% of what is optimal. Cocaine DUIPs were significantly different between cells transfected with 25% and 100% of optimal plasmid levels. Parallel WIN 35,428 saturation binding analysis confirmed that Bmax values shifted in accordance with manipulation of plasmid level (Table 5). WIN 35,428 is not expected to cross the plasma membrane of the intact cell appreciably (Chen et al., 2004), meaning that the Table 5 Bmax values should reflect DAT expression at the cell surface. Nevertheless, confocal microscopic analysis corroborated the anticipated change in COS cell surface DAT expression (Fig. 3). Again, WIN 35,428 binding affinity was unaffected even though cocaine DUIP was altered (Table 5).
Table 5.
Dopamine uptake inhibition potency of cocaine and binding affinity of WIN 35,428 at COS-7 cells as a function of amount of WT DAT plasmid introduced
| WT DAT Plasmid (% of Optimal) | |||
|---|---|---|---|
| 25 | 50 | 100 | |
| [3H]-DA uptake inhibition | IC50 (nM) | ||
| Cocaine | 228 ± 7 | 353 ± 20 | 558 ± 75a |
| [3H]-WIN 35,428 binding | |||
| Kd (nM) | 12 ± 2 | 15 ± 2 | 14 ± 2 |
| Bmax (pmol/mg) | 0.8 ± 0.4 | 2.0 ± 0.5 | 4.0 ± 0.2a |
P < 0.05 versus "25% plasmid" cells for that assay (one-way ANOVA, Newman-Keuls post hoc test)
Transient transfections were carried out using 25%, 50% or 100% of the optimal amount of WT DAT - pIRES plasmid in 35 mm wells containing confluent monolayers of COS-7 cells controlled for passage number. Cocaine inhibition of [3H]-dopamine uptake as well as [3H]-WIN 35,428 binding were measured under identical assay conditions 48 hours after COS cell transfections. Mean ± SEM for 3 experiments.
Fig. 3.

Confocal microscopy of COS-7 cells transiently transfected with varying amounts of WT DAT cDNA plasmid. DAT protein was visualized with Alexa Fluor 488 (green signal; Panels A and B); cortical F-actin, a marker at the cell plasma membrane, was visualized with rhodamine phalloidin (red signal; Panels C and D). Shown are representative confocal images of four independent experiments. Panel E: Cell membrane signal intensities were quantitated for 10 fields corresponding to each of the 4 scenarios represented by Panels A–D. Scale bar = 8.00 μm for all images.
CHO and COS cells are non-neuronal; to better address the physiological significance of the cocaine DUIP fluctuation phenomenon, a neuronal cell line was tested. Naive N2A neuroblastoma cells were transiently transfected with the WT DAT plasmid using amounts 25 or 100% of optimal. The pharmacologic profile mimicked that from the analogous COS cell experiment in that cocaine DUIP was significantly altered by the plasmid titration; binding affinities for cocaine and WIN 35,428 were unaffected (Fig. 4 and Table 6). Again, Bmax values from parallel WIN 35,428 saturation binding analysis indicated that DAT expression was predictably shifted by the manipulation of plasmid level (Table 6). The findings verify that at least one neuronal cell line possesses the machinery to manifest the DUIP fluctuation phenomenon.
Fig. 4.

Cocaine inhibition of [3H]-dopamine uptake (left) or [3H]-WIN 35,428 binding (right) under identical conditions at N2A neuroblastoma cells transiently transfected with 25% (squares) or 100% (circles) of the optimal amount of WT DAT cDNA plasmid. The data are representative of at least 3 independent experiments.
Table 6.
Dopamine uptake inhibition potency and binding affinity of cocaine at N2A neuroblastoma cells as a function of amount of WT DAT plasmid introduced
| WT DAT Plasmid (% of Optimal) | ||
|---|---|---|
| 25 | 100 | |
| IC50 (nM) | ||
| [3H]-DA uptake inhibition | ||
| Cocaine | 476 ± 46 | 789 ± 64a |
| IC50 (nM) | ||
| [3H]-WIN 35,428 inhibition | ||
| Cocaine | 229 ± 34 | 196 ± 11 |
| [3H]-WIN 35,428 binding | ||
| Kd (nM) | 25 ± 2 | 18 ± 3 |
| Bmax (pmol/mg) | 1.3 ± 0.4 | 2.8 ± 0.5a |
P < 0.05 versus "25% plasmid" cells for that assay (one-way ANOVA, Newman-Keuls post hoc test
Transient transfections were carried out using 25% or 100% of the optimal amount of WT DAT - pIRES plasmid in 35 mm wells containing confluent monolayers of N2A neuroblastoma cells controlled for passage number. Cocaine inhibition of [3H]-dopamine uptake and [3H]-WIN 35,428 binding, as well as direct [3H]-WIN 35,428 binding, were measured under identical assay conditions 48 hours after N2A cell transfections. Mean ± SEM for 3 experiments.
3. Discussion
A relationship between DAT inhibitor DUIP and WT DAT CHO cell passage number was initially investigated only because cocaine DUIP appeared to vary with the number of weeks in cell culture. It is unclear whether increasing passage number of a cultured cell line can be correlated to cell aging. Moreover, the number of available DAT sites in the brain decreases with aging (Bannon and Whitty, 1997; Salvatore et al., 2003), in contrast with the present CHO cell findings. For each of the classical DAT blockers cocaine, mazindol, methylphenidate and benztropine, DUIP IC50 values differed significantly between “low passage” and “high passage” WT DAT CHO cells. Regardless of the number of uptake inhibition assays conducted, amphetamine did not display a statistically significant DUIP difference between these low and high passage cells, although a trend toward decreasing potency with higher passage may be present. In contrast, the binding affinities (measured by displacement of [3H]-WIN 35,428) of all 5 inhibitors were clearly not altered as a function of cell passage number (Table 1). The uptake inhibition:binding affinity constant ratio for amphetamine differed strikingly from that of the 4 nonsubstrate inhibitors (Table 2), possibly related to the fact that a nonsubstrate inhibitor ([3H]-WIN 35,428) was displaced in the binding assay. High passage cells displayed a Bmax value twice that of low passage cells in [3H]-WIN 35,428 saturation binding experiments, and this increase appeared to solely account for a concomitant dopamine uptake Vmax increase; dopamine transport turnover rate remained unchanged (Table 3 and Fig. 2).
It might be argued at this point that the increase in accessible DAT binding sites for high passage cells necessitated higher levels of DAT blocker for dopamine uptake inhibition, increasing the DUIP IC50 value. This explanation for the DAT blocker potency decrease can be ruled out by the observation of unvarying DUIP values for WT DAT CHO cells even though cell density differed by over 7 fold (Table 4; net [3H]-dopamine uptake differed by several fold between 20% and 150% confluent cells, proportional to the expected difference in surface DAT number - data not shown). Manipulating surface DAT levels for an individual cell, in contrast, did correlate with a shift in cocaine DUIP (Table 5 and Fig. 3), suggesting that DAT expression level has a direct bearing on the DUIP of cocaine, and probably that of the other classical DAT blockers. Reports that cocaine increases surface DAT levels (Little et al., 2002; Daws et al., 2002) while amphetamine decreases surface DAT levels (Saunders et al., 2000) are probably irrelevant to the present DUIP pattern differences (Table 1), which did not employ the high psychostimulant concentration and drug preincubation period necessary to induce DAT trafficking. On the other hand, amphetamine preincubation initially (within a minute or less) increases surface DAT levels (Johnson et al., 2005), a time course consistent with the present experimental conditions.
The fact that cocaine DUIP IC50 values fluctuate with cell state seems inconsistent with the idea that a single cocaine binding site of the DAT dictates dopamine uptake inhibition. More plausible is the possibility that two or more cocaine-recognizing DAT conformations or populations contribute to the uptake inhibition pharmacologic profile (Wang et al., 2003; Ukairo et al., 2005). The DAT adopts several transitional conformations in the course of one translocation cycle, which consists of binding and shuttling dopamine, Na+ and Cl− across the cell membrane into the cytoplasm and resetting the conformation so that the substrate pore is again extracellularly accessible (Rudnick, 1997). The distribution of DAT conformations can be altered by DAT mutation (Loland et al., 2002); similarly, this distribution may depend on cell state. Cocaine may bind to multiple DAT conformations, each with its own affinity for the drug. Thus, an IC50 value for dopamine uptake inhibition may reflect simultaneous inhibition at several DAT conformations. The scenario is also consistent with the invariance of cocaine binding affinity. The inhibitor binding assay only differs from the uptake inhibition assay in its lack of dopamine; thus, the DAT translocation cycle should be arrested at the Na+ and/or Cl− bound conformation that immediately precedes loading of dopamine. All DAT molecules would be suspended in this conformation, yielding an unvarying binding affinity constant. The situation is less clear regarding amphetamine. As a substrate, the translocation cycle would not arrest as with cocaine; the amphetamine DUIP value may simply reflect the concentration at which dopamine access to the initial DAT conformation has been reduced (by displacement) by 50%. If so, the DUIP would not be expected to change for amphetamine, or any substrate that similarly inhibited dopamine uptake. Testing whether the amphetamine DUIP pattern extends to other DAT substrates is a future direction of the work.
The DAT conformation distribution hypothesis alone is insufficient, however, in explaining why DUIP values vary as a function of cell state (e.g., age, as defined by passage number) or DAT Bmax value. The hypothesis is perhaps more satisfying with the amendment that the relative distribution of cocaine-binding DAT conformations is controlled by an unknown factor linked to cell state. The percentage of a given cocaine-binding DAT conformation could be altered, for example, by a cell state-specific DAT posttranslational modification or DAT complex formation/dissociation. The IC50 value would shift depending on the potency of cocaine at the dominant DAT conformation during a particular cell state; the dominant conformation would be stabilized by DAT modification or change in oligomerization state. It may be more accurate in this case to refer to DAT populations, as DAT molecules with varying degrees of posttranslational modification or that form homo- or hetero-oligomeric complexes may coexist, each imparting its own influence on the observed IC50 value. The DAT is N-terminally phosphorylated (Foster et al., 2002; Granas et al., 2003), an event that may be linked to cell surface trafficking alterations (Holton et al., 2005). Interestingly, prevention of DAT N-glycosylation altered cocaine, but not mazindol, DUIP at intact cells. Cocaine binding affinity in the same study (assessed for a membrane preparation) was independent of glycosylation state (Li et al., 2004). It is conceivable that DAT glycosylation controls transporter function by influencing the extent of DAT complex formation (Li et al., 2004). DAT homo-oligomerization impacts transporter function, and its extent may differ depending on whether a DAT substrate or blocker is present (Sorkina et al., 2003; Hastrup et al., 2003). Several proteins directly interact with the DAT and bear on its cell surface expression or function, including synuclein, the SNARE proteins syntaxin 1A and SNAP-25, and the kinase-associated proteins PICK1 and RACK1 (reviewed in Torres, 2006). In explaining a relationship between surface DAT levels and cocaine DUIP fluctuation, the relative distribution of DAT populations differing in posttranslational modifications, oligomerization extent, or association with DAT-interacting factors may be critical to DUIP. Ever-changing cell conditions that affect expression of the DAT, a relevant enzyme mediating posttranslational modifications, or a DAT-interacting factor could shift this DAT population distribution, in turn shifting the DUIP of cocaine.
The DUIP fluctuation phenomenon was found to extend to the neuronal N2A cell line (Table 6 and Fig. 4), suggesting physiological relevance. Future studies will investigate whether the DUIP fluctuation occurs in brain tissue, and will determine conditions necessary to support the phenomenon. The latter may involve characterizing DAT cell surface levels, discriminating coexisting DAT species varying in posttranslational modification, or determining which DAT modifiers or interacting factors are present in a given cell or tissue and testing for a correlation between cocaine DUIP and the presence or activity of the modifier/factor. The possibility that certain DAT conformations or populations are more relevant to the mechanism(s) of action of cocaine, amphetamines and other psychostimulants could be a new and exciting research direction for neurotransmitter transporter structure-function studies. Characterization of such conformations/populations may identify new cellular targets for anti-addiction medications development.
4. Experimental Procedure
4.1. Materials
[3H]-WIN 35,428 (~85 Ci/mmol) and [3H]-dopamine (~23.5 Ci/mmol) were obtained from PerkinElmer Life Sciences (Boston, MA). Nonradioactive WIN 35,428, cocaine, methylphenidate, and (+)-amphetamine were obtained from Research Triangle Institute (Research Triangle Park, NC) via the National Institute on Drug Abuse Division of Basic Research. Nonradioactive dopamine and ascorbic acid were obtained from Sigma Chemical Co. (St. Louis, MO); mazindol and benztropine were obtained from RBI/Sigma (Natick, MA). Scintillation counting materials and cell culture media and sera were from Fisher Scientific (Pittsburgh, PA). PolyFect transfection reagent was obtained from Qiagen (Los Angeles, CA). COS-7 and CHO-K1 cell lines were obtained from American Type Culture Collection (Manassas, VA). Naive N2A neuroblastoma cell lines were a gift of Dr. Margaret Gnegy (Univ. Michigan). GVA mounting solution was from Zymed (San Francisco, CA) and rat monoclonal anti-DAT antibody (MAB369) was from Chemicon (Temecula, CA); all other confocal microscopy reagents were obtained from Molecular Probes (Eugene, OR).
4.2. Cell culture and transfections
CHO-K1 cells stably transfected with WT DAT were prepared as previously described (Wang et al., 2003). These cells were grown at 37°C and 5% CO2 in F12K medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 mg/ml streptomycin and 100 μg/ml G-418. COS-7 and N2A neuroblastoma cells were employed for experiments requiring transient transfection of plasmid DNA. Both cell lines were maintained in “complete” DMEM media (supplemented with 10% FBS, 100 units/ml penicillin, 100 mg/ml streptomycin, and 20 mM L- glutamine). Cells were grown in 75 cm2 flasks and subcultured twice weekly.
Transient transfections were conducted via the PolyFect method with modification of the manufacturer’s protocol. A day before the transfection procedure, cells were seeded in 6-well plates (35 mm diameter wells) and incubated overnight at 37°C and 5% CO2 such that the cells would be 50–80% confluent on the day of transfection. Prior to transfection, cell monolayers were washed with 1 ml phosphate buffered saline (PBS), 1.5 ml complete DMEM media was added to the monolayer, and cells were maintained at 37°C and 5% CO2 until addition of DNA. Separately, 1500 ng wild type rDAT cDNA plasmid was diluted with 100 μl DMEM (containing no serum or antibiotics) and mixed by vortex for 10 seconds. (In experiments manipulating DAT expression level, the suboptimal amounts of 375 ng/well and 750 ng/well plasmid were employed). Ten μl PolyFect reagent was added to the DNA solution, followed by mixing and incubation at room temperature for 8 min to allow DNA-PolyFect complex formation. Subsequently, 600 μl complete DMEM was added to each tube and mixed by pipetting, the entire volume was immediately transferred to the cell monolayer, and cells were allowed to incubate at 37°C and 5% CO2 for 48 hours more before use in in vitro assays. The calcium phosphate transfection method (Graham and van der Eb, 1973) was found to be more effective, and employed, for N2A neuroblastoma cells.
4.3. Immunocytochemistry and confocal microscopy
COS-7 cells were seeded on coverslips placed in 6-well plates and grown to 40–60% confluence. Cells were transiently transfected on the following day with WT DAT plasmid or the vector control plasmid. After 48 hours, cells were fixed in 4% paraformaldehyde solution in PBS at room temperature for 15 min, rinsed once with PBS, and incubated with blocking-permeabilizing solution (5% goat serum, 1% BSA, and 0.1% Triton X-100 in PBS buffer solution) for 45 min at room temperature. Cells were next incubated with rat monoclonal anti-DAT antibody at 1:1000 dilution for 1 hr. The anti-DAT antibody solution was aspirated and cells were washed five times with PBS containing 0.1% Triton X-100 (TPBS), and incubated with a mixture of secondary antibody (goat anti-rat Alexa Fluor 488) at 1:500 dilution and rhodamine phalloidin at 1:250 dilution for 1 hr. After three washes in TPBS followed by two washes in PBS, coverslips were mounted on slides using GVA solution and left to dry overnight in the dark at 4°C. DAT protein was visualized using a Leica TCS-SP2 confocal laser microscope with an oil immersion 100x objective. Alexa 488 was excited at 488 nm with an argon/krypton laser and emission photons from 500–600 nm were accumulated by the photomultiplier tube. Rhodamine phalloidin was excited at 543 nm with a helium/neon laser and emission photons from 550 to 650 nm were accumulated. Images were quantitated using the accompanying Leica confocal software. Ten individual cells were selected randomly, and the mean intensity values for a defined pixel area for 10 different regions within the plasma membrane were determined.
4.4. [3H]-Dopamine uptake assays
Assays were conducted with cell monolayers in 6-well plates, at 22°C. Before substrate uptake commenced, the monolayer was washed 2 x 2 ml with “KRH buffer” (25 mM HEPES, pH 7.3, 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM Mg2SO4, 1.2 mM KH2PO4, 5.6 mM glucose). In uptake inhibition assays, the DAT inhibitor was added 10 min before initiation of uptake with 10 nM [3H]-dopamine and 50 mM ascorbic acid (AA) in KRH (1 ml) to duplicate cell monolayers. Five min after addition of the radiosubstrate, uptake was quenched by washing the monolayer with 2 x 2 ml KRH/AA. Cell monolayers were solubilized in 0.5 ml of 1% SDS, and transferred to scintillation vials for determination of incorporated tritium. Nonspecific uptake was assessed by inclusion of 10 μM mazindol, or 30 μM cocaine if mazindol was the drug to be assessed. Uptake inhibition experiments included nonradioactive DAT inhibitors at the following concentration ranges: Cocaine, 1 nM - 100 μM; WIN 35,428, 3 nM - 10 μM; mazindol, 3 nM - 10 μM; methylphenidate, 3 nM - 10 μM; benztropine, 0.1 nM–30 μM; (+)amphetamine, 1 nM–10 μM. In dopamine uptake saturation assays, cell monolayers were washed with KRH/AA, then incubated with a final dopamine concentration of 0.5–16 μM for 5 min; [3H]-dopamine was diluted with nonradioactive dopamine to obtain a specific activity of ~0.1 Ci/mmol. Total protein levels (Bradford) of monolayers prepared in parallel were determined. Km and Vmax values for transport and IC50 values for uptake inhibition were determined with GraphPad Prism 3.0 software (San Diego, CA). Tabular values reflect the mean ± s.e.m. for at least 3 independent experiments.
4.5. Ligand binding assays
[3H]-WIN 35,428 was the radioligand employed for all binding experiments. Assays establishing affinities of nonradioactive DAT inhibitors were conducted exactly as described above for the dopamine uptake inhibition assay except that [3H]-dopamine was replaced with 1 nM [3H]-WIN 35,428, and radioligand and nonradioactive competitor were incubated with cells for 15 min (the same incubation period allowed for an uptake blocker in the uptake assay) at 22°C. It was previously determined that in the absence of competitor, specific [3H]-WIN 35,428 binding did not increase appreciably after 15 minutes (data not shown). Nonradioactive competitor concentrations were as indicated above for uptake inhibition. Nonspecific binding was assessed by addition of 10 μM mazindol except when mazindol was the drug tested, in which case 30 μM cocaine was substituted. WIN 35,428 saturation binding experiments employed a nonradioactive WIN 35,428 concentration range of 1 nM - 10 μM, combined with 1 nM [3H]-WIN 35,428. Data were analyzed with GraphPad Prism 3.0 software to obtain Kd, Ki and Bmax values. Tabular values reflect the mean ± s.e.m. for at least 3 independent experiments.
Acknowledgments
We thank Yurong Huang and Corry D. Bondi for assistance with pharmacologic assays, Dr. John Pollock (Duquesne University) for useful discussions on aspects of the confocal microscopy experiments, and Dr. Margaret Gnegy (Univ. Michigan) for the generous gift of N2A neuroblastoma cells. Several of the drugs employed were generously provided by NIDA Drug Supply. This work was supported by grants from the National Institutes of Health (DA16604) and the Samuel and Emma Winters Foundation (to C.K.S.). Confocal microscopy support was provided by the National Science Foundation (Grant No. 0400776, to Duquesne University).
Abbreviations
- WIN 35
428, (−)-3β-(4-fluorophenyl)tropan-2β-carboxylic acid methyl ester tartrate
- KRH
Krebs/Ringer/HEPES buffer
- SDS
sodium dodecyl sulfate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bannon MJ, Whitty CJ. Age-related and regional differences in dopamine transporter mRNA expression in human midbrain. Neurology. 1997;48:969–977. doi: 10.1212/wnl.48.4.969. [DOI] [PubMed] [Google Scholar]
- Boja JW, Markham L, Patel A, Uhl G, Kuhar MJ. Expression of a single dopamine transporter cDNA can confer two cocaine binding sites. Neuroreport. 1992;3:247–248. doi: 10.1097/00001756-199203000-00007. [DOI] [PubMed] [Google Scholar]
- Boja JW, Rahman MA, Phillip A, Lewin AH, Carroll FI, Kuhar MJ. Isothiocyanate derivatives of cocaine: irreversible inhibition of ligand binding at the dopamine transporter. Mol Pharmacol. 1991;39:339–345. [PubMed] [Google Scholar]
- Chen N, Zhen J, Reith MEA. Mutation of Trp84 and Asp313 of the dopamine transporter reveals similar mode of binding interaction for GBR12909 and benztropine as opposed to cocaine. J Neurochem. 2004;89:853–864. doi: 10.1111/j.1471-4159.2004.02386.x. [DOI] [PubMed] [Google Scholar]
- Daws LC, Callaghan PD, Moron JA, Kahlig KM, Shippenberg TS, Javitch JA, Galli A. Cocaine increases dopamine uptake and cell surface expression of dopamine transporters. Biochem Biophys Res Commun. 2002;290:1545–1550. doi: 10.1006/bbrc.2002.6384. [DOI] [PubMed] [Google Scholar]
- Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Vaughan RA. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem. 2002;277:25178–25186. doi: 10.1074/jbc.M200294200. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gracz LM, Madras BK. [3H]WIN 35,428 ([3H]CFT) binds to multiple charge-states of the solubilized dopamine transporter in primate striatum. J Pharmacol Exp Ther. 1995;273:1224–1234. [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Granas C, Ferrer J, Loland CJ, Javitch JA, Gether U. N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J Biol Chem. 2003;278:4990–5000. doi: 10.1074/jbc.M205058200. [DOI] [PubMed] [Google Scholar]
- Hastrup H, Sen N, Javitch JA. The human dopamine transporter forms a tetramer in the plasma membrane: cross-linking of a cysteine in the fourth transmembrane segment is sensitive to cocaine analogs. J Biol Chem. 2003;278:45045–45048. doi: 10.1074/jbc.C300349200. [DOI] [PubMed] [Google Scholar]
- Holton KL, Loder MK, Melikian HE. Nonclassical, distinct endocytic signals dictate constitutive and PKC-regulated neurotransmitter transporter internalization. Nat Neurosci. 2005;8:881–888. doi: 10.1038/nn1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane upon amphetamine stimulation. Neuropharmacology. 2005;49:750–758. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Li LB, Chen N, Ramamoorthy S, Chi L, Cui XN, Wang LC, Reith MEA. The role of N-glycosylation in function and surface trafficking of the human dopamine transporter. J Biol Chem. 2004;279:21012–21020. doi: 10.1074/jbc.M311972200. [DOI] [PubMed] [Google Scholar]
- Little KY, Elmer LW, Zhong H, Scheys JO, Zhang L. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol. 2002;61:436–445. doi: 10.1124/mol.61.2.436. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Norregaard L, Litman T, Gether U. Generation of an activating Zn2+ switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transporter cycle. Proc Natl Acad Sci USA. 2002;99:1683–1688. doi: 10.1073/pnas.032386299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madras BK, Spealman RD, Fahey MA, Neumeyer JL, Saha JK, Milius RA. Cocaine receptors labeled by [3H]2 beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane. Mol Pharmacol. 1989;36:518–524. [PubMed] [Google Scholar]
- Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: Dissociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol. 1994;45:125–135. [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Reith MEA, Coffey LL. Structure-activity relationships for cocaine congeners in inhibiting dopamine uptake into rat brain synaptic vesicles and bovine chromaffin granule ghosts. J Pharmacol Exp Ther. 1994;271:1444–1452. [PubMed] [Google Scholar]
- Rothman RB, Becketts KM, Radesca LR, de Costa BR, Rice KC, Carroll FI, Dersch CM. Studies of the biogenic amine transporters. II. A brief study on the use of [3H]DA-uptake-inhibition to transporter-binding-inhibition ratios for the in vitro evaluation of putative cocaine antagonists. Life Sci. 1993;53:PL267–PL272. doi: 10.1016/0024-3205(93)90602-y. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Cadet JL, Akunne HC, Silverthorn ML, Baumann MH, Carroll FI, Rice KC, de Costa BR, Partilla JS, Wang JB. Studies of the biogenic amine transporters. IV Demonstration of a multiplicity of binding sites in rat caudate membranes for the cocaine analog [125I]RTI-55. J Pharmacol Exp Ther. 1994;270:296–309. [PubMed] [Google Scholar]
- Rudnick G. Mechanisms of biogenic amine neurotransmitter transporters. In: Reith MEA, editor. Neurotransmitter Transporters: Structure, Function, and Regulation. Humana Press; Totowa, NJ: 1997. pp. 73–100. [Google Scholar]
- Salvatore MF, Apparsundaram S, Gerhardt GA. Decreased plasma membrane expression of striatal dopamine transporter in aging. Neurobiol Aging. 2003;24:1147–1154. doi: 10.1016/s0197-4580(03)00129-5. [DOI] [PubMed] [Google Scholar]
- Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LMF, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: An internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci USA. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C. Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem. 1998;71:1289–1297. doi: 10.1046/j.1471-4159.1998.71031289.x. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A. Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J Biol Chem. 2003;278:28274–28283. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- Spealman RD, Madras BK, Bergman J. Effects of cocaine and related drugs in nonhuman primates. II Stimulant effects on schedule-controlled behavior. J Pharmacol Exp Ther. 1989;251:142–149. [PubMed] [Google Scholar]
- Torres GE. The dopamine transporter proteome. J Neurochem. 2006;97:3–10. doi: 10.1111/j.1471-4159.2006.03719.x. [DOI] [PubMed] [Google Scholar]
- Ukairo OT, Bondi CD, Newman AH, Kulkarni SS, Kozikowski AP, Pan S, Surratt CK. Recognition of benztropine by the dopamine transporter (DAT) differs from that of the classical dopamine uptake inhibitors cocaine, methylphenidate and mazindol as a function of a DAT transmembrane 1 aspartic acid residue. J Pharmacol Exp Ther. 2005;314:575–583. doi: 10.1124/jpet.105.085829. [DOI] [PubMed] [Google Scholar]
- Wang W, Sonders MS, Ukairo OT, Scott H, Kloetzel MK, Surratt CK. Dissociation of high-affinity cocaine analog binding and dopamine uptake inhibition at the dopamine transporter. Mol Pharmacol. 2003;64:430–439. doi: 10.1124/mol.64.2.430. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. Brain mechanisms of drug reward and euphoria. Psychiatr Med. 1985;3:445–460. [PubMed] [Google Scholar]